
Abstract
Background
Recent studies have shown that plant genomes have potentially undergone rampant horizontal gene transfer (HGT). In plant parasitic systems HGT appears to be facilitated by the intimate physical association between the parasite and its host. HGT in these systems has been invoked when a DNA sequence obtained from a parasite is placed phylogenetically very near to its host rather than with its closest relatives. Studies of HGT in parasitic plants have relied largely on the fortuitous discovery of gene phylogenies that indicate HGT, and no broad systematic search for HGT has been undertaken in parasitic systems where it is most expected to occur.
Results
We analyzed the transcriptomes of the holoparasite Rafflesia cantleyi Solms-Laubach and its obligate host Tetrastigma rafflesiae Miq. using phylogenomic approaches. Our analyses show that several dozen actively transcribed genes, most of which appear to be encoded in the nuclear genome, are likely of host origin. We also find that hundreds of vertically inherited genes (VGT) in this parasitic plant exhibit codon usage properties that are more similar to its host than to its closest relatives.
Conclusions
Our results establish for the first time a substantive number of HGTs in a plant host-parasite system. The elevated rate of unidirectional host-to- parasite gene transfer raises the possibility that HGTs may provide a fitness benefit to Rafflesia for maintaining these genes. Finally, a similar convergence in codon usage of VGTs has been shown in microbes with high HGT rates, which may help to explain the increase of HGTs in these parasitic plants.
Similar content being viewed by others


Background
Recent studies have shown that plant genomes have potentially undergone rampant horizontal gene transfer (HGT) [1–6]. In plant parasitic systems HGT appears to be facilitated by the intimate physical association between the parasite and its host [7–12]. HGT in these systems has been invoked when a DNA sequence obtained from a parasite is placed phylogenetically very near to its host rather than with its closest relatives [7]. Studies of HGT in parasitic plants have focused largely on single or few genes, and relied mostly on the fortuitous discovery of gene phylogenies that indicate HGT. No broad, systematic, genome-wide search for HGT has been undertaken in parasitic systems where it is most expected to occur. One parasitic plant clade that appears to be prone to HGT is Rafflesiaceae sensu stricto, which belong to the order Malpighiales [7, 13, 14] and whose members produce the largest flowers in the world. Rafflesiaceae are endophytic holoparasites, which lack leaves and stems. They parasitize a small number of species of Tetrastigma (i.e., members of the grapevine family, Vitaceae) hosts, on which they rely exclusively for their nutrition. The many reported host-to-parasite gene transfers from Tetrastigma to Rafflesiaceae make this system especially intriguing for more in-depth investigation [7, 9, 10]. Moreover, the association of Tetrastigma and Rafflesiaceae provides one of the best opportunities to study HGT in plant parasitic systems because i.) the parasites have a very narrow host specialization range, ii.) complete genomes are available for close relatives of the parasite (Manihot esculenta Crantz [15], Populus trichocarpa Torr. & Gray [16], and Ricinus communis L. [17]; Malpighiales) and its host (Vitis vinifera L. [18]; Vitaceae), and iii.) the host and parasite are separated by at least 115 million years of evolution [19]. These three factors make it easier to distinguish horizontally from vertically inherited gene regions.
To better understand HGT in this host-parasite plant system, we generated transcriptomic data for both the parasite (Rafflesia) and its obligate host (Tetrastigma). These data were analyzed using phylogenomic approaches that included whole-genome sequences from nine other plant model organisms. Our results show that several dozen actively transcribed, largely nuclear, genes are of host origin. These results are above false positive rates and establish for the first time a substantive number of HGTs in a plant host-parasite system. Moreover, we find that hundreds of vertically inherited genes in these parasitic plants exhibit codon usage properties that are more similar to their hosts than to their closest relatives, which may help to explain the increase of HGTs in these parasitic plants.
Results and discussion
Phylogenomic evidence for elevated rates of HGT in Rafflesia
We constructed and sequenced [20–22] complementary DNA (cDNA) libraries for Rafflesia cantleyi Solms-Laubach Additional file ( 1 :Figure S1) and its obligate host, Tetrastigma rafflesiae Miq. [23] Additional file ( 2: Figure S2). These cDNA transcripts were analyzed with protein-coding DNA sequences from nine other species whose whole genomes have been sequenced (Figure 1, see also Additional file 3: Table S1; Aquilegia coerulea James [15], Arabidopsis thaliana Heynh. [24], Manihot esculenta Medicago truncatula Gaertn. [15], Mimulus guttatus DC. [15], Oryza sativa L. [25], Populus trichocarpa Ricinus communis, and Vitis vinifera). Each Rafflesia and Tetrastigma transcript was placed into one of the following three categories on the basis of its phylogenetic position and support: vertical gene transfer (VGT), HGT, or unassigned. Transcripts whose placements were concordant with accepted species tree relationships were best explained as a result of VGT. VGT was inferred when Rafflesia or Tetrastigma transcripts were placed with ≥50% bootstrap support (BS) with their closest organismal relatives (i.e., Rafflesia with Manihot/Populus/Ricinus, and Tetrastigma with Vitis). Similarly, HGT was inferred when Rafflesia or Tetrastigma transcripts were placed with Vitis and Manihot/Populus/Ricinus transcripts, respectively. Transcripts that did not meet the criteria above, and were instead placed with the remaining included taxa, were left unassigned.

