
Abstract
Background
Nitrate (NO3−) and ammonium (NH4+) are the primary forms of inorganic nitrogen (N) taken up by plant roots, and a lack of these N sources commonly limits plant growth. To better understand how NO3− and NH4+ differentially affect root system architecture, we analyzed the expression profiles of microRNAs and their targets in poplar roots treated with three forms of nitrogen S1 (NO3−), S2 (NH4NO3, normal), and S3 (NH4+) via RNA sequencing.
Results
The results revealed a total of 709 miRNAs. Among them, 57 significantly differentially expressed miRNAs and 28 differentially expressed miRNA-target pairs showed correlated expression profiles in S1 vs. S2. Thirty-six significantly differentially expressed miRNAs and 12 differentially expressed miRNA-target pairs showed correlated expression profiles in S3 vs. S2. In particular, NFYA3, a target of upregulated ptc-miR169i and ptc-miR169b, was downregulated in S1 vs. S2, while NFYA1, a target of upregulated ptc-miR169b, was downregulated in S3 vs. S2 and probably played an important role in the changes in root morphology observed when the poplar plants were treated with different N forms. Furthermore, the miRNA-target pairs ptc-miR169i/b-D6PKL2, ptc-miR393a-5p-AFB2, ptc-miR6445a-NAC14, ptc-miR172d-AP2, csi-miR396a-5p_R + 1_1ss21GA-EBP1, ath-miR396b-5p_R + 1-TPR4, and ptc-miR166a/b/c-ATHB-8 probably contributed to the changes in root morphology observed when poplar plants were treated with different N forms.
Conclusions
These results demonstrate that differentially expressed miRNAs and their targets play an important role in the regulation of the poplar root system architecture by different N forms.
Similar content being viewed by others
Background
Nitrogen (N) is one of the essential elements required by plants and plays key roles in their growth, development and morphological composition [1, 2]. In most higher plants, nitrate (NO3−) and ammonium (NH4+) are the main forms of inorganic N resources absorbed by plant roots from the soil [3, 4]. The mechanisms of NO3− and NH4+ absorption and utilization differ among plants, and different N forms can have differential effects on plant root morphology [5,6,7]. In Populus simonii × P. nigra [5] and Arabidopsis thaliana [8], NH4+ can inhibit primary root growth compared to that observed in the presence of NO3−, whereas NH4+ has a greater promoting effect than NO3− on lateral root development in A. thaliana [9]. Despite considerable research progress on the changes in root system architecture induced by different N forms [5, 8, 9], limited information is available regarding the underlying molecular mechanisms, especially the microRNA (miRNA) regulatory mechanisms underlying changes in root system architecture in the presence of different N forms.
miRNAs are highly conserved, endogenous, noncoding small RNAs with lengths of approximately 18–25 nucleotides. miRNAs inhibit posttranscriptional gene expression by inducing the cleavage of target genes or weakening translation, affecting organismal morphogenesis, development, and adaptability to environmental changes [10,11,12,13,14]. In recent years, increasing amounts of data have indicated that miRNAs are involved in herbaceous plant responses to various N stresses [6, 9, 15,16,17]. The first miRNA to be linked to the N response was miR167. In the pericycle cells of Arabidopsis roots, NO3− (5 mM) inhibits miR167 expression while promoting the expression of its target ARF8 (AUXIN RESPONSE FACTOR 8), which promotes lateral root initiation and emergence [9, 15]. In rice (Oryza sativa), miR166 targets RDD1(rice Dof daily fluctuations 1), which is related to NH4+ uptake and transport, leading to a change in root morphology [18]. These results, which were largely obtained in herbaceous plants, suggest that miRNAs are regulated by NO3− and NH4+ and in turn regulate their target genes and affect the plant root system architecture [9, 15, 19]. Nevertheless, we have a poor understanding of the miRNA regulatory mechanisms affecting the root system architecture of woody plants under different N conditions.
Poplar, which presents a large demand for N fertilizer, has become a model system for research on the molecular mechanisms of woody plant root growth, development, and responses to the environment [2]. Some progress has been achieved in understanding the root morphological and physiological characteristics of several fast-growing poplar trees (such as Populus × canescens and P. simonii × P. nigra) as well as the regulation of distinct genes in the presence of different N forms [5]. Our previous studies have mainly focused on the role of miRNAs in association with different NO3− absorption rates in different sections of poplar root tips [7]. However, few studies have focused on the links between morphological changes in roots and the expression levels of miRNAs and their targets when poplar plants are treated with different N forms (NO3− and NH4+ or both).
In this study, using a hydroponic culture system, we monitored the root system architecture and molecular changes in P. × canescens when treated with 1 mM NO3− (S1), 500 μM NH4NO3 (S2) or 1 mM NH4+ (S3) for 21 days. The primary goal of this work was to distinguish the morphological changes in poplar roots and analyze the potential miRNA-target pair regulation mechanisms of root morphological characteristics of poplar trees under different N forms. To achieve this goal, root length, lateral root initiation and lateral root density were measured. The results showed spatial variability in poplar root length and dry weight in the presence of different N forms. Through miRNA sequencing, degradome sequencing and transcriptomic sequencing, several candidate miRNA-target pairs associated with morphological changes in poplar roots were identified. The results of this work provide new ideas for the regulatory mechanisms whereby miRNA-mRNA regulatory networks change the root system architecture in response to different N forms, facilitating our understanding of the mechanism of root system architecture regulation in the presence of different N forms mediated by miRNA-target pairs.
Results
Morphological characteristics of P. × canescens roots in the presence of different N forms
P. × canescens roots showed significantly different phenotypes after 21 days of long-term hydroponic cultivation with 1 mM NO3− (S1), 500 μM NH4NO3 (S2, normal) or 1 mM NH4+ (S3). When the roots were grown in medium containing S1, root growth was significantly promoted compared to that in the presence of S2, whereas when the roots were grown in medium containing S3, root growth was significantly inhibited compared to that in the presence of S2. The length of the roots in S3 was 64% of that in S1 (Fig. 1a, b). The location of lateral root initiation in S1 was 19 mm from the root tip. The location of lateral root initiation in S2 was 10 mm from the root tip, and the location of lateral root initiation in S3 was 5 mm from the root tip (Fig. 1c). However, lateral root density was lower in S1 than in S2 and was lower in S2 than in S3 (Fig. 1d).

