
Abstract
Background
In flowering plants, apomixis (asexual reproduction via seeds) is widely believed to result from failure of key regulators of the sexual female reproductive pathway. In the past few years, both differential display and RNA-seq comparative approaches involving reproductive organs of sexual plants and their apomictic counterparts have yielded extensive lists of candidate genes. Nevertheless, only a limited number of these genes have been functionally characterized, with few clues consequently available for understanding the molecular control of apomixis. We have previously identified several cDNA fragments with high similarity to genes involved in RNA biology and with differential amplification between sexual and apomictic Paspalum notatum plants. Here, we report the characterization of one of these candidates, namely, N69 encoding a protein of the S-adenosyl-L-methionine-dependent methyltransferases superfamily. The purpose of this work was to extend the N69 cDNA sequence and to characterize its expression at different developmental stages in both sexual and apomictic individuals.
Results
Molecular characterization of the N69 cDNA revealed homology with genes encoding proteins similar to yeast and mammalian trimethylguanosine synthase/PRIP-interacting proteins. These proteins play a dual role as ERK2-controlled transcriptional coactivators and mediators of sn(o)RNA and telomerase RNA cap trimethylation, and participate in mammals and yeast development. The N69-extended sequence was consequently renamed PnTgs1-like. Expression of PnTgs1-like during reproductive development was significantly higher in floral organs of sexual genotypes compared with apomicts. This difference was not detected in vegetative tissues. In addition, expression levels in reproductive tissues of several genotypes were negatively correlated with facultative apomixis rates. Moreover, in situ hybridization observations revealed that PnTgs1-like expression is relatively higher in ovules of sexual plants throughout development, from premeiosis to maturity. Tissues where differential expression is detected include nucellar cells, the site of aposporous initials differentiation in apomictic genotypes.
Conclusions
Our results indicate that PnTgs1-like (formerly N69) encodes a trimethylguanosine synthase-like protein whose function in mammals and yeast is critical for development, including reproduction. Our findings also suggest a pivotal role for this candidate gene in nucellar cell fate, as its diminished expression is correlated with initiation of the apomictic pathway in plants.
Similar content being viewed by others

Background
Gametophytic apomixis in flowering plants refers to asexual reproduction through seeds [1]. This reproductive mode can be achieved through diverse paths [2] and is widespread in angiosperms [3]. With two major distinctions, the developmental programs governing plant sexuality typically form the basis of apomixis. The first important difference involves differentiation of one or more functional unreduced female gametophytes, which occurs within the nucellus after either meiosis failure (diplosporous type) or nucellar cell fate alteration (aposporous type). Second, female gamete fertilization is not required for seed formation, leading to the development of a maternal embryo by parthenogenesis. The endosperm originates autonomously or after fertilization of the polar nuclei (pseudogamy).
Paspalum [4], one of the largest genera within Poaceae (Gramineae), encompasses approximately 370 species classified into four subgenera (Anachyris, Ceresia, Harpostachys, and Paspalum sensu stricto) (reviewed in [5]). Paspalum notatum is a member of the subgenus Paspalum and forms an agamic complex comprising self-sterile sexual diploids and self-fertile apomictic autotetraploids [5]. Paspalum notatum apomictic genotypes reproduce through apospory. In immature ovules at the premeiotic stage, one to several companion nucellar cells surrounding the megaspore mother cell enlarge, undergo a series of mitoses, and finally differentiate into non-reduced embryo sacs termed aposporous embryo sacs (AESs). AESs may coexist with the single meiotically-derived embryo sac (MES), or alternatively outcompete it and occupy the entire volume of the mature ovule [5]. The structure of a Paspalum notatum MES is typical of Gramineae species: one egg cell, two synergids, one bi-nucleated central cell, and a mass of proliferating antipodals [6]. On the other hand, AESs exhibit a distinct pentanucleate morphological structure (Paspalum-type embryo sacs) characterized by the presence of one egg cell, two synergids, and one bi-nucleated central cell but no antipodals; this structure allows straightforward classification by cytoembryological analysis of clarified ovules [7]. At anthesis, MESs undergo typical angiosperm double fertilization to produce viable seeds. In contrast, AESs usually develop into seeds after fertilization of the central cell with parthenogenetic embryo formation (pseudogamy). Nearly 100% of ovules of obligate aposporous individuals contain only AESs, whereas facultative aposporous individuals display variable proportions of mature mixed ovules that include both a MES and one or more AES. Although fully sexual polyploid individuals do not exist in nature, some genotypes have been produced artificially through colchicine-induced chromosome doubling of sexual diploids [8] or by crossing facultative apomicts [9].
Apomixis in Paspalum notatum is inherited as a dominant, monogenic trait, with a distorted segregation ratio, and is associated with a single genomic region, the Apospory Controlling Region (ACR) [7],[10]-[13]. Mapping approaches have revealed strong suppression of recombination within the Paspalum notatum ACR and disomic inheritance, whereas the remaining genomic regions show polysomic inheritance [12],[13]. Partial resolution of the ACR has unveiled a rather complex genomic structure comprising genomic sectors syntenic to rice chromosomes 2 and 12 but extensively rearranged through inversion, translocation, and/or insertion of low- and high-copy number retroelements [10],[11],[13]-[15]. These features are strongly consistent with the lack of recombination detected by genetic mapping and the distorted segregation ratios against apospory observed in some progenies. Whether the peculiar ACR genomic structure is pivotal to the transcriptomic changes required to switch from sexuality to apospory is nevertheless unknown. Interestingly, our current knowledge suggests that although gene-poor, the ACR contains several sequences putatively encoding proteins, including an MT-A70-like candidate (mRNA N6-adenosine-methyltransferase) [14]. This enzyme catalyzes N6-adenosine methylation in nascent mRNA and plays key roles in cell fate decision in multiple eukaryote systems [16]. In particular, MT-A70 loss of function in the model plant Arabidopsis thaliana leads to early embryo development failure [17]. Another interesting sequence mapped onto the ACR is a K homology (KH) domain-containing protein, which is an RNA-binding protein implicated in mRNA stability and regulation of gene expression at the post-transcriptional level [18],[19]. KH proteins have been associated with maintenance of an inactive chromatin state in knox genes located within the peripheral zone of the shoot apical meristem required for proper leaf development in maize [20].