Accepted relationships between the twelve taxa included in our phylogenomic analyses [[26]]. The nine reference taxa for which complete genome sequences are available are labeled in black. Holoparasitic Rafflesia cantleyi is a member of Malpighiales (clade shown in red), and its obligate host Tetrastigma rafflesiae is a member of Vitaceae (clade shown in blue). The approximate divergence time between the parasite and host clade is 115 Ma [19]. Open flower of Rafflesia cantleyi shown in left inset (~0.5 m in diameter); floral bud in right inset shown attached to Tetrastigma rafflesiae host vine with leaves of the latter in foreground.
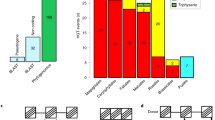
Not unexpectedly, the largest fraction of Rafflesia and Tetrastigma transcripts were found to have phylogenetic placements consistent with VGT (85.4% [n = 1979] and 96.9% [n = 1610], respectively). We also found dozens of Rafflesia transcripts that have phylogenetic placements consistent with HGT (49 transcripts, 2.1% of the observed transcripts), but far fewer Tetrastigma transcripts that have placements consistent with HGT (13 transcripts, 0.8%; Figure 2, see also Additional file 4: Figure S3). We further examined five different BS thresholds between 50% and 70% (i.e., 50%, 55%, 60%, 65%, and 70%); the percentages of putative HGT transcripts inferred for Rafflesia and Tetrastigma are similar across all thresholds (Additional file 5: Figure S4). Thus, our results are robust and are not sensitive to different levels of phylogenetic support.

Percentage of HGT transcripts in parasitic Rafflesia cantleyi and its obligate host Tetrastigma rafflesiae . These species are similarly contrasted against two autotrophic species that are closely related to Rafflesia, Manihot esculenta and Ricinus communis. Placements consistent with HGT in the latter two non-host, non-parasitic malpighialean taxa as defined for Rafflesia provide an estimate of the rate of non-HGT related factors that contribute to phylogenetic discordance. Species belonging to the Malpighiales and Vitaceae clade are shown in red and blue, respectively. The total number of transcripts used as denominators to calculate percentages are shown in parentheses.
Although striking discordance between gene trees and species trees can be indicative of HGT, gene trees can be incongruent with species trees due to a variety of factors other than HGT, including incomplete lineage sorting, incorrect orthology assessment (either due to inadequate paralog sampling or differential gene loss), small taxon sampling, and elevated substitution rates [27]. In order to assess the contribution of these factors, we first estimated the percentage of transcripts for Manihot esculenta and Ricinus communis that have placements consistent with HGT as defined above for Rafflesia (i.e., with Vitis). These two species are the closest relatives of Rafflesia and are not expected a priori to be prone to rampant HGT. Thus, they provide an estimate of the non-HGT related factors that may contribute to phylogenetic discordance. Our results indicate that only 0.3% and 0.4% of the transcripts in Manihot and Ricinus, respectively, have placements defined as HGT (Figure 2). Thus, the very small number of Tetrastigma transcripts whose phylogenetic placements are consistent with HGT is comparable to background rates in these non-host, non-parasitic taxa. We also compared nucleotide substitution rates between Rafflesia HGT and VGT transcripts, and with their homologous sister lineages (i.e., Rafflesia HGT with Vitis; Rafflesia VGT with Manihot and Ricinus). Although Rafflesia transcripts show elevated substitution rates overall (Additional file 6: Figure S5), Rafflesia HGT transcripts are not evolving significantly faster than Rafflesia VGT transcripts (p-value = 0.08 in Welch’s t test), nor to homologous Vitis transcripts (p-value = 0.42). Both the background HGT rate assessment and nucleotide substitution rate assessment indicate that our estimates of HGT in Rafflesia are robust.
