
Abstract
Background
Jatropha curcas L. is a potential biofuel plant. Application of exogenous cytokinin (6-benzyladenine, BA) on its inflorescence buds can significantly increase the number of female flowers, thereby improving seed yield. To investigate which genes and signal pathways are involved in the response to cytokinin in J. curcas inflorescence buds, we monitored transcriptional activity in inflorescences at 0, 3, 12, 24, and 48 h after BA treatment using a microarray.
Results
We detected 5,555 differentially expressed transcripts over the course of the experiment, which could be grouped into 12 distinct temporal expression patterns. We also identified 31 and 131 transcripts in J. curcas whose homologs in model plants function in flowering and phytohormonal signaling pathways, respectively. According to the transcriptional analysis of genes involved in flower development, we hypothesized that BA treatment delays floral organ formation by inhibiting the transcription of the A, B and E classes of floral organ-identity genes, which would allow more time to generate more floral primordia in inflorescence meristems, thereby enhancing inflorescence branching and significantly increasing flower number per inflorescence. BA treatment might also play an important role in maintaining the flowering signals by activating the transcription of GIGANTEA (GI) and inactivating the transcription of CONSTITUTIVE PHOTOMORPHOGENIC 1 (COP1) and TERMINAL FLOWER 1b (TFL1b). In addition, exogenous cytokinin treatment could regulate the expression of genes involved in the metabolism and signaling of other phytohormones, indicating that cytokinin and other phytohormones jointly regulate flower development in J. curcas inflorescence buds.
Conclusions
Our study provides a framework to better understand the molecular mechanisms underlying changes in flowering traits in response to cytokinin treatment in J. curcas inflorescence buds. The results provide valuable information related to the mechanisms of cross-talk among multiple phytohormone signaling pathways in woody plants.
Similar content being viewed by others


Background
Jatropha curcas L. (Euphorbiaceae) is a perennial bush or small tree that is widely cultivated in tropical and subtropical climates. The oil content of J. curcas seeds is 30–40%, and J. curcas grows well on marginal lands, avoiding competition with food production. Thus, J. curcas is a potential biofuel plant [1],[2]. However, its potential as a biofuel plant is limited by its poor seed yield [3]. Research into the biological and genetic factors that contribute to seed production in J. curcas is necessary for genetic improvement by conventional and molecular breeding approaches [4]-[6]. J. curcas is a monoecious plant with unisexual flowers: both male and female flowers are borne on the same racemose inflorescence. Each inflorescence has approximately 15 female flowers and 13 pieces of fruit under normal growth conditions [7]. Therefore, to improve the seed yield of J. curcas, generating sufficient female flowers is crucial. A previous study [7] applied exogenous cytokinin (6-benzyladenine, BA) to J. curcas inflorescence buds and obtained a 9.4-fold increase in the number of female flowers per inflorescence and a 2.3-fold increase in seed yield, providing a promising strategy for improving the seed yield of J. curcas.
Cytokinins are an important class of phytohormones that were first discovered to promote cell division in tobacco tissues in 1955 [8]. Cytokinins are involved in many important aspects of plant growth and development, e.g. promoting vascular cambium activity, controlling organ development, and regulating shoot and root branching, as well as responding to biotic and abiotic stresses [9]-[11]. They also play important roles in maintaining the activity and function of the shoot apical meristem (SAM) [9],[12]. The SAM comprises a small population of dividing cells located at the shoot tip, and is responsible for the initiation of all the aerial parts of plants, including the reproductive organs [13]. At least three possible pathways have been identified for maintaining the homeostasis of stem cells, which is necessary for meristem activity in Arabidopsis [14]. A number of genes, such as SHOOT MERISTEMLESS (STM), WUSCHEL (WUS), CLAVATA (CLV), LONELY GUY (LOG), AINTEGUMENTA (ANT), ANT-like 6 (AIL6), ANT-like 7 (AIL7), are involved in this process [14]-[22].
KNOTTED1-like homeobox (KNOXI) increases cytokinin biosynthesis by promoting the expression of ISOPENTENYL TRANSFERASE (IPT), which encodes a rate-limiting enzyme in cytokinin biosynthesis. The application of exogenous cytokinin or the expression of a cytokinin biosynthesis gene rescued a stm mutant [22],[23]. WUS directly represses the expression of ARABIDOPSIS RESPONSE REGULATOR 5, 6, 7, and 15 (ARR5, 6, 7, and 15), which are negative regulators in the cytokinin signaling pathway, and cytokinin signaling activated the expression of WUS through both CLV-dependent and CLV-independent pathways [24]-[26]. LOG catalyzes the final step of cytokinin biosynthesis within the rice meristem, and log mutants have defects in shoot meristem function, showing small panicles, and abnormal floral organs and branching patterns [22]. Cytokinin oxidase/dehydrogenase (CKX) catalyzes the degradation of cytokinin to regulate the activity of reproductive meristems in Arabidopsis. A ckx3ckx5 double mutant produced larger inflorescences and floral meristems, and had an approximately 55% higher seed yield [27],[28]. In rice, a mutant with a reduced expression of OsCKX2 accumulated cytokinin in inflorescence meristems, which contributed to an increased number of spikelets and reproductive organs [29]. Therefore, endogenous cytokinins are assumed to promote the size of reproductive meristems and the number of reproductive organs by initiating more floral primordia in the SAM [28]. In addition, cytokinins influence the switch from the vegetative to the reproductive phase and are involved in the regulation of floral development [30]-[34]. The application of cytokinin promoted Arabidopsis flowering by activating TWIN SISTER OF FT (TSF), FD, and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1). By contrast, FLOWERING LOCUS T (FT) was not required, suggesting that FT and TSF belong to distinct floral signal pathways that respond to different environmental and internal signals [11].
Our previous study showed that exogenous application of BA significantly increased the total number of flowers and the proportion of female flowers in J. curcas [7]. To understand the molecular mechanism of cytokinin action in J. curcas inflorescence buds, we analyzed the dynamic changes in gene expression at different time points after BA treatment using a microarray. Differentially expressed genes involved in the metabolism and signaling of cytokinin and other phytohormones, flowering and floral organ development, and cell division were identified. Our results provide a basis for determining the mechanism of cytokinin action on floral development in J. curcas.
Results and discussion
Effects of 6-benzyladenine (BA) on flowering and fruiting in J. curcas
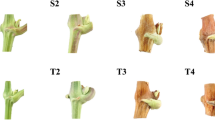
The application of BA changed the flowering characteristics of J. curcas and remarkably increased the number of female flowers to improve seed yield [7]. To select a suitable concentration of BA for this study, we treated J. curcas inflorescence buds with 0, 0.5, 1.0, 2.0, or 4.0 mM of BA, and then surveyed the total flower number, female flower number, ratio of female to male flowers, fruit number, fruiting rate, seed number, seed yield, weight of 100 seeds, and seed oil content per inflorescence. We confirmed that the application of BA is an effective way to significantly increase the number of female flowers and fruits, resulting in an increased seed yield in J. curcas (Figures 1 and 2). The total flower number, female flower number, ratio of female to male flowers, fruit number, seed number, and seed yield all increased with BA concentration from 0.5 to 4.0 mM, while the fruiting rate, weight of 100 seeds, and seed oil content decreased (Table 1). The numbers of female flowers, fruits, and seeds per inflorescence were 7.7-, 4.4-, and 4.0-fold higher, respectively, in the 4.0 mM BA treatment than in the control. As shown in Figure 3, 1.0 mM BA was a transition point in the biological response, and therefore was selected for use in subsequent experiments. Indeed, the effect of a single 1.0 mM BA treatment employed in this study was similar to that of three consecutive treatments at 1-day intervals with 160 mg/L (0.71 mM) BA [7]: both resulted in a 2.3-fold increase in final seed yield (Table 1).