Morphological parameters of P. × canescens roots treated with different N forms for 21 days. Phenotypes of P. × canescens cultured with 1 mM NO3−, 500 μM NH4 NO3 or 1 mM NH4+ for 21 days (a). Root length (b), lateral root initiation (c) and lateral root density (d). Data are presented as the mean ± SEs (n = 18). a, b and c indicate significant differences based on one-way ANOVA and Duncan’s test (P < 0.05)
Identification of novel and known miRNAs
Sequencing yielded approximately 11.96 million raw reads per library. Redundant reads were filtered to obtain clean reads. A total of 6 724 370, 5 799 345 and 5 653 606 valid reads corresponded to 2 066 888, 1 621 481 and 1 586 438 unique reads in the S1, S2 and S3 libraries, respectively (Supplementary Table 2). The sequences of low-quality and those ≤ 18 nt were removed, and the sequences of 18–25 nt were retained. The proportions of total and unique small RNAs (sRNAs) ranging from 18–25 nt are listed in Supplementary Fig. 1, among which sRNAs of 24 nts accounted for the highest proportion.
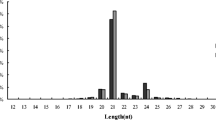
In total, 465 known miRNAs were identified in three libraries (Table 1, Supplementary Table 3), derived from 474 miRNA precursors belonging to 57 already documented miRNA families (Supplementary Table 4). Additionally, 29 novel miRNAs, corresponding to 26 miRNA precursors, were identified (Table 1, Supplementary Table 5). Among these miRNAs, 197 miRNAs were found in all three libraries, and 77, 5 and 19 miRNAs existed exclusively in S1, S2 and S3, respectively (Fig. 2a). The proportions of all identified miRNAs ranging from 18–25 nt are summarized in Fig. 2b, among which miRNAs of 21 nts accounted for the highest proportion.

Venn diagrams of the detected miRNAs (a) and the lengths of the miRNAs (b) in three P. × canescens libraries
Differentially expressed miRNAs in the presence of different N forms
In total, 54 known miRNAs belonging to 35 miRNA families and 3 novel miRNAs were significantly differentially expressed in S1 vs. S2 (Supplementary Table 6). Among these miRNAs, 44 upregulated and 13 downregulated miRNAs were identified in S1 vs. S2 (Fig. 3). The most upregulated miRNA identified in this comparison was PC-3p-86649_80 (15.13-fold), while the most downregulated was ptc-MIR169n-p5_2ss19TC21AC (5.79-fold). Additionally, 33 known miRNAs belonging to 19 miRNA families and 3 novel miRNAs were significantly differentially expressed in S3 vs. S2 (Supplementary Table 6). Twenty upregulated and sixteen downregulated miRNAs were identified in S3 vs. S2 (Fig. 3). Among them, the expression of PC-3p-86649_80 presented the greatest increase (9.05-fold), while the expression of ptc-miR6425a-5p showed the greatest decrease (10.66-fold).

Significantly differentially expressed miRNAs in S1 vs. S2 and S3 vs. S2 of P. × canescens. Red and blue indicate up- and downregulated, respectively
We also compared the 57 significantly differentially expressed miRNAs in S1 vs. S2 with the 36 significantly differentially expressed miRNAs in S3 vs. S2 identified above, and the comparison is shown in Fig. 3. Eight significantly differentially expressed miRNAs were shared by both S1 vs. S2 and S3 vs. S2. Among the eight common significantly differentially expressed miRNAs, four (mdm-miR169b_R-1, ptc-miR398a, ptc-miR6459a-3p_R-1 and PC-3p-86649_80) were upregulated in both S1 vs. S2 and S3 vs. S2, one miRNA (sbi-MIR398-p3_2ss5GA18AG) was downregulated in both S1 vs. S2 and S3 vs. S2, one miRNA (ptc-miR1444a_R+1) was upregulated in S1 vs. S2 but had the opposite expression pattern (downregulated) in S3 vs. S2, and two miRNAs (ptc-miR403c-5p and ptc-MIR6425e-p3) were downregulated in S1 vs. S2 but had the opposite expression pattern (upregulated) in S3 vs. S2. These results suggest that the regulatory mechanisms of these five miRNAs could be conserved between S1 vs. S2 and S3 vs. S2, while sbi-MIR398-p3_2ss5GA18AG, ptc-miR403c-5p and ptc-MIR6425e-p3 may undergo a different evolution under treatment with different N forms.
Validation of miRNAs sequence
To validate the miRNA sequence results, we selected twenty-two miRNAs in S1 vs. S2 and fifteen miRNAs in S3 vs. S2 for qRT–PCR (Supplementary Fig. 2). log2(FC) could be fitted by linear equations in both qRT–PCR and miRNA-seq. The slopes were 0.34 in S1 vs. S2 (Supplementary Fig. 2a) and -0.002 in S3 vs. S2 (Supplementary Fig. 2b). These values showed that log2(FC) in qRT–PCR was positively correlated with miRNA-seq in S1 vs. S2 and S3 vs. S2, indicating that miRNA-seq results were reliable.
Prediction of miRNAs targets by degradome sequencing
To verify the targets of these miRNAs, three mixed degradome library sequencing analyses were performed to identify miRNA targets using the same total RNA samples. Through degradome sequencing, 78 470 784 raw reads representing 20 632 172 unique raw reads were generated from the three mixed degradome libraries. After the adapter-missing reads were removed, 78 109 423 (99.54% of all reads) sequences that were mapped to 6 812 869 unique transcript reads were successfully mapped against the P. × canescens transcriptomic data. According to the signature abundance of each occupied transcript site, all transcripts that could be cleaved were divided into five categories. A total of 1826 targets of 78 known miRNA families were identified from degradome sequencing in S1 vs. S2. We also identified 11 targets of novel miRNAs, which targeted 89 transcripts according to degradome sequencing in S1 vs. S2 (Supplementary Table 7). Additionally, a total of 1813 targets of 76 known miRNA families were identified from the degradome in S3 vs. S2. We also identified 8 targets of novel miRNAs, which included 79 transcripts according to degradome sequencing in S3 vs. S2 (Supplementary Table 7).