Transcriptomic surveys have enabled the identification of numerous candidate genes associated with aposporous apomixis in plant species such as Brachiaria brizantha, Poa pratensis, Paspalum notatum, Panicum maximum, Boechera spp., Paspalum simplex and Hieracium spp. [21]-[27]. Comparative transcriptomic analysis of Paspalum notatum has suffered from several major drawbacks commonly found in apomictic systems: lack of genuine near-isogenic apomictic and sexual lines, high heterozygosity, and limited genomic resources (reviewed in [5]). RNA profiling assays based on differential display have been designed to overcome these difficulties and have been used to identify a set of 45 candidates usually down-regulated during apomictic development compared with sexual formation [23],[28]. Interestingly, two of these genes (N4 and N69) show significant similarity to genes encoding RNA methyltransferases [23].
The identification of an RNA-N6-adenosine-methyltransferase gene and an RNA-binding protein gene within the Paspalum notatum ACR, coupled with two additional RNA methyltransferases differentially expressed in flowers of sexual and apomictic plants, prompted us to explore the possible role of RNA methylation in aposporous reproductive development. As part of this effort, we sought to infer the possible function of the N69 cDNA candidate in reproductive development. To accomplish this goal, we characterized the candidate, identified its putative orthologs in model species, and analyzed its expression in reproductive tissues of sexual and apomictic genotypes at key developmental steps.
Results
N69 sequence characterization
The deduced amino acid sequence of the 888-nucleotide N69 cDNA fragment originally isolated from Paspalum notatum [23] showed strong homology with the RNA-cap guanine-N2 methyltransferase domain (PF09445; E-value: 1.4 × 10−46) [23]. In yeast, mammalian, and Drosophila genomes, PF09445 is encoded by a single gene, TGS1 (trimethylguanosime synthase), a conserved nucleolar methyl transferase responsible for the conversion of m(7)G cap of sn-, sno- and telomerase RNAs to m(2,2,7)G, as well as for nucleolar assembly and splicing of meiotic pre-mRNAs [29]-[31]. On the other hand, plant genomes contain two different genes encoding TGS1-like proteins. The first one, that is conserved across Eukaryotes, includes only the RNA-cap guanine-N2 methyltransferase domain (GRMZM2G151887, OS06T0187100, and AT1G30550, respectively in maize, rice and A. thaliana) while the second one displays a plant-specific architecture, with the RNA methyltransferase domain associated with a WW domain involved in protein-protein interactions (GRMZM2G347808, OS03T0396900 and AT1G45231, respectively in maize, rice and A. thaliana). By taking advantage of Roche 454 RNA sequencing data obtained from reproductive tissues of sexual and apomictic plants, which we combined with RACE experiments using Marathon cDNA libraries, we assembled a single contig representing the N69-extended cDNA consensus sequence covering the whole N69 CDS (see Methods and Figure 1). BLAST analyses further demonstrated that it derived from transcripts of a Paspalum notatum gene homologous to a plant specific member (WW + AdoMet donains) of theTGS1 family, that we consequently named PnTgs1-like. Note that the N69-extended sequence contains the complete CDS of PnTgs1-like but that we could not resolve the full 5′ and 3′ UTRs.

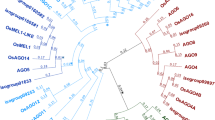
N69 partial cDNA derives from a plant-specific gene encoding a trimethyl guanosine synthase. (A) A consensus cDNA sequence was assembled using N69 cDNA, a N69-extended fragment obtained by RACE (black bars) and reads from RNA 454 sequencing in sexual (C4-4x) and apomictic (Q4117) reproductive tissues (white bars). This sequence contains a 2004 bp predicted ORF coding for a product sharing homology with yeast TRIMETHYL GUANOSINE SYNTHASE 1 (TGS1). (B) Primers were designed in order to amplify 4 different sequence fragments (F1, F2, F3, F4), which were amplified and sequenced for validation, from reproductive tissues of apomictic (Q4117) and sexual (C4-4x) genotypes. (C) Phylogenetic analysis of proteins similar to yeast TGS1 from several yeast and plant model species revealed that the Paspalum notatum predicted product is highly similar with plant specific members of the TGS1 protein family defined by the association of the RNA-cap guanine-N2 methyltransferase domain with a WW domain. Domains are indicated as colored ticks. ClustalW alignments are shown on the right (arrows indicate Pasalum notatum sequences).
Using this information, we designed a set of primers to amplify and sequence both overlapping fragments (F1, F2, F3) and the complete sequence (F4) from mRNA samples extracted from flowers of Q4117 (apomictic) and C4-4x (sexual) plants (Figure 1; Table 1). We recovered two complete sequences (ApoPnTgs1 and SexPnTgs1) with synonymous variant sites, including SNPs and a few INDELs (deposited in GeneBank under accession numbers BankIt1742582 ApoPnTgs1 KM114904 and BankIt1742582 SexPnTgs1 KM114905) but sharing 96.9% identity at the amino acid level, thus suggesting that PnTGS1-like is functional in both sexual and apomictic plants. The construction of a phylogenetic tree including 16 total TGS1-like protein sequences originated from yeast and plants revealed a higher similarity with plant-specific sequences, containing both WW + AdoMet domains (Figure 1).