In addition, most putative Rafflesia HGT transcripts showed their closest affinity to Vitis in our BLAST searches (Additional file 7: Table S2), which is consistent with these phylogenetic conclusions. Thus, we conclude that the observed Rafflesia transcripts show evidence that they originated via HGT from their obligate hosts. This is indicative of elevated rates of unidirectional host-to- parasite gene transfer in this system. Furthermore, our results suggest that rates of HGT in this eukaryotic parasitic system are on par with some prokaryotic organisms [28]. Moreover, our estimates of HGT are likely to be conservative because our transcriptome was built from a single organ from one developmental stage.
Genomic integration of Rafflesia HGT transcripts
Since host-to-parasite exchange of RNA is known to occur via translocation in the phloem [29], we used a multi-pronged approach to confirm that our results cannot be attributed to host contamination (see also Methods). Importantly, if our sample preparations were contaminated, we would not expect the high degree of sequence divergence we observed between putative HGT transcripts of Rafflesia and homologous transcripts of Tetrastigma. Of the 49 HGT transcripts from Rafflesia that could be directly compared with Tetrastigma, sequence divergence ranged from 0.05%–39.7% (mean divergence, 22.5%). This indicates that some period of evolution has elapsed since the time of gene transfer. Furthermore, assuming synteny between Tetrastigma and Vitis, the Rafflesia HGT transcripts most likely originated from multiple chromosomes in the host (Additional file 7: Table S2), which suggests a series of episodic HGT events.
To further verify genomic integration of these Rafflesia HGT transcripts, we also sequenced the genomic DNA (gDNA) from the same floral organ used for Rafflesia cDNA library construction. Our next-generation sequencing of Rafflesia gDNA verified a large percentage of the transgenes (63.3%, 31 of 49 HGT transcripts) identified from our cDNA analyses, independent of the bootstrap support threshold (Additional file 8: Figure S6) or transcript length (Additional file 9: Figure S7). The percentage of HGT transcripts verified to be integrated into the genome of Rafflesia was nearly identical to the percentage of verified VGTs (61.5%, 1218 of 1979 transcripts). Thus, our identification of putative HGTs does not appear to be due to contamination of the mRNA pool. Additionally, of the 31 Rafflesia HGT transcripts verified from our next-generation sequencing of genomic DNA, 16 show the presence of an intron (Additional file 7: Table S2 and Additional file 10: Figure S8). Although only two introns were covered in their entirety by our gDNA sequencing, all introns possess the characteristic GT or AG splice site [30]. This suggests that the source of many of these integrated HGTs at the time of transfer was likely genomic DNA, rather than processed mRNA transcripts. Together, these results indicate that the HGTs identified here are indeed integrated into the Rafflesia genome.
Gene location and function of Rafflesia HGT transcripts
Most previous reports of parasitic plant HGTs appear to involve only the movement of gene regions that reside in the mitochondrial genome [2]. Our results confirm recent findings in the parasitic plant Striga that some horizontally transferred genes likely reside in the nucleus [31] and indicate that perhaps many of these transgenes originated from the nuclear genomes of their hosts. Homologues of nearly all HGT regions inferred in Rafflesia are localized in the nuclear genomes of the other reference genomes examined here (47 are nuclear and two are mitochondrial; Additional file 7: Table S2).
These HGTs represent a wide range of cellular functions as determined by gene annotation data, including roles related to respiration, metabolism, mitochondrial translation, and protein turnover, to name a few (Additional file 7: Table S2). A natural extension of these results is to examine the extent to which these transgenes are functional in their recipient species. Many previously reported transgenes in plants appear to be non-functional and often coexist with a native, functional homologue [1]. Although some studies have shown that a small number of transgenes are likely transcribed [9], none have been convincingly demonstrated to be functional in their recipient lineages. In contrast, we find that Rafflesia HGTs are expressed at levels comparable to VGTs (Additional file 11: Figure S9). This suggests that these HGTs likely have functional promoters and therefore may play a role in cellular function. In addition, only 11 of the 49 HGTs had homologous VGTs expressed in the same transcript pool (Additional file 7: Table S2), which suggests that the HGT may have replaced the VGT function in some cases. The observed expression levels of the HGTs further rules out the possibility that the HGTs are due to contaminating RNA, which would not be present at native levels, and confirms that HGTs have been integrated into the Rafflesia genome.
