
Abstract
Background
Karst caves are considered as extreme environments with nutrition deficiency, darkness, and oxygen deprivation, and they are also the sources of biodiversity and metabolic pathways. Microorganisms are usually involved in the formation and maintenance of the cave system through various metabolic activities, and are indicators of changes environment influenced by human. Zhijin cave is a typical Karst cave and attracts tourists in China. However, the bacterial diversity and composition of the Karst cave are still unclear. The present study aims to reveal the bacterial diversity and composition in the cave and the potential impact of tourism activities, and better understand the roles and co-occurrence pattern of the bacterial community in the extreme cave habitats.
Results
The bacterial community consisted of the major Proteobacteria, Actinobacteria, and Firmicutes, with Proteobacteria being the predominant phylum in the rock, soil, and stalactite samples. Compositions and specialized bacterial phyla of the bacterial communities were different among different sample types. The highest diversity index was found in the rock samples with a Shannon index of 4.71. Overall, Zhijin cave has relatively lower diversity than that in natural caves. The prediction of function showed that various enzymes, including ribulose-bisphosphate carboxylase, 4-hydroxybutyryl-CoA dehydratase, nitrogenase NifH, and Nitrite reductase, involved in carbon and nitrogen cycles were detected in Zhijin cave. Additionally, the modularity indices of all co-occurrence network were greater than 0.40 and the species interactions were complex across different sample types. Co-occurring positive interactions in the bacteria groups in different phyla were also observed.
Conclusion
These results uncovered that the oligotrophic Zhijin cave maintains the bacterial communities with the diverse metabolic pathways, interdependent and cooperative co-existence patterns. Moreover, as a hotspot for tourism, the composition and diversity of bacterial community are influenced by tourism activities. These afford new insights for further exploring the adaptation of bacteria to extreme environments and the conservation of cave ecosystem.
Similar content being viewed by others

Background
The adaptation of life to extreme environments, which are steady or fluctuating habitats and not conducive for human, has recently become a hot topic for research. Extremophiles can colonize the extreme environment, and they are the sources of novel biomolecules and metabolic pathways [1, 2]. Karst caves are the terrain formed by the process of soluble rock dissolution [3]. Meantime, it is one of the extreme habitats with harsh conditions, for instance, nutrient-limitation, darkness, and high humidity [4]. Microorganisms can also act as the primary producers and sustain the cycles of substance and energy by chemoautotrophic and photosynthetic activities, which are normally found in various cave habitats [4,5,6,7]. These microorganisms participate in the geochemical cycles through inorganic chemical reactions in the cave ecosystems [8, 9]. However, our understanding of the microbial diversity, distribution patterns, and the roles of microorganisms in Karst caves is incomplete [3]. Exploring the microbial diversity and composition of Karst cave is particularly important for understanding the ecosystem and biodiversity in such extreme environment.
Previous studies about microorganisms in caves mostly focused on the microbial community composition, including describing and comparing the number of taxa, relative abundances, and alpha diversity [10], or investigating the microbial communities from different sample types such as wall surface [11], sediments [12], water [13,14,15], rock, and air [15]. However, the co-occurrence patterns of complex microbial communities are primarily unclear [16]. The co-occurrence network analysis allows us to explore the interactions between coexisting taxa in complex and diverse microbial communities [10, 17, 18]. Recently, it has been used to analyze microbial communities in complex habitats from gut intestine [19] to cave [15, 16] and ocean [20]. Network analysis approaches provide new insights for the interaction networks, structure, and niches distribution of communities in the Karst caves [10, 17, 21].