Differentially expressed miRNAs and their targets involved in the regulation of the poplar root system architecture in the presence of different N forms
To better understand the functions of significantly differentially expressed miRNAs, we predicted 227 and 126 target genes of these miRNAs in S1 vs. S2 and S3 vs. S2, respectively (Supplementary Table 8). MapMan was used to assign these targets to functional categories (Supplementary Table 8). Several functional categories, including RNA regulation of transport, transcription and development, were associated with the regulation of the poplar root system architecture in the presence of different N forms (Supplementary Table 8). Among the identified target genes, AAP7 (an amino acid transmembrane transporter), which is targeted by gra-MIR8654c-p3_2ss12TG18AG family members, is responsible for the transport of amino acids. Several targets, including NFYA1/3 (nuclear transcription factor Y subunit A), ARFs (auxin response factors), AP2/EREBP (APETALA2/ethylene-responsive element binding protein) family members and NAC (NAC transcription factor) transcription factors, which belong to the miR169, miR160, miR172 and miR6445 families, accounted for the largest proportion of targets involved in the RNA-mediated regulation of transcription (Supplementary Table 8). These transcription factors are closely related to plant development and the response to nitrogen treatments [7, 9, 15, 20,21,22,23].
The functions of these 227 and 126 targets were further revealed by GO functional classification analysis. These identified target genes are predominantly involved in biological processes, cellular components and molecular functions. Twenty-five biological processes were identified, with the most frequent category being ‘transcription, DNA-templated’. Among the 15 cellular component categories, the most representative was ‘nucleus’. Finally, there were 10 molecular function categories, with the most abundant being ‘sequence-specific DNA binding transcription factor activity’ in S1 vs. S2 and S3 vs. S2 (Fig. S3a, b). This finding suggested that P. × canescens adopted complex and broad responsive approaches to accommodate the challenges of different N form conditions. KEGG analysis showed that the maximum target gene categories were differentially expressed for plant signal transduction pathways in both S1 vs. S2 and S3 vs. S2 (Fig. S4a, b). These pathways could be related to plant N physiological processes and root growth or development.
Correlations between miRNAs and their targets
Twenty-eight significantly differentially expressed miRNA-target pairs were identified in S1 vs. S2 (Table 2). Among them, seventeen miRNA-target pairs were negatively correlated. In contrast, the expression levels of the other 11 miRNA-target pairs were positively correlated. Meanwhile, twelve significantly differentially expressed miRNA-target pairs were identified in S3 vs. S2 (Table 2). The expression levels of 4 miRNA-target pairs displayed negative correlations. In contrast, the expression levels of the other 8 miRNA-target pairs showed positive correlations. The correlations between these miRNAs and their targets were verified by qRT–PCR (Fig. 4).

Validation of significantly differentially expressed miRNAs and their targets in P. × canescens in the presence of different N forms by sRNA-seq and qRT–PCR
Validation of the miRNA-target pairs
To validate the miRNA-target pairs predicted via degradome sequencing, transient coexpression experiments were performed in Nicotiana benthamiana leaves. Two randomly selected miRNA-mRNA pairs ptc-miR169i_1ss15TA and its target NFYA3 (Potri.018G064700.2) and stu-miR166b_R-1_1ss19CT and its target eIF (eukaryotic translation initiation factor) (Potri.007G119200.2) were selected for transient coexpression experiments. The NFYA3 transcript level was significantly decreased after NFYA3 and ptc-miR169i_1ss15TA coexpression in N. benthamiana leaves for 2 days, and the expression of the target gene eIF was significantly decreased under eIF and stu-miR166b_R-1_1ss19CT coexpression; thus, the expression levels of the target genes NFYA3 and eIF were decreased considerably by ptc-miR169i_1ss15TA and stu-miR166b_R-1_1ss19CT, respectively, in comparison with the expression levels observed under transient expression of the NFYA3 and eIF target genes alone (Fig. 5). These results suggest that NFYA3 and eIF are the targets of ptc-miR169i_1ss15TA and miR166b_R-1_1ss19CT, respectively.

Degradation effect of miRNAs on target genes. In the graph, validation of ptc-miR169i-NFYA3 (a) and ptc-miR166b-eIF (b) using transient coexpression assays in N. benthamiana leaves. The expression levels were quantified using qRT–PCR. Bars indicate means ± SE (n = 4). Different letters on the error bars indicate significant differences
miRNA-target pairs involved in the regulation of the poplar root system architecture in the presence of different N forms
A BLASTN search of Arabidopsis and P. × canescens genomes was performed to annotate these significantly differentially expressed target genes of significantly differentially expressed miRNAs in S1 vs. S2 and S3 vs. S2 (Supplementary Table 9). We examined their annotation and found that nine miRNA-target pairs in S1 vs. S2 were strongly associated with the regulation of the poplar root system architecture (Fig. 6). These miRNA target pairs included ptc-miR169i/b and their target NFYA3, ptc-miR169i/b and their target D6PKL2 (serine/threonine-protein kinase D6PKL2), ptc-miR393a-5p and its target AFB2 (AUXIN SIGNALING F-BOX2), ptc-miR6445a and its target NAC14, ptc-miR172d and its target AP2, ath-miR396b-5p_R + 1 and its target TPR4 (topless-related protein 4-like), and csi-miR396a-5p_R + 1_1ss21GA and its target EBP1 (ERBB-3 BINDING PROTEIN 1). Four miRNA-target pairs in S3 vs. S2 had close relationships with the regulation of the poplar root system architecture (Fig. 6). These miRNA target pairs included ptc-miR169b and its target NFYA1 and ptc-miR166a/b/c and their target ATHB-8 (homeobox-leucine zipper protein ATHB-8). In particular, NFYA3, a target of upregulated ptc-miR169i and ptc-miR169b, was downregulated in S1 vs. S2, while NFYA1, a target of upregulated ptc-miR169b, was downregulated in S3 vs. S2, indicating that miR169-NFYA pairs probably had an important and complex regulatory effect on poplar root system architecture under different N forms.