Correlation of PnTgs1-like expression with reproductive behaviors
We first measured PnTgs1-like expression levels during reproductive development of apomictic (Q4117) and sexual (Q4188) plants using RNA samples extracted from spikelets collected at premeiosis, meiosis, postmeiosis, and anthesis. As shown in Figure 2A, quantitative analysis revealed significantly higher expression levels in the sexual genotype at all developmental stages. In addition, expression in the sexual genotype at anthesis was increased around 5-fold compared with that measured at premeiosis. Such an increase was not observed in the apomictic genotype.

Quantitative real-time PCR analysis of PnTgs1-like transcripts in sexual and apomictic Paspalum notatum plants. (A) Flowers were collected at premeiosis (pre), meiosis (mei), postmeiosis (post), and anthesis (ant) from Q4188 (sexual) and Q4117 (apomictic). (B) Expression was measured in three sexual and three apomictic individuals of an F1 population derived from Q4188 × Q4117 [12],[13].
To determine whether this differential expression was genuinely associated with the reproductive mode, we investigated PnTgs1-like expression in spikelets collected at anthesis from three sexual and three apomictic F1 plants derived from a Q4188 × Q4117 cross. All genotypes showed PnTgs1-like expression levels similar to those observed in the corresponding sexual and apomictic progenitors (Figure 2B).
We next verified whether PnTgs1-like expression levels were correlated with facultative apomixis rates previously recorded for Paspalum notatum. For this purpose, we used several genotypes whose apospory/sexuality expression ratios have been reported by Quarin et al. [9], Stein et al. [12], and Espinoza et al. [32] (Table 2). Interestingly, the expression level of PnTgs1-like increased with the degree of sexuality of the tested genotypes (Figure 3A). Moreover, we detected a positive correlation between PnTgs1-like expression levels and the percentage of ovules carrying MESs (R2 = 0.889); conversely, expression levels were negatively correlated with the percentage of ovaries carrying AESs (R2 = 0.889) (Figure 3B and C).

Correlation of PnTgs1-like expression at anthesis with reproductive behaviors. (A) Relative quantitative expression in sexual, facultative apomictic, and fully apomictic plants categorized according to the percentage of ovules carrying meiotic and aposporous embryo sacs (MES and AES, respectively) as shown in Table 2. PnTgs1 expression was oppositely correlated with the percentage of ovules carrying AESs (B) compared with MESs (C). B and C show plots of fitted values from linear regressions obtained using the lm command of the R program. Error bars indicate ranges of qPCR replicates
Finally, we detected similar PnTgs1-like expression levels in vegetative tissues (leaves and roots) of sexual and apomictic plants (Additional file 1). PnTgs1expression was detectable at levels that were lower but did not differ significantly from those measured in floral tissues. These results indicate that this gene might be performing a common role in non-reproductive tissues of both apomictic and sexual plants, but display a specific function in floral tissues associated with the reproductive mode.
In situ localization of PnTgs1-like expression
Using in situ mRNA hybridization, we investigated PnTgs1-like expression in Paspalum notatum spikelets sampled at two developmental stages critical to the success of apospory, late premeiosis/meiosis and anthesis, respectively concomitant with aposporous initial differentiation and embryo parthenogenesis.
During late premeiosis/meiosis, a strong signal was observed in nucellar cells and anther tapetum of the sexual genotype. Fainter signals at similar locations were detected in the apomictic genotype (Figure 4). The same expression trend was observed at anthesis: both nucellar and integumentary tissues displayed an intense signal in the sexual genotype. On the other hand, almost no signal was detected in the apomictic genotype (Figure 5).

In situ expression of PnTgs1-like in reproductive tissues of sexual (Q4188) and apomictic (Q4117) Paspalum notatum genotypes at late premeiosis/meiosis. Sexual genotype Q4188 reproductive tissues (ovaries + anthers) hybridized with an antisense PnTgs1-like RNA probe, showing expression mainly in the ovule (A, B, C). Larger images of ovules originated from sexual genotype Q4188 (D, E, F). Apomictic genotype Q4117 reproductive tissues (ovaries + anthers) displaying a lower expression of PnTgs1-like (G, H, I). Larger images of ovules originated from apomictic genotype Q4117 (J, K, L). White dotted lines indicate ovule boundaries. OV: ovule; MMC: megaspore mother cell; AI: apospory initials. Bars: 50 μm.

In situ expression of PnTgs1-like in reproductive tissues of sexual (Q4188) and apomictic (Q4117) Paspalum notatum genotypes at anthesis. Sexual genotype Q4188 ovaries hybridized with an antisense PnTgs1-like RNA probe, showing an intense signal in nucella and integuments (A, B, C). Larger images of ovules originated from sexual genotype Q4188 (D, E, F). Apomictic genotype’s (Q4117) ovaries displaying a lower expression of PnTgs1-like and showing several non-reduced embryo sacs within the ovule (G, H, I). Details of embryo sacs originated from apomictic genotype Q4117 (J, K, L). White dotted lines indicate ovule boundaries. OV: ovule; PN: polar nuclei; EA: egg apparatus. Bars: 100 μm.
These results indicate that PnTgs1-like is active in the nucellus of sexual Paspalum notatum plants from early developmental stages until maturity, whereas expression is strongly reduced throughout reproductive development in apomictic genotypes.