Effects of 6-benzyladenine (BA) on flower development in J. curcas. Each J. curcas inflorescence bud was treated with a solution of BA (0, 0.5, 1.0, 2.0, or 4.0 mM). Each group included 30 inflorescence buds.

Effects of 6-benzyladenine (BA) on fruit development in J. curcas. Each J. curcas inflorescence bud was treated with a solution of BA (0, 0.5, 1.0, 2.0, or 4.0 mM). Each group included 30 inflorescence buds.

Effects of 6-benzyladenine (BA) on flowering and fruiting in J. curcas. Values are provided as means ± standard deviations (n = 30).
Differentially expressed genes in response to the application of BA on inflorescence buds
To identify genes that responded to the application of BA, we designed 41,651 sequence-specific oligonucleotide probes representing 20,555 transcripts to monitor the transcription levels of genes in inflorescence buds at 0, 3, 12, 24, and 48 h after BA treatment using a microarray. 10,569 probes representing 5,555 transcripts changed significantly during the time course (Additional file 1). More transcripts were upregulated in inflorescence buds than were downregulated, except at the 12-h time point, when the greatest overall number of transcripts were differentially expressed among all the time points (Figure 4), indicating that this was an important phase in the response to exogenous cytokinin. Moreover, these differentially expressed transcripts (about 27%) were annotated into 32 gene ontology (GO) categories, as defined by various molecular functions and biological processes (Figure 5), indicating that determining the molecular mechanisms involved in the response to exogenous cytokinin in inflorescence buds will be difficult.

Distribution of differentially expressed transcripts at 3-h, 12-h, 24-h, and 48-h time points after 6-benzyladenine (BA) treatment in J. curcas.

Gene ontology categories of differentially expressed transcripts after 6-benzyladenine (BA) treatment in J. curcas. 5,555 differentially expressed transcripts were annotated into 32 gene ontology categories in three main categories: biological process, cellular component, and molecular function.
Expression profiles of the differentially expressed genes in response to BA treatment
To gain further insights into the genetic and biological processes involved in the response to BA application, the transcripts were clustered into 12 sets that represented distinct temporal expression patterns (Figure 6 and Additional file 2). Among these patterns, clusters 2 and 5 were upregulated and cluster 1 was downregulated at the 3-h time point, suggesting that these genes were induced early in the response to cytokinin; examples include calcium ion binding protein (CUST_18202), gibberellin 20-oxidase (CUST_5901), and gibberellin receptor GID1 (CUST_19936), respectively. The expressions of genes in clusters 4, 6, 9, 10 and 11 changed significantly from the 3-h to 24-h time points, indicating that significant transcriptional regulation occurred during this phase. Gene expression in clusters 7 and 8 changed distinctly from the 3-h to 12-h time points and remained constant until the 48-h time point. Moreover, the expression profiles of clusters 3 and 12 changed obviously between the 24-h and 48-h time points, suggesting that they might represent downstream genes in the BA response pathway.

Clustering analysis of differentially expressed transcripts with significant expression profile changes revealed by the microarray and qRT-PCR analysis. All differentially expressed transcripts were clustered into 12 distinct temporal change patterns according to their expression profiles. The expressions of 12 selected transcripts representing the 12 expression patterns from the microarray analysis were confirmed by qRT-PCR.
To validate the results of the microarray analysis, we performed quantitative real-time reverse transcription PCR (qRT-PCR) analysis on 12 selected transcripts representative of the 12 clusters representing distinct expression patterns (Figure 6). The qRT-PCR expression profiles agreed with the profiles of their respective clusters; however, the sizes of the changes in expression at some time points were larger than in the clusters (Figure 6), suggesting that qRT-PCR was more sensitive than the microarray analysis. Based on the qRT-PCR results, we concluded that the expression profiles of transcripts in the clusters (Figure 6) accurately reflected temporal changes in the expressions of genes involved in the response to exogenous cytokinin.
Functional analysis of the genes differentially expressed in response to BA application
To understand the biological functions of the differentially expressed genes, those that changed ≥2-fold were categorized by GO analysis (Figure 7). Among all post-application time points, the numbers of transcripts in the “anatomical structure formation”, “cellular component biogenesis”, and “reproductive process” categories were highest at the 3-h time point (28%, 16% and 8%, respectively) and those in the “metabolic process”, “cellular process”, “localization” and “response to stimulus” categories were the lowest. However, the “cell”, “biological regulation”, “pigmentation”, “organelle”, “electron carrier activity” and “transporter activity” categories were similar at four time points. The results indicated that genes involved in the “anatomical structure formation”, “cellular component biogenesis”, and “reproductive process” functions were especially expressed at the 3-h time point to promote cell generation in the inflorescence, which might be the main factor driving the increase in flower number. We hypothesized that these specifically expressed genes were first induced by cytokinin, and then they in turn activated genes in the “metabolic process”, “cellular process”, “location” and “response to stimulus” categories to influence the growth and flowering of J. curcas.

Comparative analysis by gene ontology category of differentially expressed transcripts at different time points after 6-benzyladenine (BA) treatment in J. curcas. The highest numbers of transcripts at the 3-h time point were in the “anatomical structure formation”, “cellular component biogenesis” and “reproductive process” categories. The numbers of transcripts were lowest in the “metabolic process”, “cellular process”, “localization” and “response to stimulus” categories. The numbers of transcripts were similar at four time points in the “cell”, “biological regulation”, “pigmentation”, “organelle”, “electron carrier activity” and “transporter activity” categories.
Transcriptional analysis of genes related to flower development
The application of BA had a significant effect on the flowering characters of J. curcas, which in turn resulted in an increased seed yield. Thirty-one transcripts in our dataset were homologous to genes related to flowering and flower development in Arabidopsis (Table 2). The expressions of nine genes were significantly differentially regulated (≥2-fold) by BA treatment. CUP-SHAPED COTYLEDON 1 (CUC1) and GIGANTEA (GI) were upregulated, and APETALA3 (AP3), CONSTITUTIVE PHOTOMORPHOGENIC 1 (COP1), NGATHA 2 (NGA2), SEPALLATA 1, 2, and 3 (SEP1, 2, and 3), and SEEDSTICK (STK) were downregulated over the time course of the experiment.
Interestingly, GI, a clock-associated protein that is involved in the control of circadian rhythms and regulating flowering time [35],[36], was induced quickly by BA treatment (by the 3-h time point) and was expressed 88 times higher at the 12-h time point than at the 0-h time point. CUC1, belonging to the NAC family, which contributes to the formation of the SAM and the separation of cotyledons by activating STM in Arabidopsis [37],[38], was induced between the 12-h and 48-h time points, indicating that CUC1 helps to promote and maintain SAM formation to generate more floral primordia. However, CUC2 and CUC3 were insensitive to BA treatment in J. curcas. This is in contrast to observations in Arabidopsis, where CUC2 and CUC3, but not CUC1, were upregulated by cytokinin in inflorescence meristems [39].