A simple model illustrating the effect of miRNA-target pairs on the morphological changes of poplar roots under treatment with different N forms
Discussion
NO3 − treatment causes lateral root elongation, while NH4 + treatment tends to increase lateral root initiation and differentiation
Plant roots display high plasticity in response to different N forms, and the morphological characteristics of plant roots are distinctly different when NO3− or NH4+ is used as the sole N source [5, 6, 24]. Previous studies have shown that the supply of NO3− to plants mainly promotes the elongation of lateral roots, whereas the supply of NH4+ increases the initiation and branching of lateral roots [5, 25, 26]. For example, the root length of NO3−- or NH4NO3-treated P. simonii × P. nigra seedlings was greater than that of NH4+-treated seedlings after 21 days of treatment [5]. In Arabidopsis, the local supply of NH4+ increased lateral root initiation and lateral root branching [27, 28], in accord with our results. In this study, the root lengths of NO3− - or NH4NO3-treated roots were greater than those of NH4+-treated plants, and NH4+ treatment resulted in earlier lateral root initiation than that observed under NO3− treatment. These results suggest that poplar roots display high plasticity in response to different N forms. We propose that the greater root length observed under the influence of NO3− could be a result of the increased supply of NO3− due to the upregulation of nitrate transporters (NRTs) in NO3−-treated roots (S3), since NRTs are NO3− transporters that enhance nitrate-dependent root elongation [14]. On the other hand, single or excessive NH4+ treatment can have adverse effects on plants, including altered root metabolism, plant ion imbalances, and foliar oxidative stress [29], leading to the growth and development of shorter roots. Liu et al. also speculated that high concentrations of NH4+ supplied as the sole nitrogen source would cause toxic symptoms and inhibit root growth in many plant species [8]. This may be the reason for the growth of shorter roots after NH4+ treatment.
miRNAs can regulate targets participating in the uptake and assimilation of different N forms to induce changes in root development, including that of lateral roots
The morphological changes caused by different N forms are closely related to changes at the molecular level. To better understand the effects of different N forms on root morphology and growth, we detected the differentially expressed miRNAs and their targets in poplar roots treated with different N forms. In this study, 57 and 36 miRNAs showed differential expression patterns in the NO3− and NH4+ libraries, respectively. These results suggested that NO3− treatment elicits stronger miRNA responses than NH4+ treatment. The difference may reflect the fact that NO3− supplied during long-term cultivation (21 days) can trigger a markedly different miRNA response in P. × canescens, which is consistent with previous research in rice [6]. In rice, the number of differentially expressed miRNAs identified in long-term NO3−-treated plants (16 miRNAs) was greater than that in NH4+-treated plants (11 miRNAs) [6]. As expected, the number of differentially expressed target genes observed under NO3− treatment was greater than that under NH4+ treatment (data not shown), which was consistent with previous findings in P. simonii × P. nigra [5]. The 227 differentially expressed genes identified by RNA-seq under NO3− treatment were greater than the 126 differentially expressed genes identified under NH4+ treatment in P. simonii × P. nigra [5]. Considering that NO3− is an important signaling molecule [30,31,32], it is reasonable to speculate that the effect of NO3− treatment on miRNA expression may be more pronounced than that of NH4+ treatment.
Some miRNA-target pairs were also involved in the changes in plant root morphology observed in the presence of different N forms [6, 7]. Several studies have shown that miR169 family members and their target NFYA transcripts play a role in the control of root growth in A. thaliana [22, 33], wheat [34], maize [35] and poplar [36]. In wheat, the overexpression of TaNFYA-B1 promoted the expression of TANRT1.1 and TANRT2.1, increased root NO3− influx and promoted lateral root growth [34]. NFYA3, which is homologous to TaNFYA-B1, showed a significantly downregulated expression profile in S1 vs. S2 and inhibited lateral root growth. In rice, miR169o-overexpressing plants are taller than WT plants under either normal or limiting NO3− conditions, and the predicted OsNFYA1/4 target genes display an exactly opposite expression pattern to that of osa-miR169o in response to NO3− deficiency [14]. NFYA1 was homologous to OsNFYA4, and it showed a significantly downregulated expression profile in S3 vs. S2 and inhibited primary root elongation. These results indicate that ptc-miR169 family members and their target NFYA genes might participate in the alteration of poplar root morphology in response to different N forms. Interestingly, the expression of ptc-miR169i_1ss15TA and ptc-miR169b_R-1 exhibited another target, D6PKL2, in S1 vs. S2. D6PKL2 kinases may directly phosphorylate PIN-FORMED (PIN) proteins [37], and d6pk mutants show defects in lateral root initiation, and this phenotype is correlated with a reduction in auxin transport in Arabidopsis [38]. These results suggest that members of the same miRNA family may have different targets that perform different functions affecting root morphology in response to different N forms in P. × canescens. Thus, a complex mechanism may modulate the expression profiles of miRNA family members and their targets.
miR393 targets transcripts that encode the auxin receptors TIR1 (transport inhibitor response protein 1), AFB1, AFB2, and AFB3. AtAFB3 is induced by NO3− in Arabidopsis root organs, which inhibits primary root elongation and promotes lateral root initiation [19]. In our research, AFB2, which is homologous to an AtAFB3 sequence, was reduced in root organs after 21 days of exposure to NO3− treatment, promoting primary root elongation and inhibiting lateral root initiation. This result differed from previous findings in Arabidopsis [19], possibly for the following reasons. First, compared with the Arabidopsis experiment, the treatment environment and conditions were different in this experiment. Second, the AFB gene family has different functions in annual herbs and perennial woody plants. Third, in Arabidopsis, AFB3 was induced after 1 h of NO3− treatment, but miR393 was also induced after NO3− treatment, which led to the degradation of its target gene AFB3 and reset AFB3 expression to basal levels. Therefore, AFB3 was induced by NO3− treatment and degraded by miR393 to achieve negative feedback regulation [19]. However, in poplar trees, whether the miR393-AFB2 pair has a negative feedback regulation mechanism under NO3− treatment needs to be further studied.
The target gene of ptc-miR6445a is the NAC14 transcription factor. A poplar homologous gene of AtNAC4 acts downstream of AFB3 to control the root response to NO3−, while nac4 mutants exhibit changes in lateral root growth in A. thaliana [39, 40]. In wheat, TaNAC2-5A can be directly bound to the promoter regions encoding nitrate transporters and glutamine synthetase genes, which affects root growth and the nitrate influx rate [41]. ptc-miR172d was markedly upregulated in S1 vs. S2. The target gene of ptc-miR172d was the AP2/ERF transcription factor, a poplar homologous gene of AtAP2/EREBP involved in lateral root formation in Arabidopsis [42]. The target of csi-miR396a-5p_R + 1_1ss21GA was EBP1. A poplar homologous gene of AtEBP1 is involved in dividing tissues during root development in Arabidopsis [43]. Another member of the miR396 family, ath-miR396b-5p_R + 1, shows the target gene TPR4. A poplar homologous gene of AtTPR4 was recruited by WUSCHEL RELATED HOMEOBOX 5 (WOX5) to inhibit target genes that promote differentiation to ensure the maintenance of stem cells [44, 45]. These results suggest that the csi-miR396a-5p_R + 1_1ss21GA-EBP1 and ath-miR396b-5p_R + 1-TPR4 miRNA-target pairs might participate in changes in P. × canescens root morphology by regulating the cell differentiation involved in NO3− treatment. The target of ptc-miR166a/b/c was ATHB8, and both miR166 and the target gene ATHB8 were upregulated in response to NH4+ treatment. A poplar homologous gene of AtATHB8 affects vascular development [46] and may be related to lateral root initiation and differentiation [46, 47].