Discussion
The N69 cDNA fragment was initially recovered during screening to identify transcriptional differences between spikelets collected from sexual and apomictic Paspalum notatum plants [23]. Subsequent sequence analysis revealed that this fragment belongs to a gene encoding PIMT (PRIP-interacting protein with methyltransferase domain)/TGS1, a methyltransferase involved in sn(o)RNA biogenesis, mRNA splicing, and coactivation of PPAR (peroxisome proliferator activated receptor)-regulated gene expression (reviewed in [33]).
PIMT/TGS1, first isolated as a transcriptional co-activator PRIP-interacting protein from Saccharomyces cerevisiae [34], has been extensively studied in yeast, flies, and mammals (reviewed in [33]) but remains poorly characterized in plants. Interestingly, all eukaryotes possess a single tgs1 copy—except for plant genomes, which usually carry at least two copies. PIMT/TGS1 typically contains a methyltransferase domain and two binding domains; this structure allows interactions with RNA and S-adenosyl-L-methionine, the methyl donor in the methyl transfer reaction [34]. The post-transcriptional conversion of 7-methylguanosine caps (m7G) into 2,2,7-trimethylguanosine (m3G) catalyzed by PIMT/TGS1 plays a central role in the biogenesis of sn(o)RNAs and telomerase RNAs [30],[35]. In addition, PIMT/TGS1 is pivotal for transcriptional modulation in several contexts. It interacts with and co-localizes to the nucleus along with histone acetyl transferase (HAT)-containing transcriptional coactivators such as CBP/Ep300 and non-HAT-containing coactivators such as the Mediator subunit Med1 (PPAR binding protein; PBP/TRAP220/DRIP205) and PRIP [34],[36],[37]. PIMT has been proposed to serve as a molecular bridge between HAT- and non-HAT-containing transcriptional complexes and to control nuclear receptor-mediated transcription. Moreover, ERK2 phosphorylation at Ser298 of PIMT/TGS1 activates transcriptional activity at some promoters, suggesting a direct role for signal transduction pathways in modulating transcription [38].
The abolishment of PIMT/TGS1 function causes a wide range of phenotypic defects in different eukaryotic non-plant model systems. In S. cerevisiae, these alterations consist of cold-sensitive splicing defects, growth delay at low temperatures, loss of nucleolar structural organization, pre-rRNA processing deficiency, and meiotic failure after aberrant splicing of key regulators [29],[31],[35]. In mammals, PIMT/TGS1 loss of function leads to alteration of cell cycle progression, embryo lethality, and increased hepatic gluconeogenesis [38]-[40]. In Drosophila, embryo lethality in the early pupal stage has been reported [41].
Interestingly, comparative transcriptomic analyses of sexual vs. apomictic reproduction in plants, including Paspalum notatum, have provided sets of modulated genes enriched in ontological families strongly associated with PIMT/TGS1 function in eukaryotes. A major set consists of ribosomal RNAs and ribosomal protein genes [22],[23],[27], an observation consistent with the role of PIMT/TGS1 in both nucleolar organization and pre-rRNA processing [29]. Other TGS1-related functional classes that are differentially expressed in sexual and apomictic plants are proteasome-related proteins, cytoskeletal proteins, and ERK2 cascade member genes [22],[23],[27]. With regards to the body of data collected in non-plant species for TGS1 function, our observations for reduced expression of PnTgs1-like during female reproductive development in apomictic genotypes suggests that the transition from sexuality to aposporous development might depend on alterations of the splicing-machinery operating in conjunction with ERK2-mediated transcriptional regulation. Such differential expression was not observed in vegetative tissues. Whether a reduction of PnTgs1-like expression is causal for apomixis and which mechanisms are responsible for the differential expression in reproductive tissues definitively require further investigations, including trimethylguanosine synthase activity assays, RNA methylation profiling, and mutant analysis in model plant species.
Comparative transcriptional analyses and mutant characterization have revealed that apomictic developments likely emerged after alterations in the prevailing transcriptional dynamics of reproductive tissues or cells of sexual plants [42]. In particular, expression patterns in female reproductive tissues or cells are modulated during development by specific RNA-dependent DNA methylation pathways and, interestingly, some chromatin-remodeling enzymes participating in these pathways are down-regulated in maize-Tripsacum apomictic hybrids; their loss of function in maize causes profound reshaping of transcriptional activities and developmental heterochronicity partially mimicking apomictic developments [43],[44]. Defining the nature of the sequences targeted by these silencing pathways is critical to resolve two issues: the precise identification of these pathways’ roles in the evolution of apomictic reproduction from sexuality, and the elucidation of the mechanisms responsible for their chronological and spatial inhibition. With regard to the latter question, we believe that the function of TGS1 in both RNA biology and transcriptional pattern regulation offers the basis for an attractive model involving altered RNA processing to explain coordinated loss of function of key regulators.
Conclusions
Our results indicate that PnTgs1-like shows high expression in nucellar cells of sexual plants while its representation is significantly reduced in aposporous plants. Moreover, PnTgs1 expression levels are negatively correlated with the occurrence of AESs. At the same time, genes usually associated with PIMT function (such as rRNA and ribosomal protein genes) have been reported by our research group [23] and others [22],[27] to be differentially expressed between sexual and apomictic plants. These findings suggest that PnTGS1-like may participate in the repression of AES formation in nucellar cells surrounding the legitimate germ cell lineage. To confirm our hypothesis of a pivotal role for methylation-dependent RNA processing in the emergence of asexual reproductive pathways, we are currently using a transformation platform recently established in our laboratory [45] to assess the effects of the plant specific TGS1-like protein on the reproductive development of plant model species and Paspalum notatum.