Among the floral organ-identity genes, AP1, an A-class gene in Arabidopsis, and AP3 (B-class), SEP1, SEP2, and SEP3 (E-class) [40] were downregulated at the 12-h time point, while AP2 (A-class) and AG (C-class) [41] were insensitive to BA application. These results implied that BA treatment could suppress the expressions of A-, B- and E-class genes, which agreed with our observation that the flowering duration of inflorescences treated with BA was longer than that in the control (data not shown). Recently, AP1 was observed to act upstream of cytokinin, regulating cytokinin levels by directly suppressing the cytokinin biosynthetic gene LOG1 and activating the cytokinin degradation gene CKX3 to suppress meristem activity in sepal axils [42].
We also verified the expressions of SOC1, LEAFY (LFY), and TERMINAL FLOWER 1b (TFL1b) in inflorescence buds of J. curcas after BA treatment by qRT-PCR. The transcript levels of SOC1 and LFY were upregulated at the 12-h time point, and TFL1b was downregulated during the experiment (Figure 8). We hypothesized that BA treatment might contribute to the maintenance of the flowering signals by activating the expressions of GI, LFY, and SOC1 and inactivating COP1 and TFL1b. BA treatment may also delay the formation of floral organs by inhibiting the transcription of the A-, B- and E-class of floral organ-identity genes, which would allow more time to generate more floral primordia in inflorescence meristems, along with activating the expression of CUC1, which would result in a significant increase in flower number in J. curcas.

Expression analysis of LFY, SOC1 and TFL1b transcripts by qRT-PCR after 6-benzyladenine (BA) treatment in J. curcas. LFY and SOC1 were upregulated at the 12-h time point, and TFL1b was downregulated over the time course of the experiment. The GHPDA gene was used as an internal control.
Transcriptional analysis of genes involved in sex determination in J. curcas
As shown in Table 1, BA treatment significantly increased the ratio of female to male flowers, indicating that the cytokinin could affect the differentiation of male and female flowers in J. curcas. Among the oligonucleotide probes used in this study, CUST_36773 (Table 2) was homologous to the sex determination gene TASSELSEED2 (TS2) of maize, which encodes a short-chain alcohol dehydrogenase that is required for carpel abortion in maize [43]. Upon BA treatment of inflorescence buds, the J. curcas TS2 homolog (CUST_36773) was downregulated by 2.3-fold, 3.4-fold and 3.2-fold at the 12-h, 24-h and 48-h time points, respectively (Table 2). This result suggested that BA treatment activates the development of arrested pistil primordia in male flowers by repressing the expression of the TS2 homolog in J. curcas, which resulted in an increase in the ratio of female to male flowers. Sex determination in plants, however, is a complex and dynamic process, and is influenced by genetic, hormonal and environmental conditions [44],[45]. Further studies are required to elucidate the mechanism of sex determination in J. curcas.
Transcriptional analysis of genes involved in cytokinin signaling in inflorescence buds
Endogenous cytokinins regulate many essential aspects of plant growth and development, and play a critical role in the formation and maintenance of the SAM. To further investigate the response mechanism of J. curcas inflorescences to exogenous BA, we analyzed 27 transcripts in our dataset that were homologous to Arabidopsis genes involved in the cytokinin signal transduction pathway (Additional file 3). The expressions of ARR3 and ARR8 were upregulated, and IPT5 was downregulated during the experiment, implying that exogenous cytokinin promoted increased ARR expression, which contributed to signal perception and transmission, and repressed the activation of IPTs that catalyze the first key reaction in endogenous cytokinin biosynthesis in the cytokinin signaling pathway. These findings were consistent with the results of other studies involving cytokinin treatment [46]-[48]. However, some genes that play important roles in cytokinin signaling, such as ARABIDOPSIS HISTIDINE KINASE 3 (AHK3), ARABIDOPSIS HISTIDINE-CONTAINING PHOSPHOTRANSFER 2 (AHP2), ARR1, HOMEOBOX PROTEIN KNOTTED-1 LIKE 3 (KNAT3), STM, and IPT9, showed only small changes in their transcript levels, indicating that only small changes in the expressions of these genes are required to carry out their functions, rather than them being insensitive to exogenous BA. Many transcription factors are expressed at low levels; therefore, it was difficult to accurately assess their changes using the microarray method [46],[49]. In addition, ACETYL-COA CARBOXYLASE 1 (ACC1), ARABIDOPSIS HEMOGLOBIN 2 (AHB2), ARABIDOPSIS THALIANA HOMEOBOX PROTEIN 2 (ATHB2), and QUASIMODO2 (QUA2) were also induced by the application of BA; however, their roles in cytokinin signaling are unclear.
Cross-talk of cytokinin with other phytohormones
Eight major types of phytohormones coordinate plant growth and development by modulating various cellular processes in response to intrinsic and environmental cues: abscisic acid (ABA), auxins, brassinosteroids (BRs), cytokinins (CKs), ethylene, gibberellins (GAs), jasmonic acid (JA) and salicylic acid (SA) [47]. To understand the roles of other phytohormones in inflorescence buds in response to BA treatment, we identified homologous genes involved in various hormonal regulation pathways in Arabidopsis by sequence comparison between our set and the Arabidopsis Hormone Database [48]. In addition to the 27 cytokinin-related genes, 104 transcripts were identified in our dataset, including 23 abscisic acid-related genes, 32 auxin-related genes, 11 brassinosteroid-related genes, 11 ethylene-related genes, nine gibberellin-related genes, 11 jasmonic acid-related genes, and seven salicylic acid-related genes (Additional file 3).
In the ABA signaling pathways, ABSCISIC ACID RECEPTOR (ABAR) and PYRABACTIN RESISTANCE-LIKE 4 (PYL4) encode two ABA receptors that are involved in perceiving ABA signals [50],[51]. In this study, the transcript levels of PYL4 and ABAR were downregulated by 3.5-fold at the 3-h time point and by 20.6-fold at the 12-h time point, respectively. ABSCISIC ACID 1 (ABA1), which encodes a zeaxanthin epoxidase, which catalyzes the conversion of zeaxanthin to antheraxanthin and violaxanthin to generate the epoxycarotenoid precursor in the ABA biosynthetic pathway [52], was downregulated at the 12-h time point. Also, 1-DEOXY-D-XYLULOSE-5-PHOSPHATE SYNTHASE (DXS), which encodes a key enzyme catalyzing a limiting step in the biosynthesis of plastidic isoprenoids (the carotenoid precursors for ABA biosynthesis) [53], was downregulated 4-fold at the 3-h time point. Moreover, 9-CIS-EPOXYCAROTENOID DIOXYGENASE 3 (NCED3), which encodes a key enzyme in the ABA biosynthesis pathway [54], was downregulated 3.8-fold at the 12-h time point. These results showed that BA treatment inhibited ABA signaling by repressing the expression of genes involved in ABA biosynthesis and ABA perception. This indicated that endogenous cytokinins might play similar roles in inhibiting the effects of ABA and maintaining reproductive growth during flower bud development in J. curcas.