Zhijin cave exhibits the complex evolutionary process and the pattern of the plateau Karst in Guizhou province since the Paleogene. Meanwhile, it is a typical developmental zone and the microcosm of the plateau Karst in China [22]. Due to a large number of rare stalactites and the fantastic Karst landscape, Zhijin cave has become a tourist cave as well since 1983 and a global geological park. With the presence of tourists, the cave ecosystems such as the CO2 concentration, temperature, composition of microbial communities have affected by human activities [23,24,25,26]. However, the changes of microbial communities resulted in the loss of pigment on the surface of the walls and sediments in caves [27,28,29]. Hence, exploring the composition of bacterial community is important to conservation of cave. Recently, it was verified that the environment conditions in Zhijin cave have been changed, including the upward trend of the CO2 concentration and air temperature [30, 31], decreased relative humidity [31]. Moreover, the culturable bacterial composition in water has been impacted by tourism activities in Zhijin cave [32]. However, so far, the bacterial diversity and roles in the Zhijin cave are still unclear. To better understand the function and co-occurrence pattern of the bacterial community in the oligotrophic Karst cave, further reveal the potential impacts of tourism activities on bacterial diversity and composition in cave habitats, in this study, 16S rRNA high-throughput sequencing technology was used to analyze multiple different sample types, which would better reflect the composition of bacteria communities in Zhijin cave ecosystem. The bacterial composition, network structure, and function of the bacterial communities were further compared. We focus on the following objectives: (i) the composition characteristics of bacterial communities in the Karst tourism cave, (ii) their potential functions and metabolism pathways across different sample types, and (iii) the co-occurrence network patterns of bacterial communities in Zhijin cave.
Results
Sequence data
A total of 900,491 reads for 16S ribosomal RNA sequencing were successfully obtained from all the samples collected from seven sites in Zhijin cave (Supplemental file 1: Table S1). After removing low-quality, replicate, and potential chimera tags, we obtained 827,376 tags ranging from 39,403 to 93,528 per sample site. Based on 97% similarity, 27,421 OTUs were obtained across all samples.
Bacterial community composition
A total of 54 phyla, 750 genera, and 407 species were determined in three sample types. We selected the top 10 most abundant phyla in each sample type to further measure the composition of bacterial community. Their relative abundances were shown in Fig. 1a. We found that 9 phyla (Proteobacteria, Actinobacteria, Firmicutes, Acidobacteria, Nitrospirae, Bacteroidetes, Planctomycetes, Chloroflexi, and Gemmatimonadetes) were shared by all sample types. Among the top 10 predominant phyla in samples, Chlorobi was not detected in soil samples; Verrucomicrobia was not found in rock samples; Thaumarchaeota was only obtained in stalactite samples. The Proteobacteria had the highest relative abundance in the bacterial community across all sample types.

Bacterial community composition. a Relative abundances of the 10 most abundant phyla in each sample. The relative abundance not shown in chart if fewer than 4%. b Non-metric multidimensional scaling (NMDS) of bacterial community in three sample types
The relative abundances of Proteobacteria were 63.41, 65.15, and 69.79% in rock, soil, and stalactite samples, respectively. In both rock and stalactite samples, Actinobacteria remained to be the second abundant phylum in the bacterial communities (16.53 and 8.53%, respectively), and then followed the phylum Firmicutes (4.16 and 5.77%, respectively). Whereas, in soil samples, the second abundant phylum in bacterial community was Firmicutes (17.58%), which was more abundant than that in rock and stalactite samples. The Abundances of other phyla were less than 4% across the three sample types.
At the species level, the NMDS ordination method showed that bacterial communities were separately different among the three sample types (Fig. 1b). The results were consistent with the significant test that the means of the distances were considered as different (P < 0.01) among the three sample types. Besides, the results from ANOSIM showed that the differences of bacterial community compositions were significant between rock and soil samples (R = 0.70, P < 0.01), rock and stalactite samples (R = 0.74, P < 0.01), as well as stalactite and soil samples (R = 0.79, P < 0.01).
Bacterial groups with statistical differences
The LEfSe analysis was performed for comparing bacterial communities to find the specialized bacterial groups within each type of the samples. The cladogram (Fig. 2a) showed that 2 phyla, 2 classes, 8 orders, 15 families, and 26 genera were significantly variable across the three sample types. From phylum to species, there were 15, 9, and 17 groups of bacteria enriched in rock, soil, and stalactite samples, respectively. Indicator groups represented the abundance differentiation of the bacterial group (LDA value of 3 or higher) among the three sample types (Fig. 2b). There were 9 differentially abundant bacterial groups in soil samples (e.g. Bacillales, Psychrobacillus, and Planococcaceae). A total of 4 bacterial groups (e.g. Acinetobacter, Moraxellaceae, and Rhodocyclaceae) were significantly more abundant in rock samples, and 4 taxa (e.g. Salinisphaeraceae and Lactobacillales) were overrepresented in stalactite samples.