Conclusions
Above all, these results identified several miRNA-target pairs that are involved in the assimilation of different N forms in P. × canescens and probably play important roles in poplar root morphogenesis. According to the present results, we propose a simple model of the response to different N forms in P. × canescens roots (Fig. 6). There were significant differences in the morphological characteristics of poplar roots among the three N form treatments after 21 days. Our global sRNA and transcriptome analysis showed that different N forms induce distinct miRNA-target pairs to regulate poplar root morphology, and regulation by these miRNA-target pairs forms the basis for the adaptation of poplar root morphology to different N forms. Notably, the ptc-miR169i/b-NFYA1/3, ptc-miR169i/b-D6PKL2, ptc-miR393a-5p-AFB2, ptc-miR6445a-NAC14, ptc-miR172d-AP2, csi-miR396a-5p_R + 1_1ss21GA-EBP1, ath-miR396b-5p_R + 1-TPR4, and ptc-miR166a/b/c-ATHB-8 pairs probably play key roles in the changes in poplar root morphology under different N forms. In future works, we suggest the selection of functional target genes associated with root morphology and functional assignment of the transcription factors identified under treatment with different N forms. These results indicate that the miRNA-target pairs that mediate global sRNA and transcriptomic reprogramming play an important role in the morphological acclimation of the roots of P. × canescens in response to different N forms.
Materials and methods
Plant cultivation and N treatments
Plantlets of P. tremula × P. alba (INRA 717-IB4 clone) were obtained by the micropropagation method. This plant stock was obtained and granted by Prof. Andrea Polle’s laboratory at the University of Göttingen in Germany. Experimental research on these plants complied institutional and national guidelines. Seedlings were grown in a climate chamber for 30 days. Thereafter, the plants were placed in plastic pots (10 L) filled with fine sand and cultivated in a growing chamber with conditions similar to those of the climate chamber. Every other day, 50 mL of modified Long Ashton (LA) solution was provided to each plant for 14 days, with the modified LA solution configured according to P Dluzniewska et al [48]. Subsequently, the plants were transplanted into N-free medium for 3 days, a time determined by reference to the results of S Balazadeh et al [49]. The plants were then transferred to LA nutrient solution for 21 days of hydroponics, in which 500 μM NaNO3 in LA solution was replaced by 1 mM NaNO3 (S1), 500 μM NH4NO3 (S2) and 1 mM NH4Cl (S3). The nutrient solution was changed every other day. At each treatment level, eighteen plants showing similar growth performance were used for further study.
Root measurements
Root lengths (length of taproots from the stem end to the root tip) were measured with a ruler. The root tip was compressed with a transparent agent (chloral hydrate: water: glycerin = 8 g: 2 ml: 1 ml) and observed and measured under a microscope. The location of lateral root initiation was measured under a microscope. The number of lateral roots within a range of 1 cm from the start site of lateral root initiation was counted under a microscope as an indicator of lateral root density. Six plants were measured at each treatment level, and three biological replicates were performed.
Harvesting
The whole roots of each plant were carefully cleaned with a modified LA solution containing 1 mM NaNO3, 500 μM NH4NO3 and 1 mM NH4Cl. Each root sample was wrapped in aluminum foil and snap-frozen in liquid nitrogen. A ball mill (MM400, Retsch, Haan, Germany) was used to grind each root sample into fine powder, which was stored at -80 °C. The same amounts of fine powder were extracted from the root samples of six plants and mixed evenly to form a mixed sample. Thus, three mixed samples were obtained.
sRNA RNA library construction and sequencing
Total RNA from the samples was isolated using TruSeq Small RNA Prep Kits (RS-200, Illumina, CA, USA) to obtain a small RNA library. Three small RNA libraries for each treatment level were constructed and sequenced on an Illumina HiSeq 2500 sequencer (Illumina, CA, USA) at LC Science (Hangzhou, China) according to the protocol recommended by the supplier. All of the small RNA sequences are available in the Sequence Read Archive (SRA) under project ID PRJNA631845.
Small RNA data analysis and miRNA identification
Known and novel miRNAs were identified according to the procedure proposed by J Zhou et al [7]. Raw Illumina sequencing data were further processed using the ACGT101-miR program (LC Sciences, Houston, Texas, USA) to remove adapter dimers, junk sequences, sequences of low complexity, and common RNA family members (rRNA, tRNA and snRNA). Then, the remaining clean and unique reads with lengths of 18–25 nt were aligned to the sequences from the miRBase 22.0 database to identify known miRNAs, with one mismatch allowed per sequence. Novel miRNA candidates were identified based on their genome location and stem–loop hairpin structure, and secondary structures were predicted using RNAfold software as suggested by S Griffiths-Jones et al [50]. In this study, pre-miRNAs with a stable hairpin structure and a minimal folding free energy index (MFEI) greater than 0.9 were considered novel miRNAs.
The frequency of the miRNAs in each sample was normalized to transcripts per million (TPM) values. The log2(fold change) of the differentially expressed miRNAs was determined by the TPM ratio of S1 vs. S2 or S3 vs. S2. P values of less than 0.05 were considered significantly differentially expressed miRNAs. Cytoscape software (3.5.1) was used to construct heatmaps with significantly differentially expressed miRNAs. S1_1, S1_2 and S1_3 represent three biological repeats of S1, S2_1, S2_2 and S2_3 represent three biological repeats of S2, and S3_1, S3_2 and S3_3 represent three biological repeats of S3.
Degradome sequencing and target identification
A total RNA purification kit (TRK1001, LianChuan (LC) Science, Hangzhou, China) was used to extract total RNA from the samples, and a NanoDrop 2000 spectrophotometer (Thermo, Wilmington, DE) was used to assess the RNA quality according to a 260/280 nm ratio > 2.0. Three independent RNA samples of equal quantities at the same treatment level were mixed to produce one degradation library. Then, three degradome samples (S1, S2 and S3) were sequenced on the Illumina HiSeq2500 platform (LC-BIO, Hangzhou, China). Degradome library construction was performed according to Xu X et al [51]. Data analysis was performed using CleaveLand 3.0 software with specific methods referred to C Addo-Quaye et al [52]. The degradome sequencing data were also submitted to the SRA under project ID PRJNA631839.