Convergent coding properties of rafflesia VGT transcripts
Codon and dinucleotide usage patterns (i.e., coding properties) are phylogenetically conserved within clades [32], but it has been demonstrated that the successful integration of foreign genes may depend on the extent to which they possess compatible coding properties with a recipient genome at the time of transfer [33]. In light of this, we investigated whether genomic convergence may have also occurred in Rafflesia. We characterized coding properties in all VGTs for Malpighiales (Manihot [n = 5093], Populus [n = 3423], Rafflesia [n = 1979], and Ricinus [n = 4781]). We found that a significant percentage of Rafflesia VGT transcripts (29.8% [n = 590]; exact binomial test, p-value < 1×10−5) have coding properties more similar to a Vitis gene in the same cluster than to any gene from their closest relatives, Manihot or Ricinus (Figure 3). In contrast, Manihot and Ricinus are most similar to Vitis only 9.6% and 7.0% of the time, respectively. Additionally, the expression level of Rafflesia VGTs is positively correlated with similarity to Vitis-like coding patterns (p-value = 1.4×10−7), suggesting that the convergent host-like coding patterns we identify in some Rafflesia VGTs are biologically meaningful. Although results from these analyses should be interpreted cautiously, especially due to our inability to assess causality, one explanation of our results is that a substantial number of VGTs in Rafflesia may have evolved convergently to more closely match their hosts’ translational requirements. Strikingly similar patterns of convergent host-parasite codon usage have also recently been reported for honeybees and their associated viral pathogens [34].

Percentage of VGT transcripts from Rafflesia cantleyi , Manihot esculenta , and Ricinus communis that exhibit coding properties (nucleotide, codon, and dinucleotide usage) more similar to Vitis vinifera than to each other. The coding affinity for each gene was determined by calculating the smallest χ2 distance to genes from other genomes in the cluster. The total number of VGT transcripts used as denominators to calculate percentages are shown in parentheses.
Conclusions
The elevated rate of unidirectional gene transfer from Tetrastigma to Rafflesia and the apparent pattern of convergent host codon usage in vertically inherited Rafflesia genes raises the possibility that there may be a fitness benefit to Rafflesia for maintaining genes that are more host-like. A critical component of a parasite’s ability to maximize resource extraction is to minimize the ability of the host to detect and mount a defense response to the parasite. The bacterium that causes Legionnaires’ disease in humans appears to have acquired dozens of eukaryotic proteins via HGT that alter host cell functions to its advantage [35]. Similarly, in plants the obligate bacterial pathogen of citrus trees encodes a gene that was horizontally transferred; its product mimics a host protein that has been shown to limit the host’s defense response [36]. Our demonstration of HGT and VGT host codon usage convergence in Rafflesia raises the possibility that it may similarly express host-like genes to manipulate its host to its advantage. Our hypothesis that Rafflesia may be engaged in such genomic deception requires further experimental work and examination in Rafflesiaceae and in other plant parasitic systems.
Methods
Molecular techniques and next-generation sequencing
Total RNA and gDNA were obtained from the holoparasite, Rafflesia cantleyi from peninsular Malaysia. Total RNA was extracted using the RNAqueous and Plant RNA Isolation Aid kits (Ambion, Inc.), and treated with the TURBO DNA- free kit (Ambion, Inc.) at 37°C for 4 hours to remove residual DNA. The cDNA library was synthesized from total RNA following the protocols of Novaes et al. [20]. gDNA was extracted using the DNeasy Plant Mini kit (Qiagen, Inc.), and treated with RNAase A at 60°C for 1.5 hours to remove any residual RNA contamination. Illumina paired-end libraries were prepared for both cDNA and gDNA following the protocols of Bentley et al. [21]. Each library was sequenced in one lane of the Genome Analyzer II (Illumina, Inc.) with paired-end 150 base pairs (bp) read lengths at the FAS Center for Systems Biology at Harvard University (http://sysbio.harvard.edu/csb/resources/instrumentation/sequencing_illumina.html). Leaf, tendril, and stem tissue was obtained from an unparasitized specimen of Tetrastigma rafflesiae at the Missouri Botanical Garden (St. Louis, Missouri, USA). The cDNA library was prepared in a similar way as above and sequenced separately on a GS-FLX (Roche, Inc.) at the Environmental Genomics Core facility at the University of South Carolina (http://engencore.sc.edu/) following the protocols of Margulies et al. [22]. Tremendous care was taken to avoid and/or detect host or lab contamination in our sample preparation. First, a single perigone lobe from an unopened floral bud of Rafflesia was used for extractions of total RNA and gDNA. These perigone lobes are well protected by the outer bracts and are far from the zone of direct physical contact with tissue of Tetrastigma. Second, our extractions of total RNA and gDNA from Rafflesia were performed separate from any extractions of Tetrastigma; thus, laboratory contamination of our Rafflesia RNA or DNA with Tetrastigma can be eliminated. Third, we performed a PCR assay to assess the integrity of our Rafflesia and Tetrastigma gDNAs. We PCR-screened for the presence of the plastid genes matK and rbcL, which are universally present in autotrophic plants like Tetrastigma, but are absent in Rafflesia[37]. Both genes were easily amplified from Tetrastigma gDNA, but not from our Rafflesia gDNA, indicating that our Rafflesia gDNA extraction was likely free of host contamination. In addition, we PCR-screened gDNAs for the mitochondrial gene matR from Rafflesia and Tetrastigma, which is present in both species [7, 13, 14]. This gene amplified easily from our gDNAs and direct Sanger sequencing of PCR-products produced unique (i.e., singular) sequences from the host and parasite, respectively. There was no ambiguity in the matR sequence chromatograms for these samples, which would be expected if they were cross- contaminated. The cDNA reads from next-generation sequencing were first assembled using Oases v0.1.21 (http://www.ebi.ac.uk/~zerbino/oases/) with default settings, and then translated into amino acid sequences with prot4EST v2.2 [38]. Transcripts shorter than 30 amino acids were deemed too short for analysis and were discarded [20]. All sequence data have been deposited at the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) with accession #SRA052224.