The results of LEfSe analysis. a Cladograms indicating the phylogenetic distribution of bacterial lineages associated with the samples. b Indicator bacterial group significantly differentiated across the three sample types with LDA values higher than 3
Venn diagram and bacterial diversity
To further obtain insight into the differences of bacterial communities across the three sample types, the Venn analysis of the OTUs was performed, which demonstrated that OTUs differed across the three sample types (Supplemental file 2: Figure S1). The number of site-specific OTUs ranged from 175 (soil samples) to 435 (rock samples), and a total of 322 OTUs were shared among all three sample types. According to the OTUs identified at different levels of taxon, the Simpson’s index, Shannon’s index, and Simpson evenness were calculated. The Simpson’s index (0.87–0.91) and Shannon’s index (3.63–4.71) indicated that the level of diversity varied among three sample types. The average Simpson indices were 0.91, 0.89, and 0.87 in rock, soil, and stalactite samples, respectively. The average Shannon’s indices were 4.71, 3.63, and 4.17 in rock, soil, and stalactite samples, respectively. The greatest bacterial diversity was observed in rock samples. However, the LSD test showed that the diversity indices were not significantly different across the three sample types.
Inferred bacterial function by PICRUSt
Based on the PICRUSt analysis, the results of KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway abundance in each sample were obtained (Fig. 3a). A total of 6 functional modules that represented approximately 86% of the entire dataset in samples were detected, including cellular processes (4%), environmental information processing (15%), genetic information processing (15%), human diseases (1%), metabolism (49%), and organismal systems (1%). The most abundant functional module was metabolism across the three sample types. In generally, the functional modules were richer in stalactite samples than those in other sample types.

The PICRUSt predicted function in samples. a Predicted function of bacteria among the three sample types. b The second level of KEGG pathway was shown in the heatmap
A total of 41 pathways were predicted across the three sample types (Fig. 3b). Among them, there were 37 pathways in stalactite samples (e.g. cell communication, sensory system, signaling molecules and interaction, and membrane transport), which were more abundant than those in the other two sample types. Moreover, cellular processes and organismal systems (e.g. transport and catabolism, endocrine system, and digestive system) were overrepresented in rock samples. In soil samples, only the cardiovascular diseases pathway was more abundant than that in other sample types.
In addition, to understand the potential metabolism of carbon and nitrogen reactions in which the bacteria participated in, we also aimed to detect the relative encoding genes and enzymes for carbon and nitrogen metabolisms in the samples of Zhijin cave. It showed that carbon metabolism pathways, including glycolysis pathway, pentose phosphate pathway, methanogenesis pathway, and 6 carbon fixation pathways (reductive pentose phosphate cycle, 3-hydroxypropionate bicycle, reductive citrate cycle, hydroxypropionate-hydroxybutyrate cycle, reductive acetyl-CoA pathway, and dicarboxylate-hydroxybutyrate cycle), possibly existed in Zhijin cave. The predicted relative enzymes were shown in Supplemental file 3: Table S2, e.g. hexokinase [EC:2.7.1.1], glyceraldehyde 3-phosphate dehydrogenase [EC:1.2.1.12], and pyruvate kinase [EC:2.7.1.40] involved in the glycolysis pathway; hexose-6-phosphate dehydrogenase [EC:1.1.1.47 3.1.1.31], glucose-6-phosphate isomerase [EC:5.3.1.9], and transaldolase/glucose-6-phosphate isomerase [EC:2.2.1.2 5.3.1.9] involved in the pentose phosphate pathway; heterodisulfide reductase subunit D [EC:1.8.98.1], methyl-coenzyme M reductase alpha subunit [EC:2.8.4.1], and F420-non-reducing hydrogenase large subunit [EC:1.12.99.- 1.8.98.5] involved in the methanogenesis pathway; and enzymes related to carbon fixation pathways (e.g. 4-hydroxybutyryl-CoA dehydratase [EC:4.2.1.120 5.3.3.3], ribulose-bisphosphate carboxylase [EC:4.1.1.39], and malonyl-CoA reductase [EC:1.2.1.75 1.1.1.298]).