Methods
Plant material
The following tetraploid (2n =4x =40) Paspalum notatum genotypes were used in this study: i) fully apomictic genotypes Q4117 and Q4012 [32],[46]; ii) facultative apomictic genotypes Q4064 and U47 [32]; iii) fully sexual genotypes C4-4x and Q4188 [8],[9]; and iv) three fully sexual (#43, #60, #93) and three fully apomictic (#40, #65, #74) F1 hybrids derived from a Q4188 × Q4117 cross [12],[13]. The Paspalum notatum genotypes were obtained from the IBONE live germplasm collection (Instituto de Botánica del Nordeste, IBONE-CONICET, Argentina).
cDNA sequencing
RACE experiments [47] were conducted, following the manufacturer’s instructions, on two cDNA Marathon libraries (Clontech, Mountain View, California, USA) produced from Q4117 and Q4188 mRNA samples extracted from spikelets during late premeiosis/meiosis (developmental stages I/II of Laspina et al. [23]). Primers were designed with Primer 3 Plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) [48]. PCR amplifications were performed using a DNA Engine Peltier thermal cycler (Bio-Rad, Hercules, California, USA) in 25-μl reactions containing tricine buffer (0.01 M tricine and 0.1 mM EDTA) supplemented with 1 μl of a 1:250 Marathon library dilution, 0.2 μM of each specific primer, 0.2 μM AP1 primer (matching the Marathon adapter AP1), 1× Taq-LOADTM Mastermix (MP Biomedicals), 1.5 U Taq polymerase (Promega, Madison, Wisconsin, USA), 1.5 mM MgCl2, and 200 pM dNTPs. Amplicons were electrophoresed on 1.5% (w/v) agarose gels and stained with 1% (v/v) ethidium bromide. Fragments of interest were purified with a QIAquick gel extraction kit (Qiagen, Valencia, California, USA), cloned into a p-GEM-T Easy vector (Promega), and transferred by thermal shock into Escherichia coli DH5α TOPO competent strands (Invitrogen/Life Technologies, Carlsbad, California, USA). Plasmids were purified using a QIAprep Spin Miniprep kit (Qiagen), and the amplified RACE fragments were sent to Beckman Coulter Genomics (London, UK) for sequencing.
For pyrosequencing, Q4117 (apomictic) and C4-4x (sexual) total RNA samples were extracted from balanced bulks of spikelets collected at premeiosis, meiosis, postmeiosis, and anthesis. mRNA enrichment, library preparation, emulsion PCR, 454 Genome Sequencer FLX + (Roche, Penzberg, Germany) sequencing, and bioinformatic analysis were performed by INDEAR (Instituto de Agrobiotecnología de Rosario, Rosario, Argentina).
Sequence analyses
The RACE fragment sequence was trimmed using the VecScreen algorithm at NCBI (www.ncbi.nlm.nih.gov), and the primer sequences were eliminated using Sequencher 4.1.4 (Gene Codes Corporation). The PnTgs1-like contig was then assembled in Sequencher from the RACE extended N69 sequence and the Roche 454 transcripts. BLASTN and BLASTX similarity analyses were conducted using NCBI (www.ncbi.nlm.nih.gov) and Gramene database (http://ensembl.gramene.org/genome_browser/index.html). Alignments were performed using Clustal Omega at the EBI-EMBL website (http://www.ebi.ac.uk/Tools/msa/clustalo/). Open reading frames were located with ORF finder (http://www.ncbi.nlm.nih.gov/gorf/), and translation was carried out using the ExPASy translate tool (http://web.expasy.org/translate/).
Quantitative real-time PCR
Total RNA was extracted from spikelets collected at several developmental stages (premeiosis, meiosis, postmeiosis, and anthesis) using an SV Total RNA isolation kit (Promega). cDNAs were synthesized from 1 μg of total RNA using Superscript II retrotranscriptase (Invitrogen/Life Technologies). Quantitative real-time PCR amplifications were performed in 25-μl final reaction volumes containing 200 nM gene-specific primers (N69F1 and N69R1; Table 1), 1× qPCR Real Mix (Biodynamics, Buenos Aires, Argentina), and 20 ng cDNA. Amplifications were performed in a Rotor-Gene Q thermocycler (Qiagen) programmed as follows: 2 min at 94°C followed by 45 cycles of 15 s at 94°C, 30 s at 55°C, and 40 s at 72°C. A melting curve (10-s cycles from 72 to 95°C, with the temperature increased by 0.2°C per cycle) was produced at the end of the cycling period. Quantitative real-time PCRs were performed in triplicate from cDNAs obtained from two biological replicates. Values were normalized using β-tubulin as an internal reference gene, since in former work this gene was reported to show a stable representation in flowers of sexual and apomictic plants of the same ploidy level in Poa pratensis and Paspalum notatum [22],[49],[50]. Relative expression levels were calculated using REST-RG software (Corbett Life Sciences).