In auxin signaling pathways, MASSUGU2 (MSG2/IAA19) encodes an auxin-regulated protein that regulates hypocotyl growth and the formation of lateral roots together with AUXIN RESPONSE FACTOR7 (ARF7) [55]; it showed 2-fold upregulation at the 12-h time point. IAA CARBOXYL METHYLTRANSFERASE 1 (IAMT1), which methylates indole-3-acetic acid (IAA) to form methyl-IAA ester and overexpression of which causes dramatic hyponastic leaf phenotypes [56], was downregulated 2.1-fold at the 12-h time point. SUPERROOT 1 (SUR1) encodes a C-S lyase, which is involved in indolic glucosinolate biosynthesis and whose mutant showed a high-auxin phenotype related to the accumulation of indole-3-acetaldoxime promoted IAA biosynthesis [57], was downregulated 4.3-fold at the 12-h time point. TRANSPARENT TESTA 4 (TT4) encodes a chalcone synthase that is the first enzyme in flavonoid biosynthesis and is an auxin transport inhibitor [58]; it was downregulated 2.8-fold at the 12-h time point. Jones et al. found that the application or ectopic biosynthesis of cytokinin rapidly induced an auxin increase in young shoot and root tissues and proposed that cytokinin promoted auxin synthesis by controlling the transcription of certain auxin biosynthesis genes [59]. However, our results suggested that cytokinin treatment caused the increase in auxin levels via suppression of the negative regulators of auxin biosynthesis pathways. In addition, the expression of MYB77, a positive regulator of auxin signaling transduction [60], was downregulated at the 48-h time point, indicating that auxin signaling was inhibited then, contradicting our conclusion that cytokinins promoted an increase in auxin. Therefore, we hypothesized that the downregulation of MYB77 might help the accumulation of auxin in inflorescence buds by inhibiting auxin transport, indicating that the roles of MYB77 might be different in Arabidopsis and J. curcas.
A UDP-glycosyltransferase gene (UGT73C5) encode an enzyme that catalyzes the glucosylation of BRs, causing their inactivation [61]; it was downregulated 5-fold at the 12-h time point. CONSTITUTIVE PHOTOMORPHOGENESIS AND DWARFISM (CPD/DWF3), which encodes a cytochrome P450 steroid side-chain hydroxylase that plays an essential role in BR biosynthesis [62], was upregulated 3-fold at the 3-h time point. These results indicated that cytokinin caused an increase of BRs by repressing BR glucosylation and promoting BR biosynthesis, which repressed the expression of FLC to promote flowering in Arabidopsis [63].
In the ethylene signaling pathway, MULTIPROTEIN BRIDGING FACTOR 1C (MBF1c) encodes a coactivator that enhances plant tolerance to biotic and abiotic stresses, and is involved in both salicylic acid and ethylene signaling pathways [64]; it was upregulated by more than 4-fold during the time course of the experiment, especially at the 24-h time point (37.4-fold). HIGH INDOLIC GLUCOSINOLATE 1 (HIG1/MYB51), which encodes a key transcription factor in indolic glucosinolate biosynthesis and responds to ethylene stimuli [65],[66], was upregulated at the 3-h time point, and downregulated at the 12-h time point. These results indicated that MBF1c and HIG1/MYB51 are candidate cross-talk genes in cytokinin and ethylene signaling pathways.
GIBBERELLIN 20-OXIDASE 1 (GA20ox1) was upregulated between 3 h and 48 h, and GIBBERELLIN-INSENSITIVE DWARF 1b and 1c (GID1b and 1c) were upregulated at the 48-h time point; however, GIBBERELLIN 2-OXIDASE 1 (GA2ox1) was downregulated between 12 h and 48 h. GA20ox1 encodes an enzyme that, along with gibberellin 3β-hydroxylase, catalyzes the formation of active gibberellins, and GID1s encode gibberellin receptors that are positive regulators in the gibberellin signaling pathway [67],[68]. GA2ox1 functions in a major catabolic pathway that negatively regulates gibberellin signaling [69]. The results showed that cytokinin promoted gibberellin production by increasing the transcription of GID1s and GA20ox1 and decreasing that of GA2ox in inflorescence buds.
ABNORMAL INFLORESCENCE MERISTEM 1 (AIM1), ALLENE OXIDE SYNTHASE (AOS/CYP74A), OPR3 (OXOPHYTODIENOATE-REDUCTASE 3), and SUPPRESSOR OF SA-INSENSITIVITY 2 (SSI2/FAB2) are positive regulators of jasmonic acid signaling in Arabidopsis [70]-[74]: they were all upregulated after BA treatment. An AIM1 mutant had an abnormal floral meristem phenotype with severe sterility, and a knockout mutant of the AOS gene was male sterile in Arabidopsis [70],[71], indicating that cytokinins promote an increase in jasmonic acid during J. curcas flower development. However, JMT, which encodes a jasmonic acid carboxyl methyltransferase that catalyzes the methylation of jasmonic acid [74], and is a positive regulator, was downregulated between 3 h and 48 h, implying that methyl jasmonate might play different roles in the JA signaling pathway in J. curcas and Arabidopsis. Moreover, BENZOIC ACID CARBOXYL METHYLTRANSFERASE 1 (BSMT1) catalyzes the methylation of salicylate and benzoate in the salicylic acid signaling pathway in response to biotic and abiotic stresses [75]. BSMT1 was upregulated at the 3-h time point, indicating that methyl salicylate is involved in the response to BA treatment.
Based on these results, we concluded that exogenous BA influences the effects of major phytohormones by modulating the expression levels of genes involved in various metabolic pathways in J. curcas. These phytohormones may jointly regulate the development of J. curcas flowers after BA treatment, although their exact roles in this process remain to be defined. Elucidating the mechanisms of cross-talk among multiple signaling pathways is also essential.
Conclusions
The application of BA could significantly increase flower number and seed yield in J. curcas. To elucidate the mechanism underlying this response, we performed a transcriptional analysis of J. curcas inflorescence buds after BA treatment. 5,555 differentially expressed transcripts were identified, which could be grouped into 12 clusters representing distinct regulatory patterns and belonged to 32 gene ontology categories. Based on our analysis of genes involved in flowering and phytohormone signaling pathways, we hypothesized that BA application increased flower number by activating the transcription of genes that initiate flowering and repressing that of genes involved in the formation of floral organs. Moreover, exogenous cytokinin treatment could influence the production of major phytohormones by regulating the transcription of genes involved in their metabolic pathways. BA treatment repressed endogenous cytokinin biosynthesis and abscisic acid signaling and promoted auxin, brassinosteroid, gibberellin, jasmonic acid, and salicylic acid signaling, suggesting that these plant hormones might jointly regulate the development of J. curcas flowers. Our study provides a basis for understanding the molecular mechanisms underlying changes in flowering traits in response to cytokinin treatment in J. curcas, and provides useful information on the mechanisms of cross-talk among multiple hormone signaling pathways in woody plants.