Moreover, total of 5 related nitrogen metabolism pathways, including nitrogen fixation process, denitrification pathway, dissimilatory nitrate reduction pathway, assimilatory nitrate reduction reaction, and complete nitrification pathway, were revealed. The predicted related enzymes were shown in Supplemental file 4: Table S3, e.g. nitrogenase delta subunit [EC:1.18.6.1], nitrogenase iron protein NifH [EC:1.18.6.1], and ammonia monooxygenase subunit C [EC:1.13.12.-] involved in the nitrogen fixation process; hydroxylamine dehydrogenase [EC:1.7.2.6] involved in the nitrification pathway; hydroxylamine oxidase [EC:1.7.3.4], nitrite reductase (NO-forming) [EC:1.7.2.1], and nitrate reductase (cytochrome) [EC:1.9.6.1] involved in the denitrification pathway; nitrate reductase (cytochrome) [EC:1.9.6.1] and nitrate reductase/nitrite oxidoreductase beta subunit [EC:1.7.5.1 1.7.99.-] involved in the dissimilatory nitrate reduction pathway; ferredoxin-nitrate reductase [EC:1.7.7.2] and assimilatory nitrate reductase electron transfer subunit [EC:1.7.99.-] involved in the assimilatory nitrate reduction reaction.
Bacterial co-occurrence network analysis
The correlation coefficient (r > ±0.8, P < 0.01) co-occurrence network analysis (Fig. 4 & Supplemental file 1: Table S4) showed that the edges in the network included 1127 strong positive correlations and 184 negative correlations in the rock samples; 1353 strong positive correlations and 32 negative correlations in the soil samples; and 1263 strong positive correlations and 145 negative correlations in the stalactite samples. The modularity indices of the three sample types were all greater than 0.40, which suggested that the co-occurrence networks of bacterial communities had a strong modular structure and complex species interaction across the three sample types in Zhijin cave [33]. Comparing to a randomized network, three non-random co-occurrence networks were observed in Zhijin cave (P < 0.01).

The co-occurring network analysis of the bacterial communities across the three sample types. The nodes are colored by phylum level, the size of each node is proportional to the relative abundance of specific genus level. The color of each edge is positive and negative of correlation coefficient, grey represents positive correlation, and red represents negative correlation. The thickness of each edge is proportional to the correlation coefficient (Spearman’s r > ±0.8 and P < 0.01)
In the co-occurrence network, the rock samples presented a shorter average path length (the shortest among all possible pairs of nodes) and a lower diameter (the longest of the shortest paths among all pairs of nodes) (1.98 and 2.00) than those in soil samples (3.66 and 16.00) and stalactite samples (7.84 and 23.00), which reflected a more efficient information processing and substance transmission among the species in rock samples. Furthermore, the C score (co-occurrence index) was 0.33, and the robustness (stability of community) of bacterial community was 0.86 in the rock samples. The greater values of co-occurrence index indicated that bacteria were highly exclusive, and the large robustness represented that the bacterial community was more stable. Compared with the rock and stalactite samples, the clustering coefficient (the proportion of neighboring nodes that can be reached through the nodes connecting other neighbors) was greater in the soil samples (0.97). The bacterial network had lower values of co-occurrence index (0.24) and robustness (0.77) in the soil samples. These results reflected that a relatively high rate of cooperation was formed by a higher clustering coefficient and more bacteria were co-occurred in the community with a low stability in soil samples.
The node degree (the number of ties with other nodes) reflects the role of node. According to the node degree of each node, the keystone taxa in each sample type were captured. The largest node degree was found with Salinarimonas (466 links), Cloacibacterium, and Marinospirillum (both 17 links) in rock samples; Pasteurella, Dietzia, Hungatella, and Beggiatoa (all 37 links) in soil samples; and Streptomyces, Pseudolabrys, and Ignavibacterium (all 20 links) in stalactite samples, suggesting that these groups were more prominent than the other groups with fewer links in the same network.