Identification and annotation of differentially expressed target genes
To determine the expression profiles of the target genes, three cDNA libraries from each treatment were constructed from the RNA samples, and transcriptomic sequencing was performed on an Illumina HiSeq2000/2500 sequencer (Illumina) at Lianchuan Science (Hangzhou, China) according to the protocol recommended by the supplier. The transcriptomic sequencing data were also submitted to the SRA under project ID PRJNA631840.
For each library, all sequences were processed by filtering out adaptor sequences and low-quality sequences. Subsequently, all clean tags were mapped to unigenes assembled for P. × canescens for annotation, and the degradome reads were mapped according to the P. × canescens transcriptomic data (http://aspendb.uga.edu/index.php/databases/spta-717-genome). After the miRNA, degradome sequencing and transcriptomic sequencing data were combined as described by M Pertea et al [53], the fragments per kilobase of exon per million fragments mapped (FPKM) algorithm was used to calculate the target gene expression level. The log2(fold change) values of differentially expressed target genes were calculated using the FPKMs of genes determined under S1 vs. S2 or S3 vs. S2. Differentially expressed target genes with P values of less than 0.05 and absolute log2 (fold change) values greater than 1 were considered significantly differentially expressed target genes.
Functional analysis of target genes
To clarify the functions of the target genes, a Gene Ontology (GO) functional classification [54] and KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analysis [55] were conducted. MapMan was also used to analyze the pathways [56].
Quantitative RT–PCR validation of significantly differentially expressed miRNAs and genes
To verify the significantly differentially expressed miRNAs and their target genes, the samples used for sequencing as described above were employed for qRT–PCR validation using the PrimeScript™ RT reagent Kit and TB Green qRT–PCR kit (Takara, Dalian, China) as suggested by J Zhou et al [12]. Thirty-seven significantly differentially expressed miRNAs and 12 significantly differentially expressed miRNA-target pairs were selected for qRT–PCR analysis. Each miRNA or target gene was analyzed in three replicates. The specific primers of mature miRNAs and their target genes are shown in Supplementary Table 1. Two genes, U6 and ACTIN2/7, were selected as reference genes for the validation of miRNAs and target genes, respectively [12].
Validation of miRNA-target pairs
To validate the cleavage of target genes by miRNAs, transient coexpression assays were performed using Nicotiana benthamiana leaves according to the methods of J Zhou et al [7]. Two selected miRNA-target pairs were chosen. In brief, fragments of the target genes were cloned and ligated into the pCAMBIA1300 vector for mRNA overexpression. Similarly, genomic DNA fragments encoding miRNA precursors were amplified using genomic DNA from root samples and sequence-specific primers (Supplementary Table 1). The amplified miRNA precursor fragments were inserted into the pCAMBIA2300 vectors for miRNA overexpression. Then, the two vectors were transformed into Agrobacterium tumefaciens strain GV3101 by electroporation. The Agrobacterial cell cultures were inoculated overnight at 28 °C. As previously described [57], equal amounts of Agrobacterial cell cultures containing miRNAs and their target genes were mixed and infiltrated into N. benthamiana leaves. After incubation in the dark for two days, the infiltrated leaves were harvested for qRT–PCR. analysis with gene-specific primers (Supplementary Table 1). Tubulin genes (Supplementary Table 1) served as the reference genes for target genes.
Statistical analysis
Statistical analysis of the data was performed using Statgraphics software (STN, St Louis, MO, USA). Before statistical analysis, the data were tested for a normal distribution. All the data were analyzed by one-way analysis of variance (ANOVA), and the different N forms tested were included as a factor. When the P value of the ANOVA F test was less than 0.05, the differences between the means were considered significant.
Availability of data and materials
All of the small RNA sequences are available in the Sequence Read Archive (SRA) under project ID PRJNA631845 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA631845). The degradome sequencing data were also submitted to the SRA under project ID PRJNA631839 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA631839). The transcriptomic sequencing data were also submitted to the SRA under project ID PRJNA631840 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA631840).
References
Correa R, Baum DA. Evolutionary transgenomics: prospects and challenges. Front Plant Sci. 2015;6:858.
Zhang C, Meng S, Li Y, Zhao Z. Net NH4+ and NO3− fluxes, and expression of NH4+ and NO3− transporter genes in roots of Populus simonii after acclimation to moderate salinity. Trees. 2014;28(6):1813–21.
Jackson LE, Burger M, Cavagnaro TR. Roots, nitrogen transformations, and ecosystem services. Plant Biol. 2008;59(1):341.
Rewald B, Kunze ME, Godbold DL. NH4: NO3 nutrition influence on biomass productivity and root respiration of poplar and willow clones. GCB Bioenergy. 2016;8(1):51–8.
Qu CP, Xu ZR, Hu YB, Lu Y, Yang CJ, Sun GY, Liu GJ. RNA-SEQ Reveals Transcriptional Level Changes of Poplar Roots in Different Forms of Nitrogen Treatments. Front Plant Sci. 2016;7:51.
Li H, Hu B, Wang W, Zhang Z, Liang Y, Gao X, Li P, Liu Y, Zhang L, Chu C. Identification of microRNAs in rice root in response to nitrate and ammonium. J Genet Genomics. 2016;43(11):651–61.
Zhou J, Lu Y, Shi WG, Deng SR, Luo ZB. Physiological characteristics and RNA sequencing in two root zones with contrasting nitrate assimilation of Populus × canescens. Tree Physiol. 2020;40(10):1392–404.
Yu H, Jiao B, Lu L, Wang P, Chen S, Liang C, Liu W. NetMiner-an ensemble pipeline for building genome-wide and high-quality gene co-expression network using massive-scale RNA-seq samples. PLoS One. 2018;13(2):e0192613.
Gifford ML, Dean A, Gutierrez RA, Coruzzi GM, Birnbaum KD. Cell-specific nitrogen responses mediate developmental plasticity. PNAS. 2008;105(2):803–8.
Ren Y, Sun F, Hou J, Chen L, Zhang Y, Kang X, Wang Y. Differential profiling analysis of miRNAs reveals a regulatory role in low N stress response of Populus. Funct Integr Genomics. 2015;15(1):93–105.
Tang G, Reinhart BJ, Bartel DP, Zamore PD. A biochemical framework for RNA silencing in plants. Genes Dev. 2003;17(1):49–63.