Materials and methods
Plant materials and treatments
Jatropha curcas L. is a cultivated plant in Yunnan Province, China [76]. One-year-old J. curcas plants were grown in a field at a density of 2 × 2 m per plant at Xishuangbanna Tropical Botanical Garden of the Chinese Academy of Sciences, located at Menglun town in Mengla County, Yunnan Province, China (21°54' N, 101°46' E, 580 m asl). To select a suitable concentration of the synthetic cytokinin 6-benzylaminopurine (BA) for treating J. curcas, 180 inflorescence buds (about 0.5–1 cm in diameter) were selected and distributed into five treatment groups, each of which included 36 inflorescence buds. Working solutions of various concentrations of BA (0, 0.5, 1.0, 2.0, or 4.0 mM) were sprayed onto inflorescence buds with a hand sprayer, wetting the inflorescence buds to the point of run-off. Tween-20 (Polysorbate-20, Shanghai Sangon Biological Engineering Technology & Services Co., Ltd., Shanghai, China) was added to the BA working solutions at a final concentration of 0.05% (v/v) as a wetting agent. The total flower number, female flower number, ratio of female to male flowers, fruit number, fruiting rate, seed number, seed yield per inflorescence, and weight of 100 seeds and seed oil content were surveyed during the development period.
For the microarray analysis, inflorescence buds were treated with 1.0 mM BA solution containing 0.05% Tween-20. Inflorescence buds were collected at 0, 3, 12, 24, and 48 h after BA treatment. The collected samples of inflorescence buds were frozen immediately in liquid nitrogen and stored at -80°C until RNA extraction. Three biological replicates were performed for each time point. The experiments were carried out in May 2010.
Collection of sequences and design of probes
41,735 genetic sequences were collected (Additional file 4). Among them, 30,184 expressed sequence tags (ESTs) were generated in our laboratory by sequencing cDNA libraries of J. curcas flower buds and embryos [77]. The other sequences were publicly available from NCBI (up to 2010). 8,157 unigenes were produced from 16,875 ESTs derived from different J. curcas tissues [78],[79] and 3,394 unigenes from 5,619 ESTs from castor bean flowers. For the J. curcas transcript data set, 41,651 sequence-specific probes of 60-bp oligonucleotides were designed using the Agilent eArray software (Additional file 5).
RNA extraction, hybridization, microarray data acquisition, normalization and analysis
Total RNA was extracted from inflorescence buds using TRIzol (Invitrogen, Carlsbad, CA, USA). The RNA concentration was determined using a NanoDrop ND-1000 (Thermo Scientific, Waltham, MA, USA). RNA integrity was confirmed with an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA), and the total RNA was purified with a Qiagen RNeasy kit (Venlo, Netherlands). Two micrograms of RNA were reverse-transcribed to cDNA using a one-step method. The cDNA was transcribed into RNA by T7 RNA polymerase, modified by aa-UTP at 40°C, purified with a Qiagen RNeasy mini kit and quantified using the Bioanalyzer. Four micrograms of cRNA were labeled with Cy3 fluorescence dye at 25°C and purified with a Qiagen RNeasy mini kit. Eight hundred and seventy-five nanograms of Cy3 cRNA were fragmented and hybridized with 4 × 44 k arrays. After hybridization, the arrays were washed according to the manufacturer’s instructions and scanned twice using an Agilent scanner, with 10% and 100% photo multiplier tubes (PMT). Raw data from arrays were normalized by log2 transformation and analyzed using GeneSpring GX software (Agilent). Differentially expressed probes with fold-change thresholds ≥2 and corrected P-values ≤0.05 were selected. Hierarchical clustering analysis was performed using Cluster software, and the results viewed using the Java TreeView software [80],[81].
Assembly and annotation of sequences
Sequences corresponding to differentially expressed probes were assembled by CAP3 (Sequence Assembly Program) software [82]. The differentially expressed unigenes were annotated using Interproscan (version 4.8) [83], and the GO annotation results were plotted by WEGO (Web Gene Ontology Annotation Plot) [84].
Validation of gene expression by qRT-PCR
qRT-PCR was performed on a LightCycler 480 II (Roche, Penzberg, Germany) using the SYBR green fluorescent label. The cDNA was synthesized from total RNA using a PrimeScript RT Reagent Kit (Takara, Otsu, Japan). The relative expression levels of genes were calculated by the 2−ΔΔ CT method. All quantitative PCRs were repeated in 2–3 biological replications. The primers used for qRT-PCR are listed in Additional file 6.
Availability of supporting data
Oligonucleotide microarray data have been deposited into the Gene Expression Omnibus (GEO) Database under accession number GSE54366 at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE54366. All additional data files supporting the results of this article are available in the LabArchives repository and are accessible via http://dx.doi.org/10.6070/H4P848WW.
Additional files
References
Fairless D: Biofuel: the little shrub that could–maybe. Nature. 2007, 449 (7163): 652-655. 10.1038/449652a.
Sato S, Hirakawa H, Isobe S, Fukai E, Watanabe A, Kato M, Kawashima K, Minami C, Muraki A, Nakazaki N: Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res. 2011, 18 (1): 65-76. 10.1093/dnares/dsq030.
Sanderson K: Wonder weed plans fail to flourish. Nature. 2009, 461 (7262): 328-329. 10.1038/461328a.
Marques DDA, Siqueira WJ, Colombo CA, Ferrari RA: Breeding and Biotechnology of Jatropha curcas . Jatropha, Challenges for a New Energy Crop Volume 2: Genetic Improvement and Biotechnology. Edited by: Bahadur B, Sujatha M, Carels N. New York:Springer; 2013: 457-478. 10.1007/978-1-4614-4915-7_23.
Kumar N, Reddy M, Sujatha M: Genetic Transformation of Jatropha curcas: Current Status and Future Prospects. Jatropha, Challenges for a New Energy Crop Volume 2: Genetic Improvement and Biotechnology. Edited by: Bahadur B, Sujatha M, Carels N. New York: Springer; 2013: 535-546. 10.1007/978-1-4614-4915-7_28.
Sujatha M, Nithianantham S, Reddy MP: Plant Regeneration and Genetic Transformation in Jatropha . Biotechnology of Neglected and Underutilized Crops. Edited by: Jain SM, Dutta Gupta S.Netherlands: Springer; 2013: 319-342. 10.1007/978-94-007-5500-0_13.
Pan B-Z, Xu Z-F: Benzyladenine treatment significantly increases the seed yield of the biofuel plant Jatropha curcas . J Plant Growth Regul. 2011, 30 (2): 166-174. 10.1007/s00344-010-9179-3.
Hall RH, De Ropp RS: Formation of 6-furfurylaminopurine from DNA breakdown product. J Am Chem Soc. 1955, 77 (23): 6400-6400. 10.1021/ja01628a115.
Werner T, Schmulling T: Cytokinin action in plant development. Curr Opin Plant Biol. 2009, 12 (5): 527-538. 10.1016/j.pbi.2009.07.002.
Perilli S, Moubayidin L, Sabatini S: The molecular basis of cytokinin function. Curr Opin Plant Biol. 2009, 13 (1): 21-26. 10.1016/j.pbi.2009.09.018.
D'Aloia M, Bonhomme D, Bouche F, Tamseddak K, Ormenese S, Torti S, Coupland G, Perilleux C: Cytokinin promotes flowering of Arabidopsis via transcriptional activation of the FT paralogue TSF. Plant J. 2011, 65 (6): 972-979. 10.1111/j.1365-313X.2011.04482.x.