Discussion
Composition of bacterial community in different sample types
The environment of Zhijin cave was occupied by diverse bacteria. Though the Karst cave is an extreme habitat with nutrition deficiency, light limitation, and oxygen deprivation [3, 4], our results showed that 54 phyla with 750 genera were detected from the rock, stalactite, and soil samples, indicating a rich bacterial diversity in the cave. The Proteobacteria was the predominant group across all the three sample types. Proteobacteria, as a group of microbes responding to unstable carbon sources, was found to be the most abundant in some other cave ecosystems and various environments as well [25, 34,35,36,37]. The Firmicutes was the second abundant phylum found in soil samples in Zhijin cave, possibly due to their resistance towards nutrient stress and capability of survival under most extreme habitats [25, 38]. For other bacteria groups found in samples, the Actinobacteria is considered as the production source of bioactive compounds, but knowledge of their diversities in caves is very limited [4]. The Bacteroidetes and Acidobacteria are capable of decomposing organic compounds in environments [39, 40], and the Acidobacteria, specifically, as oligotrophic organism was negatively correlated with nutrient levels [41, 42]. As autotrophic green non-sulfur bacteria, the Chloroflexi, which can fix CO2, was a prevalent phylum and frequently found in caves [43, 44]. The presence of the Nitrospirae is crucial for the nitrogen cycle, since several nitrite oxidizers are present in Nitrospirae, carrying out nitrification and supplying nitrogen for oligotrophic environments [45]. In addition, the Thaumarchaeota, containing ammonia-oxidizing archaea [46], could obtain energy by oxidizing ammonia and fixing carbon in oligotrophic environments [47, 48].
Cave habitat is the important factor affecting bacteria survival, as a result of which, different bacterial communities were found in various niches in cave environments [49]. The observed divergent compositional structure and diversity of bacterial communities in different extreme cave environments might be due to the differences in cave environments, sampling mediums, and analysis methods, to a certain extent [50]. The indicator bacteria were significantly different among three sample types. The soil samples had the most abundant indicator taxa (e.g. Bacillales, Psychrobacillus, and Planococcaceae), and the LEfSe analysis also revealed more specific bacterial groups in soil samples than those in the other sample types. These indicator taxa in samples implied that these bacterial groups specifically functioned in their special habitats. For example, Bacillales is the common group in soil [51] and on mineral surface [52], and some Bacillales [53, 54] and Psychrobacillus [55] are able to degrade oils in contaminated soil. Salinisphaera was the indicator taxon in stalactite samples, and previous studies illustrated that several strains of Salinisphaera are halophilic bacteria [56, 57] and capable of fixing CO2 by using organic carbon sources [58]. It implied that Salinisphaera might be involved in the process of stalactite formation by fixing carbon cycle. Further, Moraxellaceae strains have the ability to bear the natural transformation and most of them usually inherently reside on the mucosal membranes in humans and other animals [59].
It was worth noting that there are some hints that the bacteria diversity and composition in Zhijin cave might be influenced by the changed ecosystem because of human activities. Firstly, we observed the relatively lower bacterial diversity indices in rock samples [11, 15], soil samples [45, 60], or stalactite samples [11] from Zhijin cave than those in many natural caves currently reported. Several studies using culture-based method also uncovered that microbial diversity in show caves was lower than that in natural caves [25, 26]. In some instances, the diversity of fungi was influenced by the level of anthropogenic disturbance, in which lower diversity was found in areas with heavy disturbance and higher diversity was found in sites with moderate disturbance [26]. It hinted that the lower bacterial diversity might be the consequence of tourism in Zhijin cave. Secondly, the predominance of Proteobacteria in Zhijin cave was consistent to the previous reports in tourist cave ecosystems [25, 34,35,36]. The Firmicutes dominated in bacterial community in natural cave or areas with less tourism influence, and conversely [25]. Lastly, several microbial groups of anthropogenic origin [61], such as Lactobacillus, Bacteroides, Staphylococcus, and Moraxella genera, also observed in our samples (data not shown). Here, we further inferred that the impact of human activities on cave ecosystem. With the human access and mass activities of tourists, certain energy and exogenous bacteria are potentially transported into the cave system following the dropping skin, hair, sweat, and clothing [25, 27, 49, 50]. The organics introduced from human activities provide new nutrients and habitats for some bacteria, moreover, resulted in the changes of bacterial community in cave. In additionally, the human activities affected the cave microenvironment and led to increased CO2 concentration, raised temperature, and decreased relative humidity [12, 26]. These further have a profound influence on the processes of the cave forming and the bacterial composition in cave. Integrating the tourism activities, the changed the environment conditions in the Zhijin cave, such as concentration CO2 [30, 31], and these traits of bacterial diversity and composition, which further confirmed that the cave ecosystem were disturbed by human activities in Zhijin cave. As indicators of cave tourism, monitoring the bacterial diversity and composition is crucial to comprehend human impacts and the changes of bacteria in cave habitats [62, 63].