Zhou J, Liu M, Jiang J, Qiao G, Lin S, Li H, Xie L, Zhuo R. Expression profile of miRNAs in Populus cathayana L. and Salix matsudana Koidz under salt stress. Mol Biol Rep. 2012;39(9):8645–54.
Sunkar R, Li YF, Jagadeeswaran G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012;17(4):196–203.
Yu C, Chen Y, Cao Y, Chen H, Wang J, Bi YM, Tian F, Yang F, Rothstein SJ, Zhou X, et al. Overexpression of miR169o, an Overlapping MicroRNA in Response to Both Nitrogen Limitation and Bacterial Infection, Promotes Nitrogen Use Efficiency and Susceptibility to Bacterial Blight in Rice. Plant Cell Physiol. 2018;59(6):1234–47.
Gutierrez RA. Systems biology for enhanced plant nitrogen nutrition. Science. 2012;336(6089):1673–5.
Zuluaga DL, De Paola D, Janni M, Curci PL, Sonnante G. Durum wheat miRNAs in response to nitrogen starvation at the grain filling stage. PLoS One. 2017;12(8):e0183253.
Jiang L, Ball G, Hodgman C, Coules A, Zhao H, Lu C. Analysis of Gene Regulatory Networks of Maize in Response to Nitrogen. Genes (Basel). 2018;9(3):151.
Iwamoto M, Tagiri A. MicroRNA-targeted transcription factor gene RDD1 promotes nutrient ion uptake and accumulation in rice. The Plant journal : for cell and molecular biology. 2016;85(4):466–77.
Vidal EA, Araus V, Lu C, Parry G, Green PJ, Coruzzi GM, Gutierrez RA. Nitrate-responsive miR393/AFB3 regulatory module controls root system architecture in Arabidopsis thaliana. PNAS. 2010;107(9):4477–82.
He X, Ma H, Zhao X, Nie S, Li Y, Zhang Z, Shen Y, Chen Q, Lu Y, Lan H, et al. Comparative RNA-Seq Analysis Reveals That Regulatory Network of Maize Root Development Controls the Expression of Genes in Response to N Stress. PLoS One. 2016;11(3):e0151697.
Vidal EA, Moyano TC, Krouk G, Katari MS, Tanurdzic M, McCombie WR, Coruzzi GM, Gutiérrez RA. Integrated RNA-seq and sRNA-seq analysis identifies novel nitrate-responsive genes in Arabidopsis thaliana roots. BMC Genomics. 2013;14(1):701.
Sorin C, Declerck M, Christ A, Blein T, Ma L, Lelandais-Briere C, Njo MF, Beeckman T, Crespi M, Hartmann C. A miR169 isoform regulates specific NF-YA targets and root architecture in Arabidopsis. New Phytol. 2014;202(4):1197–211.
Zhao M, Ding H, Zhu J-K, Zhang F, Li W-X. Involvement of miR169 in the nitrogen-starvation responses in Arabidopsis. New Phytol. 2011;190(4):906–15.
Lupini A, Mercati F, Araniti F, Miller AJ, Sunseri F, Abenavoli MR. NAR2.1/NRT2.1 functional interaction with NO3(-) and H(+) fluxes in high-affinity nitrate transport in maize root regions. Plant Physiol Biochem: PPB / Societe francaise de physiologie vegetale. 2016;102:107–14.
Ristova D, Carre C, Pervent M, Medici A, Kim GJ, Scalia D, Ruffel S, Birnbaum KD, Lacombe B, Busch W, et al. Combinatorial interaction network of transcriptomic and phenotypic responses to nitrogen and hormones in the Arabidopsis thaliana root. SCIENCE SIGNALING. 2016;9(RS13):1–10.
Drew MC. Comparison of effects of a localized supply of phosphate, nitrate, ammonium and potassium on growth of seminal root system, and shoot, in barley. New Phytol. 1975;75:479–90.
Sasaki K, Kojima S. Identification of genomic regions regulating ammonium-dependent inhibition of primary root length in Arabidopsis thaliana. Soil Science and Plant Nutrition. 2018;64(6):746–51.
Lima JE, Kojima S, Takahashi H, von Wirén N. Ammonium Triggers Lateral Root Branching in Arabidopsis in an AMMONIUM TRANSPORTER1;3-Dependent Manner. Plant Cell. 2010;22(11):3621–33.
Prinsi B, Espen L. Time-Course of Metabolic and Proteomic Responses to Different Nitrate/Ammonium Availabilities in Roots and Leaves of Maize. Int J Mol Sci. 2018;19(8):2202.
Kant S. Understanding nitrate uptake, signaling and remobilisation for improving plant nitrogen use efficiency. Semin Cell Dev Biol. 2018;74:89–96.
Zhao X, Tang Z, Zhang H, Atianjoh FE, Zhao JY, Liang L, Wang W, Guan X, Kao SC, Tiwari V, et al. A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat Neurosci. 2013;16(8):1024–31.
Undurraga SF, Ibarra-Henriquez C, Fredes I, Alvarez JM, Gutierrez RA. Nitrate signaling and early responses in Arabidopsis roots. J Exp Bot. 2017;68(10):2541–51.
Leyva-Gonzalez MAMA, Ibarra-Laclette E, Cruz-Ramirez A, Herrera-Estrella L. Functional and transcriptome analysis reveals an acclimatization strategy for abiotic stress tolerance mediated by Arabidopsis NF-YA family members. PLoS One. 2012;7(10):e48138.
Qu B, He X, Wang J, Zhao Y, Teng W, Shao A, Zhao X, Ma W, Wang J, Li B, et al. A wheat CCAAT box-binding transcription factor increases the grain yield of wheat with less fertilizer input. Plant Physiol. 2015;167(2):411–23.
Luan M, Xu M, Lu Y, Zhang Q, Zhang L, Zhang C, Fan Y, Lang Z, Wang L. Family-wide survey of miR169s and NF-YAs and their expression profiles response to abiotic stress in maize roots. PLoS One. 2014;9(3):e91369.
Lu Y, Deng S, Li Z, Wu J, Liu Q, Liu W, Yu WJ, Zhang Y, Shi W, Zhou J, et al. Competing Endogenous RNA Networks Underlying Anatomical and Physiological Characteristics of Poplar Wood in Acclimation to Low Nitrogen Availability. Plant Cell Physiology. 2019;60(11):2478–95.