Kyozuka J: Control of shoot and root meristem function by cytokinin. Curr Opin Plant Biol. 2007, 10 (5): 442-446. 10.1016/j.pbi.2007.08.010.
Laufs P, Grandjean O, Jonak C, Kieu K, Traas J: Cellular parameters of the shoot apical meristem in Arabidopsis . The Plant Cell. 1998, 10 (8): 1375-1390. 10.1105/tpc.10.8.1375.
Mudunkothge JS, Krizek BA: Three Arabidopsis AIL/PLT genes act in combination to regulate shoot apical meristem function. Plant J. 2012, 71 (1): 108-121. 10.1111/j.1365-313X.2012.04975.x.
Barton MK, Poethig RS: Formation of the shoot apical meristem in Arabidopsis thaliana: an analysis of development in the wild type and in the shoot meristemless mutant. Development. 1993, 119 (3): 823-831.
Endrizzi K, Moussian B, Haecker A, Levin JZ, Laux T: The SHOOT MERISTEMLESS gene is required for maintenance of undifferentiated cells in Arabidopsis shoot and floral meristems and acts at a different regulatory level than the meristem genes WUSCHEL and ZWILLE. Plant J. 1996, 10 (6): 967-979. 10.1046/j.1365-313X.1996.10060967.x.
Long JA, Moan EI, Medford JI, Barton MK: A member of the KNOTTED class of homeodomain proteins encoded by the STM gene of Arabidopsis . Nature. 1996, 379 (6560): 66-69. 10.1038/379066a0.
Mayer KFX, Schoof H, Haecker A, Lenhard M, Jurgens G, Laux T: Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell. 1998, 95 (6): 805-815. 10.1016/S0092-8674(00)81703-1.
Clark SE, Running MP, Meyerowitz EM: CLAVATA1, a regulator of meristem and flower development in Arabidopsis . Development. 1993, 119 (2): 397-418.
Kayes JM, Clark SE: CLAVATA2, a regulator of meristem and organ development in Arabidopsis . Development. 1998, 125 (19): 3843-3851.
Clark SE, Running MP, Meyerowitz EM: CLAVATA3 is a specific regulator of shoot and floral meristem development affecting the same processes as CLAVATA1. Development. 1995, 121 (7): 2057-2067.
Kurakawa T, Ueda N, Maekawa M, Kobayashi K, Kojima M, Nagato Y, Sakakibara H, Kyozuka J: Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature. 2007, 445 (7128): 652-655. 10.1038/nature05504.
Yanai O, Shani E, Dolezal K, Tarkowski P, Sablowski R, Sandberg G, Samach A, Ori N: Arabidopsis KNOXI proteins activate cytokinin biosynthesis. Curr Biol. 2005, 15 (17): 1566-1571. 10.1016/j.cub.2005.07.060.
To JPC, Haberer G, Ferreira FJ, Deruere J, Mason MG, Schaller GE, Alonso JM, Ecker JR, Kieber JJ: Type-A Arabidopsis response regulators are partially redundant negative regulators of cytokinin signaling. The Plant Cell. 2004, 16 (3): 658-671. 10.1105/tpc.018978.
Leibfried A, To JPC, Busch W, Stehling S, Kehle A, Demar M, Kieber JJ, Lohmann JU: WUSCHEL controls meristem function by direct regulation of cytokinin-inducible response regulators. Nature. 2005, 438 (7071): 1172-1175. 10.1038/nature04270.
Gordon SP, Chickarmane VS, Ohno C, Meyerowitz EM: Multiple feedback loops through cytokinin signaling control stem cell number within the Arabidopsis shoot meristem. Proc Natl Acad Sci USA. 2009, 106 (38): 16529-16534. 10.1073/pnas.0908122106.
Sakakibara H: Cytokinins: Activity, biosynthesis, and translocation. Annu Rev Plant Biol. 2006, 57: 431-449. 10.1146/annurev.arplant.57.032905.105231.
Bartrina I, Otto E, Strnad M, Werner T, Schmulling T: Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana . The Plant Cell. 2011, 23 (1): 69-80. 10.1105/tpc.110.079079.
Ashikari M, Sakakibara H, Lin S, Yamamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M: Cytokinin oxidase regulates rice grain production. Science. 2005, 309 (5735): 741-745. 10.1126/science.1113373.
Bernier G: The control of floral evocation and morphogenesis. Annu Rev Plant Physiol Plant Mol Biol. 1988, 39 (1): 175-219. 10.1146/annurev.pp.39.060188.001135.
Estruch JJ, Granell A, Hansen G, Prinsen E, Redig P, Onckelen H, Schwarz-Sommer Z, Sommer H, Spena A: Floral development and expression of floral homeotic genes are influenced by cytokinins. Plant J. 1993, 4 (2): 379-384. 10.1046/j.1365-313X.1993.04020379.x.
Chaudhury AM, Letham S, Craig S, Dennis ES: amp1 - a mutant with high cytokinin levels and altered embryonic pattern, faster vegetative growth, constitutive photomorphogenesis and precocious flowering. Plant J. 1993, 4 (6): 907-916. 10.1046/j.1365-313X.1993.04060907.x.
He YW, Loh CS: Induction of early bolting in Arabidopsis thaliana by triacontanol, cerium and lanthanum is correlated with increased endogenous concentration of isopentenyl adenosine (iPAdos). J Exp Bot. 2002, 53 (368): 505-512. 10.1093/jexbot/53.368.505.
Corbesier L, Prinsen E, Jacqmard A, Lejeune P, Van Onckelen H, Périlleux C, Bernier G: Cytokinin levels in leaves, leaf exudate and shoot apical meristem of Arabidopsis thaliana during floral transition. J Exp Bot. 2003, 54 (392): 2511-2517. 10.1093/jxb/erg276.
Fowler S, Lee K, Onouchi H, Samach A, Richardson K, Morris B, Coupland G, Putterill J: GIGANTEA: a circadian clock-controlled gene that regulates photoperiodic flowering in Arabidopsis and encodes a protein with several possible membrane-spanning domains. EMBO J. 1999, 18 (17): 4679-4688. 10.1093/emboj/18.17.4679.
Park DH, Somers DE, Kim YS, Choy YH, Lim HK, Soh MS, Kim HJ, Kay SA, Nam HG: Control of circadian rhythms and photoperiodic flowering by the Arabidopsis GIGANTEA gene. Science. 1999, 285 (5433): 1579-1582. 10.1126/science.285.5433.1579.
K-i H, Takada S, Tasaka M: CUC1 gene activates the expression of SAM-related genes to induce adventitious shoot formation. Plant J. 2003, 36 (5): 687-696. 10.1046/j.1365-313X.2003.01911.x.
Aida M, Ishida T, Fukaki H, Fujisawa H, Tasaka M: Genes involved in organ separation in Arabidopsis: an analysis of the cup-shaped cotyledon mutant. The Plant Cell. 1997, 9 (6): 841-857. 10.1105/tpc.9.6.841.
Li X, Su Y, Zhao X, Li W, Gao X, Zhang X: Cytokinin overproduction-caused alteration of flower development is partially mediated by CUC2 and CUC3 in Arabidopsis . Gene. 2010, 450 (1–2): 109-120. 10.1016/j.gene.2009.11.003.