Orthology assignment and phylogenomic analyses
Nine species available from whole genome sequencing projects (Additional file 3: Table S1) were included with Rafflesia and Tetrastigma to establish orthology and build our datasets for phylogenetic analysis following Dunn et al. [39]. Only those genes clusters that included at least Oryza (for outgroup rooting), Rafflesia/Tetrastigma (transcripts under investigation), Manihot/Populus/Ricinus (close Malpighiales relatives of the parasite), and Vitis (close Vitaceae relative of the host) were retained. The best maximum likelihood tree for each cluster group was inferred using RAxML v7.2.8 [40] under the GTRGAMMA nucleotide substitution model. BS values were estimated similarly using the rapid bootstrap algorithm with 100 replicates as implemented in RAxML.
Verification of genomic integration of Rafflesia HGT transcripts
Rafflesia gDNA Illumina reads were mapped onto assembled Rafflesia cDNA transcripts using Bowtie v0.12.7 [41]. To avoid complications with intron regions we first divided each 150 bp Illumina read into multiple 25 bp fragments following Kim and Salzberg [42], and then mapped each read onto the assembled cDNA transcripts with zero mismatches.
Inferring the gene location and function of Rafflesia HGT transcripts
Rafflesia HGT transcripts were BLAST searched against the NCBI nucleotide sequence database (March 1, 2011) using BLASTN v2.2.25 [43]. Searches were first performed using megablast (high similarity search). For sequences that failed to match existing sequences with high confidence, we then used more dissimilar (discontiguous megablast) searches. Most sequences had high- confidence BLAST hits in both of these searches. Where higher similarity searches retrieved no hits, we used blastn. Putative gene location and function were inferred based on the highest confidence hit with a functional annotation.
Gene expression level analysis
Rafflesia cDNA Illumina reads were re-mapped onto the assembled Rafflesia cDNA transcripts using Bowtie v0.12.7 [41] as described above. Illumina reads that re-mapped onto each transcript were summed and normalized using the formula:
where N is the number of mapped Illumina reads for a given transcript, L is the transcript length, k is the minimum number of base pairs which must overlap between each Illumina read and the transcript (k = 25 for this analysis), and l is the read length (l = 25). R was further normalized for each transcript to the standard reads per kilobase per million reads (i.e., RPKM; [44]).
Characterizing coding properties of Rafflesia VGT transcripts
The VGT transcripts were first converted into four nucleotide frequencies (i.e., A, C, G, and T), 16 dinucleotide frequencies, and 61 codon frequencies (excluding stop codons), and normalized by the length of each transcript. Next, χ2 distances were calculated between each transcript within a gene cluster [45]. Finally, we chose the smallest χ2 distance between the gene of interest and homologues from other species within the cluster (i.e., for a Rafflesia gene, whether it is closest to Manihot or Ricinus [Malpighiales], or Vitis). If the transcript was closer to Malpighiales, then it was assigned to be Mapighiales-like; otherwise, it was assigned to be Vitis-like.
Abbreviations
- bp:
-
Base pair
- BS:
-
Bootstrap support
- cDNA:
-
Complementary DNA
- gDNA:
-
Genomic DNA
- HGT:
-
Horizontal gene transfer
- RPKM:
-
Reads per kilobase per million reads
- VGT:
-
Vertical gene transfer.
References
Keeling PJ, Palmer JD: Horizontal gene transfer in eukaryotic evolution. Nat Rev Genet. 2008, 9: 605-618. 10.1038/nrg2386.