Prediction of bacterial functions using PICRUSt
To respond to the severely limited resource, chemolithotrophic microorganisms create biogenic energy and nutrients in the cave ecosystem using sulfur and metal irons [64, 65]. Besides, other metabolic processes such as nitrogen fixation, carbon fixation, and carbon mineralization allow microorganisms to maintain the cave ecosystem [63, 66]. Similarly, we found that the bacteria can survive through various metabolic processes in Zhijin cave. The co-existent bacteria could involve in a variety of complex metabolic reactions in the cave habitat, which was supported by the relative genes and enzymes for carbon and nitrogen metabolism pathways detected by PICRUSt analysis across the three sample types.
We obtained 6 functional modules in the samples, and the predominant module was the metabolism. However, due to the lack of photosynthesis in cave environments, the autotrophic bacteria may act as both the primary producer and the common energy input [67,68,69]. Carbon fixation is considered as a watershed between the heterotroph and autotroph organisms. Autotrophic organisms can fix CO2 through Calvin cycle, which are widely distributed in environments [70]. The ribulose-bisphosphate carboxylase, which is the rate-limited enzyme in Calvin cycle, was observed in the samples, suggesting that Calvin cycle appeared to exist in the environment of Zhijin cave. Moreover, bacteria may utilize several other specific metabolic pathways in extreme environments. The 4-hydroxybutyryl-CoA dehydratase is the indicator enzyme of 3-hydroxypropionate/4-hydroxybutyrate cycle [47, 71] and dicarboxylate cycle/4-hydroxybutyrate cycle [72], involving CO2 fixation in archaea [47] and Thaumarchaeota [73]. According to the previously reported study, 4-hydroxybutyryl-CoA dehydratase was shown abundant in oligotrophic environments and contributed to the processes of carbon assimilation in cave environments [74]. The 3-hydroxypropionate bicycle is another autotrophic carbon fixation pathway, as a new CO2 fixation pathway in Chloroflexus [44, 45]. The malonyl-CoA reductase is the key enzyme of 3-hydroxypropionate bicycle, which was observed in several green non-sulfur bacteria for autotrophic CO2 fixation [44].
For the nitrogen cycle, previous studies mostly focused on the role of bacteria in soil, rock, aquatic, and other oligotrophic conditions [75]. Similarly, we also detected the relative enzymes for nitrogen cycle from samples in Zhijin cave. These enzymes involved nitrogen fixation, denitrification, nitrate reduction, nitrate reduction, and nitrification were predicted. Due to the limitation of nitrogen resource in the cave ecosystems, bacteria could survive by specific strategies and metabolic pathways [34, 76,77,78]. Some autotrophic bacteria in Nitrospirae, Chloroflexi, and Chlorobi identified in Zhijin cave, might transfer the N element into the nitrogen cycle by the nitrogenase NifH or chemoautotrophic process, utilize inorganic compounds such as ammonia by the nitrification, and promote the nitrogen cycle in environment [6]. In addition, the nitrite reductase is a key enzyme in the dissimilatory denitrification [79], the bacteria catalyze the reduction of nitrite to nitric oxide in the environments by using the nitrite reductase, and may contribute to reduce nitrogen loss in oligotrophic cave [32]. The ammonia-oxidizing bacteria containing hydroxylamine oxidase probably come from the Nitrospirae and Thaumarchaeota found in Zhijin cave, are the predominant ammonia oxidizers and participate in the ammonia oxidation process which is the first key step of nitrification [5, 32, 80]. In a conclusion, the presences of these metabolic enzymes suggested that bacteria, including some autotrophic bacteria, can survive by participating in specific metabolic pathways in the nutrient-limited Zhijin cave.
In addition, the abundances of metabolic pathways were divergent across the three sample types in Zhijin cave. This probably indicated that the bacterial metabolic activities are divergent, functional bacteria involved in different dynamic activities could drastically shift in different samples and habitats in the cave system, and specific microorganisms play a key role in energy transformation and different geological cycles [81].