Pelaz S, Ditta GS, Baumann E, Wisman E, Yanofsky MF: B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature. 2000, 405 (6783): 200-203. 10.1038/35012103.
Bowman JL, Smyth DR, Meyerowitz EM: Genes directing flower development in Arabidopsis . The Plant Cell. 1989, 1 (1): 37-52. 10.1105/tpc.1.1.37.
Han Y, Zhang C, Yang H, Jiao Y: Cytokinin pathway mediates APETALA1 function in the establishment of determinate floral meristems in Arabidopsis . Proc Natl Acad Sci USA. 2014, 111 (18): 6840-6845. 10.1073/pnas.1318532111.
DeLong A, Calderon-Urrea A, Dellaporta SL: Sex determination gene TASSELSEED2 of maize encodes a short-chain alcohol dehydrogenase required for stage-specific floral organ abortion. Cell. 1993, 74 (4): 757-768. 10.1016/0092-8674(93)90522-R.
Aryal R, Ming R: Sex determination in flowering plants: papaya as a model system. Plant Sci. 2014, 217–218: 56-62. 10.1016/j.plantsci.2013.10.018.
Hartwig T, Chuck GS, Fujioka S, Klempien A, Weizbauer R, Potluri DP, Choe S, Johal GS, Schulz B: Brassinosteroid control of sex determination in maize. Proc Natl Acad Sci USA. 2011, 108 (49): 19814-19819. 10.1073/pnas.1108359108.
Bhargava A, Clabaugh I, To JP, Maxwell BB, Chiang Y-H, Schaller GE, Loraine A, Kieber JJ: Identification of Cytokinin-Responsive Genes Using Microarray Meta-Analysis and RNA-Seq in Arabidopsis. Plant Physiol. 2013, 162 (1): 272-294. 10.1104/pp.113.217026.
Wolters H, Jürgens G: Survival of the flexible: hormonal growth control and adaptation in plant development. Nature Reviews Genetics. 2009, 10 (5): 305-317. 10.1038/nrg2558.
Peng ZY, Zhou X, Li LC, Yu XC, Li HJ, Jiang ZQ, Cao GY, Bai MY, Wang XC, Jiang CF, Lu HB, Hou XH, Qu LJ, Wang ZY, Zuo JR, Fu XD, Su Z, Li SG, Guo HW: Arabidopsis Hormone Database: a comprehensive genetic and phenotypic information database for plant hormone research in Arabidopsis. Nucleic Acids Res. 2009, 37: D975-D982. 10.1093/nar/gkn873.
Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM: A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009, 10 (4): 252-263. 10.1038/nrg2538.
Shen Y-Y, Wang X-F, Wu F-Q, Du S-Y, Cao Z, Shang Y, Wang X-L, Peng C-C, Yu X-C, Zhu S-Y: The Mg-chelatase H subunit is an abscisic acid receptor. Nature. 2006, 443 (7113): 823-826. 10.1038/nature05176.
Nishimura N, Sarkeshik A, Nito K, Park S-Y, Wang A, Carvalho PC, Lee S, Caddell DF, Cutler SR, Chory J: PYR/PYL/RCAR family members are major in-vivo ABI1 protein phosphatase 2C-interacting proteins in Arabidopsis . Plant J. 2010, 61 (2): 290-299. 10.1111/j.1365-313X.2009.04054.x.
Barrero JM, Piqueras P, González-Guzmán M, Serrano R, Rodríguez PL, Ponce MR, Micol JL: A mutational analysis of the ABA1 gene of Arabidopsis thaliana highlights the involvement of ABA in vegetative development. J Exp Bot. 2005, 56 (418): 2071-2083. 10.1093/jxb/eri206.
Estévez JM, Cantero A, Reindl A, Reichler S, León P: 1-Deoxy-D-xylulose-5-phosphate synthase, a limiting enzyme for plastidic isoprenoid biosynthesis in plants. J Biol Chem. 2001, 276 (25): 22901-22909. 10.1074/jbc.M100854200.
Iuchi S, Kobayashi M, Taji T, Naramoto M, Seki M, Kato T, Tabata S, Kakubari Y, Yamaguchi-Shinozaki K, Shinozaki K: Regulation of drought tolerance by gene manipulation of 9-cis-epoxycarotenoid dioxygenase, a key enzyme in abscisic acid biosynthesis in Arabidopsis . Plant J. 2001, 27 (4): 325-333. 10.1046/j.1365-313x.2001.01096.x.
Tatematsu K, Kumagai S, Muto H, Sato A, Watahiki MK, Harper RM, Liscum E, Yamamoto KT: MASSUGU2 encodes Aux/IAA19, an auxin-regulated protein that functions together with the transcriptional activator NPH4/ARF7 to regulate differential growth responses of hypocotyl and formation of lateral roots in Arabidopsis thaliana . Plant Cell. 2004, 16 (2): 379-393. 10.1105/tpc.018630.
Qin GJ, Gu HY, Zhao YD, Ma ZQ, Shi GL, Yang Y, Pichersky E, Chen HD, Liu MH, Chen ZL, Qu LJ: An indole-3-acetic acid carboxyl methyltransferase regulates Arabidopsis leaf development. Plant Cell. 2005, 17 (10): 2693-2704. 10.1105/tpc.105.034959.
Mikkelsen MD, Naur P, Halkier BA: Arabidopsis mutants in the C-S lyase of glucosinolate biosynthesis establish a critical role for indole-3-acetaldoxime in auxin homeostasis. Plant J. 2004, 37 (5): 770-777. 10.1111/j.1365-313X.2004.02002.x.
Brown DE, Rashotte AM, Murphy AS, Normanly J, Tague BW, Peer WA, Taiz L, Muday GK: Flavonoids act as negative regulators of auxin transport in vivo in Arabidopsis . Plant Physiol. 2001, 126 (2): 524-535. 10.1104/pp.126.2.524.
Jones B, Gunneras SA, Petersson SV, Tarkowski P, Graham N, May S, Dolezal K, Sandberg G, Ljung K: Cytokinin regulation of auxin synthesis in Arabidopsis involves a homeostatic feedback loop regulated via auxin and cytokinin signal transduction. Plant Cell. 2010, 22 (9): 2956-2969. 10.1105/tpc.110.074856.
Shin R, Burch AY, Huppert KA, Tiwari SB, Murphy AS, Guilfoyle TJ, Schachtman DP: The Arabidopsis transcription factor MYB77 modulates auxin signal transduction. Plant Cell. 2007, 19 (8): 2440-2453. 10.1105/tpc.107.050963.
Poppenberger B, Fujioka S, Soeno K, George GL, Vaistij FE, Hiranuma S, Seto H, Takatsuto S, Adam G, Yoshida S, Bowles D: The UGT73C5 of Arabidopsis thaliana glucosylates brassinosteroids. Proc Natl Acad Sci USA. 2005, 102 (42): 15253-15258. 10.1073/pnas.0504279102.
Szekeres M, Németh K, Koncz-Kálmán Z, Mathur J, Kauschmann A, Altmann T, Rédei GP, Nagy F, Schell J, Koncz C: Brassinosteroids rescue the deficiency of CYP90, a cytochrome P450, controlling cell elongation and de-etiolation in Arabidopsis . Cell. 1996, 85 (2): 171-182. 10.1016/S0092-8674(00)81094-6.