Richardson AO, Palmer JD: Horizontal gene transfer in plants. J Exp Bot. 2007, 58: 1-9.
Stegemann S, Keuthe M, Greiner S, Bock R: Horizontal transfer of chloroplast genomes between plant species. Proc Natl Acad Sci USA. 2012, 109: 2434-2438. 10.1073/pnas.1114076109.
Stegemann S, Bock R: Exchange of genetic material between cells in plant tissue grafts. Science. 2009, 324: 649-651. 10.1126/science.1170397.
Bock R: The give-and-take of DNA: horizontal gene transfer in plants. Trends Plant Sci. 2010, 15: 11-22. 10.1016/j.tplants.2009.10.001.
Christin P-A, Edwards EJ, Besnard G, Boxall SF, Gregory R, Kellogg EA, Hartwell J, Osborne CP: Adaptive evolution of C4 photosynthesis through recurrent lateral gene transfer. Curr Biol. 2012, 22: 445-449. 10.1016/j.cub.2012.01.054.
Davis CC, Wurdack KJ: Host-to-parasite gene transfer in flowering plants: phylogenetic evidence from Malpighiales. Science. 2004, 305: 676-678. 10.1126/science.1100671.
Mower JP, Stefanovic S, Young GJ, Palmer JD: Gene transfer from parasitic to host plants. Nature. 2004, 432: 165-166.
Barkman TJ, McNeal JR, Lim SH, Coat G, Croom HB, Young ND, dePamphilis CW: Mitochondrial DNA suggests at least 11 origins of parasitism in angiosperms and reveals genomic chimerism in parasitic plants. BMC Evol Biol. 2007, 7: 248-10.1186/1471-2148-7-248.
Nickrent DL, Blarer A, Qiu Y-L, Vidal-Russell R, Anderson FE: Phylogenetic inference in Rafflesiales: the influence of rate heterogeneity and horizontal gene transfer. BMC Evol Biol. 2004, 4: 40-10.1186/1471-2148-4-40.
Park JM, Manen JF, Schneeweiss GM: Horizontal gene transfer of a plastid gene in the non-photosynthetic flowering plants Orobanche and Phelipanche (Orobanchaceae). Mol Phylog Evol. 2007, 43: 974-985. 10.1016/j.ympev.2006.10.011.
Davis CC, Anderson WR, Wurdack KJ: Gene transfer from a parasitic flowering plant to a fern. Proc R Soc B. 2005, 272: 2237-2242. 10.1098/rspb.2005.3226.
Davis CC, Latvis M, Nickrent DL, Wurdack KJ, Baum DA: Floral gigantism in Rafflesiaceae. Science. 1812, 2007: 315-
Barkman TJ, Lim S-K, Mat Salleh K, Nais J: Mitochondrial DNA sequences reveal the photosynthetic relatives of Rafflesia, the world’s largest flower. Proc Natl Acad Sci USA. 2004, 101: 787-792. 10.1073/pnas.0305562101.
Goodstein DM, Shu SQ, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N: Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40: D1178-D1186. 10.1093/nar/gkr944.
Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A: The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science. 2006, 313: 1596-1604. 10.1126/science.1128691.
Chan AP, Crabtree J, Zhao Q, Lorenzi H, Orvis J, Puiu D, Melake-Berhan A, Jones KM, Redman J, Chen G: Draft genome sequence of the oilseed species Ricinus communis. Nat Biotechnol. 2010, 28: 951-956. 10.1038/nbt.1674.
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C: The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007, 449: 463-467. 10.1038/nature06148.
Wikström N, Savolainen V, Chase MW: Evolution of the angiosperms: calibrating the family tree. Proc R Soc Lond B. 2001, 268: 2211-2220. 10.1098/rspb.2001.1782.
Novaes E, Drost DR, Farmerie WG, Pappas GJ, Grattapaglia D, Sederoff RR, Kirst M: High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics. 2008, 9: 312-10.1186/1471-2164-9-312.
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR: Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008, 456: 53-59. 10.1038/nature07517.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z: Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005, 437: 376-380.
Veldkamp JF: The correct name for the Tetrastigma (Vitaceae) host of Rafflesia (Rafflesiaceae) in Malesia and a (not so) new species. Reinwardtia. 2008, 12 (4): 261-265.
Swarbreck D, Wilks C, Lamesch P, Berardini TZ, Garcia-Hernandez M, Foerster H, Li D, Meyer T, Muller R, Ploetz L: The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Res. 2008, 36: D1009-D1014.