In situhybridization experiments
Spikelets of sexual (Q4188) and apomictic (Q4117) Paspalum notatum genotypes were collected and fixed in a solution of 4% paraformaldehyde/0.25% glutaraldehyde/0.01 M phosphate buffer (pH 7.2), dehydrated in an ethanol/xylol series, and embedded in paraffin. Specimens were sliced into 10-μm-thin sections and placed onto slides treated with 100 μg ml−1 poly-L-lysine. Paraffin was removed with a xylol/ethanol series. Both sense (T7) and antisense (SP6) RNA probes were produced using a plasmid containing the N69 5′ RACE clone. Probes were labeled using a Roche DIG RNA labeling kit (SP6/T7) and hydrolyzed into 150–200-bp fragments. Following prehybridization at 37°C for 10 min in 0.05 M Tris–HCl buffer (pH 7.5) containing 1 μg ml−1 proteinase K, hybridization at 37°C was performed overnight in a buffer containing 10 mM Tris–HCl (pH 7.5), 300 mM NaCl, 50% deionized formamide, 1 mM EDTA (pH 8), 1× Denhardt’s solution, 10% dextran sulfate, 600 ng ml−1 total RNA, and 60 ng of the corresponding probe. Detection was performed following the Roche DIG detection kit instructions using anti-DIG AP and NBT/BCIP as substrates.
Availability of supporting data
The data set supporting the results of this study is included within the manuscript and its additional file(s).
Additional file
References
Nogler GA: Gametophytic Apomixis. Embryology of Angiosperms. Edited by: Johri BM. 1984, Springer, Berlin, 475-518. 10.1007/978-3-642-69302-1_10.
Crane CF: Classification of Apomictic Mechanisms. The Flowering of Apomixis: From Mechanisms to Genetic Engineering. Edited by: Savidan Y, Carman JG, Dresselhaus T. 2001, CIMMYT, IRD, European Commission DG VI (FAIR), Mexico City, 24-43.
Ozias-Akins P: Apomixis: developmental characteristics and genetics. Crit Rev Plant Sci. 2006, 25: 199-214. 10.1080/07352680600563926.
Linnaeus C: Systema Naturae. 1758, Holmiæ, Impensis direct. Laurentii Salvii (Salvius publ.), Stockholm, 10
Ortiz JPA, Quarin CL, Pessino SC, Acuña C, Martínez EJ, Espinoza F, Hojsgaard DH, Sartor ME, Caceres ME, Pupilli F: Harnessing apomictic reproduction in grasses: what we have learned from Paspalum . Ann Bot London. 2013, 112: 767-787. 10.1093/aob/mct152.
Quarin CL: The nature of apomixis and its origin in Panicoid grasses. Apomixis Newsletter. 1992, 5: 8-15.
Martínez EJ, Urbani MH, Quarin CL, Ortiz JPA: Inheritance of apospory in bahiagrass, Paspalum notatum . Hereditas. 2001, 135: 19-25. 10.1111/j.1601-5223.2001.00019.x.
Quarin CL, Espinoza F, Martínez EJ, Pessino SC, Bovo OA: A rise of ploidy level induces the expression of apomixis in Paspalum notatum . Sex Plant Reprod. 2001, 13: 243-249. 10.1007/s004970100070.
Quarin CL, Urbani MH, Blount AR, Martinez EJ, Hack CM, Burton GW, Quesenberry KH: Registration of Q4188 and Q4205, sexual tetraploid germplasm lines of Bahiagrass. Crop Sci. 2003, 43: 745-746. 10.2135/cropsci2003.0745.
Martínez EJ, Hopp HE, Stein J, Ortiz JPA, Quarin CL: Genetic characterization of apospory in tetraploid Paspalum notatum based on the identification of linked molecular markers. Mol Breed. 2003, 12: 312-327. 10.1023/B:MOLB.0000006868.12568.32.
Pupilli F, Martínez EJ, Busti A, Calderini O, Quarin CL, Arcioni S: Comparative mapping reveals partial conservation of synteny at the apomixis locus in Paspalum spp. Mol Genet Genom. 2004, 270: 539-548. 10.1007/s00438-003-0949-5.
Stein J, Quarin CL, Martínez EJ, Pessino SC, Ortiz JPA: Tetraploid races of Paspalum notatum show polysomic inheritance and preferential chromosome pairing around the apospory-controlling locus. Theor Appl Genet. 2004, 109: 186-191. 10.1007/s00122-004-1614-z.
Stein J, Pessino SC, Martínez EJ, Rodríguez MP, Siena LA, Quarin CL, Ortiz JPA: A genetic map of tetraploid Paspalum notatum Flügge (bahiagrass) based on single-dose molecular markers. Mol Breed. 2007, 20: 153-166. 10.1007/s11032-007-9083-0.
Podio M, Rodriguez MP, Felitti S, Stein J, Martinez E, Siena LA, Quarin CL, Pessino SC, Ortiz JPA: Sequence characterization, in silico mapping and cytosine methylation analysis of markers linked to apospory in Paspalum notatum . Genet Mol Biol. 2012, 35: 827-837. 10.1590/S1415-47572012005000070.
Podio M, Siena LA, Hojsgaard D, Stein J, Quarin CL, Ortiz JPA: Evaluation of meiotic abnormalities and pollen viability in aposporous and sexual tetraploid Paspalum notatum (Poaceae). Plant System Evol. 2012, 298: 1625-1633. 10.1007/s00606-012-0664-y.
Jia G, Fu Y, He C: Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2012, 29: 108-115. 10.1016/j.tig.2012.11.003.
Zhong S, Li H, Bodi Z, Button J, Vespa L, Herzog M, Fray RG: MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell. 2008, 20: 1278-1288. 10.1105/tpc.108.058883.
Burd CG, Dreyfuss G: Conserved structures and diversity of functions of RNA-binding proteins. Science. 1994, 265: 615-621. 10.1126/science.8036511.
Lorkovic ZJ, Barta A: Genome analysis: RNA recognition motif (RRM) and K homology (KH) domain RNAbinding proteins from the flowering plant Arabidopsis thaliana . Nucleic Acids Res. 2002, 30: 623-635. 10.1093/nar/30.3.623.