Barbosa IC, Zourelidou M, Willige BC, Weller B, Schwechheimer C. D6 PROTEIN KINASE activates auxin transport-dependent growth and PIN-FORMED phosphorylation at the plasma membrane. Dev Cell. 2014;29(6):674–85.
Zourelidou M, Muller I, Willige BC, Nill C, Jikumaru Y, Li H, Schwechheimer C. The polarly localized D6 PROTEIN KINASE is required for efficient auxin transport in Arabidopsis thaliana. Development. 2009;136(4):627–36.
Vidal EA, Álvarez JM, Gutiérrez RA. Nitrate regulation ofAFB3andNAC4gene expression inArabidopsisroots depends on NRT1.1 nitrate transport function. Plant Signal Behav. 2014;9(6):e28501.
Guo H-S, Xie Q, Fei J-F, Chua N-H. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for Arabidopsis lateral root development. The Plant Cell Online. 2005;17(5):1376–86.
He X, Qu B, Li W, Zhao X, Teng W, Ma W, Ren Y, Li B, Li Z, Tong Y. The Nitrate-Inducible NAC Transcription Factor TaNAC2-5A Controls Nitrate Response and Increases Wheat Yield. Plant Physiol. 2015;169(3):1991–2005.
Wang R, Zou J, Meng J, Wang J. Integrative analysis of genome-wide lncRNA and mRNA expression in newly synthesized Brassica hexaploids. Ecol Evol. 2018;8(12):6034–52.
Lokdarshi A, Papdi C, Pettkó-Szandtner A, Dorokhov S, Scheres B, Magyar Z, von Arnim AG, Bögre L, Horváth BM. ErbB-3 BINDING PROTEIN 1 Regulates Translation and Counteracts RETINOBLASTOMA RELATED to Maintain the Root Meristem. Plant Physiol. 2020;182(2):919–32.
Kieffer M, Stern Y, Cook H, Clerici E, Maulbetsch C, Laux T, Davies B. Analysis of the Transcription Factor WUSCHEL and Its Functional Homologue in Antirrhinum Reveals a Potential Mechanism for Their Roles in Meristem Maintenance. Plant Cell. 2006;18(3):560–73.
Pi L, Aichinger E, van der Graaff E, Llavata-Peris CI, Weijers D, Hennig L, Groot E, Laux T. Organizer-Derived WOX5 Signal Maintains Root Columella Stem Cells through Chromatin-Mediated Repression of CDF4 Expression. Dev Cell. 2015;33(5):576–88.
Simopoulos CMA, Weretilnyk EA, Golding GB. Prediction of plant lncRNA by ensemble machine learning classifiers. BMC Genomics. 2018;19(1):316.
Liu X, Li D, Zhang D, Yin D, Zhao Y, Ji C, Zhao X, Li X, He Q, Chen R, et al. A novel antisense long noncoding RNA, TWISTED LEAF, maintains leaf blade flattening by regulating its associated sense R2R3-MYB gene in rice. New Phytol. 2018;218(2):774–88.
Dluzniewska P, Gessler A, Dietrich H, Schnitzler JP, Teuber M, Rennenberg H. Nitrogen uptake and metabolism in Populus x canescens as affected by salinity. New Phytol. 2007;173(2):279–93.
Balazadeh S, Schildhauer J, Araujo WL, Munne-Bosch S, Fernie AR, Proost S, Humbeck K, Mueller-Roeber B. Reversal of senescence by N resupply to N-starved Arabidopsis thaliana: transcriptomic and metabolomic consequences. J Exp Bot. 2014;65(14):3975–92.
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36(Issue suppl_1):D154-158.
Xu X, Yin L, Ying Q, Song H, Xue D, Lai T, Xu M, Shen B, Wang H, Shi X. High-throughput sequencing and degradome analysis identify miRNAs and their targets involved in fruit senescence of Fragaria ananassa. PLoS One. 2013;8(8):e70959.
Addo-Quaye C, Miller W, Axtell MJ. CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics. 2009;25(1):130–1.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6.
Masoudi-Nejad A, Goto S, Endo TR, Kanehisa M. KEGG bioinformatics resource for plant genomics research. Methods Mol Biol. 2007;406:437–58.
Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M. Mapman: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004;37:914–39.
He XF, Fang YY, Feng L, Guo HS. Characterization of conserved and novel microRNAs and their targets, including a TuMV-induced TIR-NBS-LRR class R gene-derived novel miRNA in Brassica. FEBS Lett. 2008;582(16):2445–52.
Acknowledgements
The platform of the facilities from the State Key Laboratory of Tree Genetics and Breeding is acknowledged.
Funding
This work was supported by the National Natural Science Foundation of China (grant nos. 32171739 and 31500507) and the Fundamental Research Funds for the Central Non-profit Research Institution of CAF (grant no. CAFYBB2016QB005).
Author information
Authors and Affiliations
Contributions
ZJ conceived and designed the project. ZJ and WJ collected samples and extracted RNA. ZJ performed sequence data analysis. ZJ wrote the paper. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable in this study.
Consent for publication
Not applicable in this study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supporting informationAdditionalsupporting information can be found in the online version of this article: Supplementary Fig.1.Lengths of total small RNAs (a) and unique small RNAs (b) in the S1, S2 and S3libraries.Supplementary Fig.2. Validation of significantly differentially expressed miRNAs. Supplementary Fig. 3.Analysis of GO functional classification of identified differentially expressedtarget genes. S1 vs. S2 (a) and S3 vs. S2 (b). Supplementary Fig. 4. KEGG pathway analysis of identified differentially expressed target genes. S1 vs. S2(a) and S3 vs. S2 (b).
Additional file 2:
Supplementary Table1. Primers used for qRT–PCR.Supplementary Table 2. Distribution of smallRNAs in different categories.Supplementary Table 3.Identified known miRNAs.Supplementary Table 4.Identified miRNAfamilies.SupplementaryTable 5. Identified novel miRNAs.Supplementary Table 6.Significantly differentially expressed known and novelmiRNAs.Supplementary Table 7.Degradome sequencing forthe identification of target genes of known and novel miRNAs.Supplementary Table 8. Target genes ofsignificantly differentially expressed miRNAs.Supplementary Table 9.Annotated analysis of the significantly differentially expressed target genesof DEmiRNAs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, J., Wu, JT. Nitrate/ammonium-responsive microRNA-mRNA regulatory networks affect root system architecture in Populus × canescens. BMC Plant Biol 22, 96 (2022). https://doi.org/10.1186/s12870-022-03482-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03482-3