Ouyang S, Zhu W, Hamilton J, Lin H, Campbell M, Childs K, Thibaud- Nissen F, Malek RL, Lee Y, Zheng L: The TIGR Rice Genome Annotation Resource: Improvements and new features. Nucleic Acids Res. 2007, 35: D883-D887. 10.1093/nar/gkl976.
APG: An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot J Linn Soc. 2009, 161: 105-121.
Felsenstein J: Inferring phylogenies. 2004, Massachusetts: Sinauer Associates, Sunderland
Ochman H, Lawrence JG, Groisman EA: Lateral gene transfer and the nature of bacterial innovation. Nature. 2000, 405: 299-304. 10.1038/35012500.
David-Schwartz R, Runo S, Townsley B, Machuka J, Sinha N: Long-distance transport of mRNA via parenchyma cells and phloem across the host-parasite junction in Cuscuta. New Phytol. 2008, 179: 1133-1141. 10.1111/j.1469-8137.2008.02540.x.
Kitamura-Abe S, Itoh H, Washio T, Tsutsumi A, Tomita M: Characterization of the splice sites in GT-AG and GC-AG introns in higher eukaryotes using full-length cDNAs. J Bioinform Comput Biol. 2004, 2: 309-331. 10.1142/S0219720004000570.
Yoshida S, Maruyama S, Nozaki H, Shirasu K: Horizontal gene transfer by the parasitic plant Striga hermonthica. Science. 2010, 328: 1128-10.1126/science.1187145.
Lawrence JG, Ochman H: Amelioration of bacterial genomes: rates of change and exchange. J Mol Evol. 1997, 44: 383-397. 10.1007/PL00006158.
Medrano-Soto A, Moreno-Hagelsieb G, Vinuesa P, Christen JA, Collado-Vides J: Successful lateral transfer requires codon usage compatibility between foreign genes and recipient genomes. Mol Biol Evol. 2004, 21: 1884-1894. 10.1093/molbev/msh202.
Chantawannakul P, Cutler RW: Convergent host-parasite codon usage between honeybee and bee associated viral genomes. J Invertebr Pathol. 2008, 98: 206-210. 10.1016/j.jip.2008.02.016.
de Felipe KS, Pampou S, Jovanovic OS, Pericone CD, Ye SF, Kalachikov S, Shuman HA: Evidence for acquisition of Legionella Type IV secretion substrates via interdomain horizontal gene transfer. J Bacteriol. 2005, 187: 7716-7726. 10.1128/JB.187.22.7716-7726.2005.
Garavaglia BS, Thomas L, Gottig N, Zimaro T, Garofalo CG, Gehring C, Ottado J: Shedding light on the role of photosynthesis in pathogen colonization and host defense. Commun Integr Biol. 2010, 3: 382-384. 10.4161/cib.3.4.12029.
Nickrent DL, Duff RJ, Colwell AE, Wolfe AD, Young ND, Steiner KE, dePamphilis CW: Molecular phylogenetic and evolutionary studies of parasitic plants. Molecular systematics of plants II: DNA sequencing. Edited by: Soltis DE, Soltis PS, Doyle JJ. 1998, Boston: Kluwer, 211-241. 2
Wasmuth JD, Blaxter ML: prot4EST: translating expressed sequence tags from neglected genomes. BMC Bioinforma. 2004, 5: 187-10.1186/1471-2105-5-187.
Dunn CW, Hejnol A, Matus DQ, Pang K, Browne WE, Smith SA, Seaver E, Rouse GW, Obst M, Edgecombe GD: Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008, 452: 745-749. 10.1038/nature06614.
Stamatakis A, Hoover P, Rougemont J: A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol. 2008, 57: 758-771. 10.1080/10635150802429642.
Langmead B, Trapnell C, Pop M, Salzberg SL: Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10: R25-10.1186/gb-2009-10-3-r25.
Kim D, Salzberg SL: TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011, 12: R72-10.1186/gb-2011-12-8-r72.
Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B: Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008, 5: 621-628. 10.1038/nmeth.1226.
Médigue C, Rouxel T, Vigier P, Hénaut A, Danchin A: Evidence for horizontal gene transfer in Escherichia coli speciation. J Mol Biol. 1991, 222: 851-856. 10.1016/0022-2836(91)90575-Q.