Buckner B, Swaggart KA, Wong CC, Smith HA, Aurnad KM, Scanlon MJ, Schnable PS, Janic-Buckner D: Expression and nucleotide diversity of the maize RIK gene. J Hered. 2008, 99: 407-416. 10.1093/jhered/esn013.
Rodrigues JC, Cabral GB, Dusi DMA, Mello LV, Rinden D, Carneiro VTC: Identification of differentially expressed cDNA sequences in ovaries of sexual and apomictic plants of Brachiaria brizantha . Plant Mol Biol. 2003, 53: 745-757. 10.1023/B:PLAN.0000023664.21910.bd.
Albertini E, Marconi G, Barcaccia G, Raggi L, Falcinelli M: Isolation of candidate genes for apomixis in Poa pratensis . Plant Mol Biol. 2004, 56: 879-894. 10.1007/s11103-004-5211-y.
Laspina NV, Vega T, Martelotto L, Stein J, Podio M, Ortiz JP, Echenique V, Quarin C, Pessino SC: Gene expression analysis at the onset of aposporous apomixis in immature inflorecences of Paspalum notatum . Plant Mol Biol. 2008, 67: 615-628. 10.1007/s11103-008-9341-5.
Yamada-Akiyama H, Akiyama Y, Ebina M, Xu Q, Tsuruta S, Yazaki J, Kishimoto N, Kikuchi S, Takahara M, Takamizo T, Sugita S, Nakagawa H: Analysis of expressed sequence tags in apomictic Guinea grass (Panicum maximum). J Plant Physiol. 2009, 166: 750-761. 10.1016/j.jplph.2008.10.001.
Sharbel TF, Voigt ML, Corral JM, Galla G, Kumlehn J, Klukas C, Schreiber F, Vogel H, Rotter B: Apomictic and sexual ovules of Boechera display heterochronic global gene expression patterns. Plant Cell. 2010, 22: 655-671. 10.1105/tpc.109.072223.
Polegri L, Calderini O, Arcioni S, Pupilli F: Specific expression of apomixis-linked alleles revealed by comparative transcriptomic analysis of sexual and apomictic Paspalum simplex Morong flowers. J Exp Bot. 2010, 61: 1869-1883. 10.1093/jxb/erq054.
Okada T, Hu Y, Tucker MR, Taylor JM, Johnson SD, Spriggs A, Tsuchiya T, Oelkers K, Rodrigues JCM, Koltunow AMG: Enlarging cells initiating apomixis in Hieracium praealtum transition to an embryo sac program prior to entering mitosis. Plant Physiol. 2013, 163: 216-231. 10.1104/pp.113.219485.
Pessino SC, Espinoza F, Martínez EJ, Ortiz JPA, Valle EM, Quarin CL: Isolation of cDNA clones differentially expressed in flowers of apomictic and sexual Paspalum notatum . Hereditas. 2001, 134: 35-42. 10.1111/j.1601-5223.2001.00035.x.
Colau G, Thiry M, Leduc V, Bordonné R, Lafontaine DLJ: The small nucle(ol)ar RNA cap trimethyltransferase is required for ribosome synthesis and intact nucleolar morphology. Mol Cell Biol. 2004, 24: 7976-7986. 10.1128/MCB.24.18.7976-7986.2004.
Franke J, Gehlen J, Ehrenhofer-Murray AE: Hypermethylation of yeast telomerase RNA by the snRNA and snoRNA methyltransferase Tgs1. J Cell Sci. 2008, 121: 3553-3560. 10.1242/jcs.033308.
Qiu ZR, Shuman S, Schwer B: An essential role for trimethylguanosine RNA caps in Saccharomyces cerevisiae meiosis and their requirement for splicing of SAE3 and PCH2 meiotic pre-mRNAs. Nucleic Acids Res. 2011, 39: 5633-5646. 10.1093/nar/gkr083.
Espinoza F, Daurelio LD, Pessino SC, Quarin CL, Valle EM: Genetic characterization of Paspalum notatum accessions by AFLP markers. Plant Syst Evol. 2006, 258: 147-159. 10.1007/s00606-005-0401-x.
Viswakarma N, Jia Y, Bai L, Vluggens A, Borensztajn J, Xu J, Reddy JK: Coactivators in PPAR-regulated gene expression. PPAR Res. 2010.
Zhu Y, Qi C, Cao WQ, Yeldandi AV, Rao MS, Reddy JK: Cloning and characterization of PIMT, a protein with a methyltransferase domain, which interacts with and enhances nuclear receptor coactivator PRIP function. Proc Natl Acad Sci U S A. 2001, 98: 10380-10385. 10.1073/pnas.181347498.
Mouaikel J, Verheggen C, Bertrand E, Tazi J, Bordonné R: Hypermethylation of the cap structure of both yeast snRNAs and snoRNAs requires a conserved methyltransferase that is localized to the nucleolus. Mol Cell. 2002, 9: 891-901. 10.1016/S1097-2765(02)00484-7.
Misra P, Qi C, Yu S, Shah SH, Cao WQ, Sambasiva Rao M, Thimmapaya B, Zhu Y, Reddy JK: Interaction of PIMT with transcriptional coactivators CBP, p300, and PBP differential role in transcriptional regulation. J Biol Chem. 2002, 277: 20011-20019. 10.1074/jbc.M201739200.
Kornberg RD: The molecular basis of eukaryotic transcription. Proc Natl Acad Sci U S A. 2007, 104: 12955-12961. 10.1073/pnas.0704138104.