Domagalska MA, Schomburg FM, Amasino RM, Vierstra RD, Nagy F, Davis SJ: Attenuation of brassinosteroid signaling enhances FLC expression and delays flowering. Development. 2007, 134 (15): 2841-2850. 10.1242/dev.02866.
Suzuki N, Rizhsky L, Liang H, Shuman J, Shulaev V, Mittler R: Enhanced tolerance to environmental stress in transgenic plants expressing the transcriptional coactivator multiprotein bridging factor 1c. Plant Physiol. 2005, 139 (3): 1313-1322. 10.1104/pp.105.070110.
Gigolashvili T, Berger B, Mock H-P, Müller C, Weisshaar B, Flügge U-I: The transcription factor HIG1/MYB51 regulates indolic glucosinolate biosynthesis in Arabidopsis thaliana . Plant J. 2007, 50 (5): 886-901. 10.1111/j.1365-313X.2007.03099.x.
Clay NK, Adio AM, Denoux C, Jander G, Ausubel FM: Glucosinolate metabolites required for an Arabidopsis innate immune response. Science. 2009, 323 (5910): 95-101. 10.1126/science.1164627.
Griffiths J, Murase K, Rieu I, Zentella R, Zhang ZL, Powers SJ, Gong F, Phillips AL, Hedden P, Sun TP, Thomasa SG: Genetic characterization and functional analysis of the GID1 gibberellin receptors in Arabidopsis . Plant Cell. 2006, 18 (12): 3399-3414. 10.1105/tpc.106.047415.
Lange T, Hedden P, Graebe JE: Expression cloning of a gibberellin 20-oxidase, a multifunctional enzyme involved in gibberellin biosynthesis. Proc Natl Acad Sci USA. 1994, 91 (18): 8552-8556. 10.1073/pnas.91.18.8552.
Thomas SG, Phillips AL, Hedden P: Molecular cloning and functional expression of gibberellin 2-oxidases, multifunctional enzymes involved in gibberellin deactivation. Proc Natl Acad Sci USA. 1999, 96 (8): 4698-4703. 10.1073/pnas.96.8.4698.
Richmond TA, Bleecker AB: A defect in beta-oxidation causes abnormal inflorescence development in Arabidopsis. Plant Cell. 1999, 11 (10): 1911-1923.
J-h P, Halitschke R, Kim HB, Baldwin IT, Feldmann KA, Feyereisen R: A knock-out mutation in allene oxide synthase results in male sterility and defective wound signal transduction in Arabidopsis due to a block in jasmonic acid biosynthesis. Plant J. 2002, 31 (1): 1-12. 10.1046/j.1365-313X.2002.01328.x.
Stintzi A: The Arabidopsis male-sterile mutant, opr3, lacks the 12-oxophytodienoic acid reductase required for jasmonate synthesis. Proc Natl Acad Sci USA. 2000, 97 (19): 10625-10630. 10.1073/pnas.190264497.
Kachroo P, Venugopal SC, Navarre DA, Lapchyk L, Kachroo A: Role of salicylic acid and fatty acid desaturation pathways in ssi2-mediated signaling. Plant Physiol. 2005, 139 (4): 1717-1735. 10.1104/pp.105.071662.
Seo HS, Song JT, Cheong J-J, Lee Y-H, Lee Y-W, Hwang I, Lee JS, Do Choi Y: Jasmonic acid carboxyl methyltransferase: a key enzyme for jasmonate-regulated plant responses. Proc Natl Acad Sci USA. 2001, 98 (8): 4788-4793. 10.1073/pnas.081557298.
Chen F, D'Auria JC, Tholl D, Ross JR, Gershenzon J, Noel JP, Pichersky E: An Arabidopsis thaliana gene for methylsalicylate biosynthesis, identified by a biochemical genomics approach, has a role in defense. Plant J. 2003, 36 (5): 577-588. 10.1046/j.1365-313X.2003.01902.x.
Zhong Z-Q: Jatropha curcas, a promising biofuel plant. Tropical Plant Research. 1984, 25: 62-65.
Chen M-S, Wang G-J, Wang R-L, Wang J, Song S-Q, Xu Z-F: Analysis of expressed sequence tags from biodiesel plant Jatropha curcas embryos at different developmental stages. Plant Sci. 2011, 181 (6): 696-700. 10.1016/j.plantsci.2011.08.003.
Costa GGL, Cardoso KC, Del Bem LEV, Lima AC, Cunha MAS, De Campos-Leite L, Vicentini R, Papes F, Moreira RC, Yunes JA: Transcriptome analysis of the oil-rich seed of the bioenergy crop Jatropha curcas L. BMC Genomics. 2010, 11 (1): 462-10.1186/1471-2164-11-462.
Gomes KA, Almeida TC, Gesteira AS, Lôbo IP, Guimarães ACR, Miranda AB, Sluys MAV, Cruz RS: Cascardo JCM. Carels N: ESTs from seeds to assist the selective breeding of Jatropha curcas L. for oil and active compounds. Genomics Insights. 2010, 3 (1): 29-56.
De Hoon MJ, Imoto S, Nolan J, Miyano S: Open source clustering software. Bioinformatics. 2004, 20 (9): 1453-1454. 10.1093/bioinformatics/bth078.
Saldanha AJ: Java Treeview - extensible visualization of microarray data. Bioinformatics. 2004, 20 (17): 3246-3248. 10.1093/bioinformatics/bth349.
Huang X, Madan A: CAP3: A DNA sequence assembly program. Genome Res. 1999, 9 (9): 868-877. 10.1101/gr.9.9.868.
Zdobnov EM, Apweiler R: InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001, 17 (9): 847-848. 10.1093/bioinformatics/17.9.847.
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L: WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34: W293-W297. 10.1093/nar/gkl031.
Acknowledgments
This work was supported by funding from the Top Science and Technology Talents Scheme of Yunnan Province (2009CI123), the Natural Science Foundation of Yunnan Province (2011FA034), the National Natural Science Foundation of China (31370595), and the CAS 135 Program (XTBG-T02) awarded to Z.-F. Xu. Computational work was performed at the HPC Center, Kunming Institute of Botany, Chinese Academy of Sciences, China. The authors gratefully acknowledge the Central Laboratory of the Xishuangbanna Tropical Botanical Garden for providing the research facilities.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MSC, BZP and ZFX designed the experiments. MSC, GJW, JN and LN performed the BA treatment and sample collection. MSC and ZFX analyzed the data and drafted the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12870_2014_318_MOESM1_ESM.zip
Additional file 1: 10,569 probes representing 5,555 differentially expressed transcripts identified using microarray analysis. (ZIP 6 MB)
12870_2014_318_MOESM6_ESM.xls
Additional file 6: Oligonucleotides primers used for qRT-PCR validation of selected transcripts from microarrays and for expressional analysis of LFY, SOC1 and TFL1b. (XLS 18 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Chen, MS., Pan, BZ., Wang, GJ. et al. Analysis of the transcriptional responses in inflorescence buds of Jatropha curcasexposed to cytokinin treatment. BMC Plant Biol 14, 318 (2014). https://doi.org/10.1186/s12870-014-0318-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-014-0318-z