Acknowledgments
We thank Stijn van Dongen, Casey Dunn, Michael Ethier, Amir Karger, Devin Locke, Sergey Lukyanov, Evandro Novaes, Alexis Stamatakis, and Jiangwen Zhang for technical assistance. We also thank William Anderson, Mark Beilstein, Andrew Knoll, Christopher Marx, Sarah Mathews, Brian O’Meara, and members of the Davis laboratory for helpful discussions. CCD was supported by the National Science Foundation (NSF) Assembling the Tree of Life grant DEB-0622764 and NSF DEB-1120243.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CCD, JSR, RKB, and KJW designed all analyses; ZX, KB, KMW, and MS collected the data and conducted the analyses; CCD, JSR, and ZX wrote the initial draft of the paper; all other authors contributed to subsequent revisions to the final draft. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2012_4201_MOESM1_ESM.pdf
Additional file 1: Figure S1. Histogram of the assembled cDNA transcript lengths from Illumina Genome Analyzer II sequencing of Rafflesia cantleyi . (PDF 367 KB)
12864_2012_4201_MOESM2_ESM.pdf
Additional file 2: Figure S2. Histogram of the assembled cDNA transcript lengths from GS-FLX 454-sequencing of Tetrastigma rafflesiae . (PDF 328 KB)
12864_2012_4201_MOESM3_ESM.pdf
Additional file 3: Table S1. Data sources of protein coding DNA sequences from whole genome sequencing used in the comparative phylogenomic analyses. (PDF 94 KB)
12864_2012_4201_MOESM4_ESM.pdf
Additional file 4: Figure S3. Bar chart showing the distribution of bootstrap support for putative VGT (dark grey) and HGT (light grey) transcripts identified for Rafflesia cantleyi . (PDF 309 KB)
12864_2012_4201_MOESM5_ESM.pdf
Additional file 5: Figure S4. Elevated inference of HGT genes in Rafflesia cantleyi is not sensitive to our bootstrap cutoff thresholds. The percentage of HGT genes in Malpighiales (Rafflesia, Manihot esculenta, and Ricinus communis) was the number of genes sister to Vitis vinifera, in proportion to the total number of genes with resolved relationships, at the specified bootstrap thresholds. For Tetrastigma rafflesiae, HGT percentage is the number of genes that are sister to Malpighiales in proportion to the total number of genes with resolved relationships, at the specified bootstrap thresholds. (PDF 346 KB)
12864_2012_4201_MOESM6_ESM.pdf
Additional file 6: Figure S5. (A) Summary of nucleotide substitution rates in Rafflesia cantleyi HGT transcripts versus homologous Vitis vinifera transcripts; (B) Summary of nucleotide substitution rates in Rafflesia cantleyi VGT transcripts versus homologous Manihot esculenta and Ricinus communis transcripts. The boxplot was truncated so that the median can be better visualized. The number of transcripts used to construct each boxplot is shown in parentheses. (PDF 694 KB)
12864_2012_4201_MOESM7_ESM.pdf
Additional file 7: Table S2. Putative gene location and function of Rafflesia cantleyi HGT transcripts based on NCBI BLAST search results. (PDF 143 KB)
12864_2012_4201_MOESM8_ESM.pdf
Additional file 8: Figure S6. Percentages of Rafflesia cantleyi HGTs (dark grey) and VGTs (light grey) for which gDNA Illumina reads can be positively mapped onto the assembled transcripts, as a function of bootstrap support for the VGT and HGT inference. (PDF 312 KB)
12864_2012_4201_MOESM9_ESM.pdf
Additional file 9: Figure S7. Percentages of Rafflesia cantleyi HGTs (dark grey) and VGTs (light grey) for which gDNA Illumina reads can be positively mapped onto the assembled transcripts, as a function of transcript length for the VGT and HGT inference. (PDF 319 KB)
12864_2012_4201_MOESM10_ESM.pdf
Additional file 10: Figure S8. Nucleotide sequence alignment for a typical intron-bearing HGT transcript identified from Rafflesia cantleyi genomic DNA sequencing. Nucleotides are denoted as dots when identical to the consensus sequence. The Rafflesia HGT transcript is printed in red, and sequences from genomic DNA are marked with asterisks. The intron is highlighted in grey for Rafflesia. (PDF 2 MB)
12864_2012_4201_MOESM11_ESM.pdf
Additional file 11: Figure S9. Summary of the number of Rafflesia cantleyi cDNA Illumina reads re-mapped onto the assembled VGT and HGT transcripts. The boxplot was truncated so that the median can be better visualized. The number of transcripts used to construct each boxplot is shown in parentheses. (RPKM = reads per kilobase per million reads). (PDF 335 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Xi, Z., Bradley, R.K., Wurdack, K.J. et al. Horizontal transfer of expressed genes in a parasitic flowering plant. BMC Genomics 13, 227 (2012). https://doi.org/10.1186/1471-2164-13-227
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-227