Kapadia B, Viswakarma N, Parsa KVL, Kain V, Behera S, Suraj SK, Babu PP, Kar A, Panda S, Zhu Y, Jia Y, Thimmapaya B, Reddy JK, Misra P: ERK2-mediated phosphorylation of transcriptional coactivator binding protein PIMT/NcoA6IP at Ser298 augments hepatic gluconeogenesis. PLoS One. 2013, 8: e83787-10.1371/journal.pone.0083787.
Enünlü I, Pápai G, Cserpán I, Udvardy A, Jeang K, Borors I: Different isoforms of PRIP-interacting protein with methyltransferase domain/trimethylguanosine synthase localizes to the cytoplasm and nucleus. Biochem Biophys Res Commun. 2003, 309: 44-51. 10.1016/S0006-291X(03)01514-6.
Jia Y, Viswakarma N, Crawford SE, Sarkar J, Sambasiva Rao M, Karpus WJ, Kanwar YS, Zhu YJ, Reddy JK: Early embryonic lethality of mice with disrupted transcription cofactor PIMT/NCOA6IP/Tgs1 gene. Mech Dev. 2012, 129: 193-207. 10.1016/j.mod.2012.08.002.
Komonyi O, Pápai G, Enünlü I, Muratoglu S, Pankotai T, Kopitova D, Maróy P, Udvardy A, Boros I: DTL, the Drosophila homolog of PIMT/Tgs1 nuclear receptor coactivator-interacting protein/RNA methyltransferase, has an essential role in development. J Biol Chem. 2005, 280: 12397-12404. 10.1074/jbc.M409251200.
Grimanelli D: Epigenetic regulation of reproductive development and the emergence of apomixis in angiosperms. Curr Opin Plant Biol. 2012, 15: 57-62. 10.1016/j.pbi.2011.10.002.
Garcia-Aguilar M, Michaud C, Leblanc O, Grimanelli D: Inactivation of a DNA methylation pathway in maize reproductive organs results in apomixis-like phenotypes. Plant Cell. 2010, 22: 3249-3267. 10.1105/tpc.109.072181.
Singh M, Goel S, Meeley RB, Dantec C, Parrinello H, Michaud C, Leblanc O, Grimanelli D: Production of viable gametes without meiosis in maize deficient for an ARGONAUTE protein. Plant Cell. 2011, 23: 443-458. 10.1105/tpc.110.079020.
Mancini M, Woitovich N, Permingeat H, Podio M, Siena LA, Ortiz JPA, Pessino SC, Felitti SA: Development of a modified transformation platform for apomixis candidate genes research in Paspalum notatum (bahiagrass). In Vitro Cell Dev Biol Plant. 2014.
Ortiz JP, Pessino SC, Leblanc O, Hayward MD, Quarin CL: Genetic fingerprint for determining the mode of reproduction in Paspalum notatum, a subtropical apomictic forage grass. Theor Appl Genet. 1997, 95: 850-856. 10.1007/s001220050635.
Chenchik A, Diachenko L, Moqadam F, Tarabykin V, Lukyanov S, Siebert PD: Full-length cDNA cloning and determination of mRNA 5′ and 3′ ends by amplification of adaptor-ligated cDNA. Biotech. 1996, 21: 526-534.
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM: Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35: 71-74. 10.1093/nar/gkm306.
Podio M, Felitti SA, Siena LA, Delgado L, Mancini M, Seijo G, González AM, Pessino SC, Ortiz JPA: Characterization and expression analysis of SOMATIC EMBRYOGENESIS RECEPTOR KINASE (SERK) genes in sexual and apomictic Paspalum notatum . Plant Mol Biol. 2014, 84: 479-495. 10.1007/s11103-013-0146-9.
Felitti SA, Seijo JG, González AM, Podio M, Laspina NV, Siena L, Ortiz JPA, Pessino SC: Expression of lorelei-like genes in aposporous and sexual Paspalum notatum plants. Plant Mol Biol. 2011, 77: 337-354. 10.1007/s11103-011-9814-9.
Acknowledgments
This work was supported by the collaborative ECOS-MINCyT France-Argentina program (Project A11B02); Agencia Nacional de Promoción Científica y Tecnológica, Argentina (Project PICT 2011–1269); Universidad Nacional de Rosario, Argentina (Project AGR189) and Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina (Project PIP 11220090100613). Authors L. Siena, J. P. A. Ortiz, and S. Pessino are research staff members of CONICET.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LAS carried out RACE experiments, in situ hybridization experiments and participated in inflorescence collection, sample preparation, cytoembryological classification, and real-time PCR assays. JPAO participated in inflorescence collection, sample preparation, and sequence analysis, and helped to draft the manuscript. CM participated in sample preparation, sequencing and cloning experiments. OL conceived the study, helped collect inflorescences, participated in experimental design and coordination, and helped to draft the manuscript. SP conceived the study, performed real-time PCR assays, participated in experimental design, and drafted the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12870_2014_297_MOESM1_ESM.pdf
Additional file 1: Figure S1.: Real-time PCR analysis of PnTgs1 expression in leaves and roots. Description of data: Expression was detected in both leaves and roots, but no significant difference was observed between apomictic and sexual genotypes. On the contrary, differential expression was observed in flowers. Error bars indicate ranges of qPCR replicates. (PDF 445 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Siena, L.A., Ortiz, J.P.A., Leblanc, O. et al. PnTgs1-like expression during reproductive development supports a role for RNA methyltransferases in the aposporous pathway. BMC Plant Biol 14, 297 (2014). https://doi.org/10.1186/s12870-014-0297-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-014-0297-0