Co-occurrence pattern of bacterial communities
The bacterial co-occurrence networks had different topological properties and complex species interactions across all the three sample types in Zhijin cave in our study. We observed that a great number of edges, high robustness, and modularity in each sample, indicating that steady and complex interactions and strong modular structure were present in the bacterial communities [33]. Moreover, the low values of average path length and diameter in rock samples illustrated that the information and the substances were quickly transmitted among species in the bacteria communities. This implied that the bacteria had a higher transmission powers in the rock samples than in the other samples [82]. Whereas, the highest co-occurrence index detected in the rock samples implied a lower co-existence degree in the bacterial communities. For the soil and stalactite samples, the clustering coefficients were 0.97 and 0.74 in the bacterial networks respectively, suggesting the relatively strong correlation of co-occurrence network [16]. The lower co-occurrence index, higher clustering coefficient, and lower negative edges all hinted that the bacterial community in the soil samples possessed higher cooperation. Previous study reported that most ecological networks had a low value of connectance [83], the same results of which were obtained in all the samples from Zhijin cave as well in this study. As a matter of fact, networks with low connectance indicate a power-law distribution [83, 84].
Though the keystone taxa were divergent in different sample types, the bacterial groups had a number of links with the other groups in the same communities. It implied that these bacteria taxa perform key roles or are responsible for the interactions among the communities through specific metabolic activities. For example, Salinarimonas rosea sp. nov. is a halotolerant bacterium and capable of reducing the nitrate [85]; Pasteurella could oxidize organic compounds and assimilate sulfur compounds [86]; and Streptomyces may produce various and complex secondary metabolites [87]. These keystone taxa involve in complex metabolic cycles and supply primary or secondary metabolic products for cooperators of the communities in cave.
Furthermore, the networks were consisted of a number of positive edges but fewer negative edges in each sample type in Zhijin cave. This species co-occurrence patterns were displayed by the co-occurring and positive interactions among different phyla. Several previous reports uncovered that metabolic exchanges were detected in nutrients-limited environments [88,89,90]. These examples indicated that the metabolic cooperation could drive the co-occurrence pattern of bacteria and shape the compositions of communities [88]. Thereby, the different co-occurrence network structures of the bacterial communities could be explained by the interdependencies in the microorganisms from the collected samples. The consistent result was revealed by the network descriptors with disparate values of networks and the compositions of the communities in Zhijin cave.
Conclusions
Network analysis allows us to explore the composition and interaction of a community. In this study, our results confirmed that the tourism activities could influence the bacterial diversity and composition in Zhijin cave. In the dark and oligotrophic cave, the bacteria could co-exist through positive interactions and cooperation by participating in diverse metabolic pathways. This study develops a better understanding of the adaptation and interaction patterns of bacterial communities in extreme habitats, and provides the evidence for the development and conservation of cave system.
Methods
Study sites and sample collection
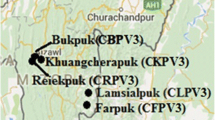
Zhijin cave is located in Guizhou Province in China (26°38′31″-26°52′35″N, 105°44′42″-106°11′38″E). The average annual temperature is 18–20 °C, the average annual humidity is 90%, and the average CO2 concentration is 0.20%. Three types of samples, including rock, soil, and stalactite, were collected in December 2016. Each type of sample was collected from five different sites (Fig. 5). To collect the samples on the rocks and stalactites, forty-five swabs moistened with sterile deionized H2O were used to swab the surface (4 cm2 per swab) of the rocks or stalactites at individual sampling site [74]. Soil samples (200 g) were collected from the surface soil (0–10 cm) with a spade at each sampling site. The swabs and soils were placed in tubes, which were then capped, placed on ice, and immediately transported back to store at − 20 °C for further DNA extraction in the laboratory.

Distribution of sampling sites. A, J, S, T, W, X, Y represent different sampling sites
DNA extraction and sequencing
Genomic DNA was extracted [12] from 30 swabs or 1 g soil. The sample material was resuspended in 1500 μL of TES buffer (0.3 M sucrose, 25 mM Tris-HCl pH 8.0, 0.25 mM EDTA pH 8.0) supplemented with 50 μL Lysozyme (20 mg/mL). The mixture was vortexed for one minute and incubated for 1 h at 37 °C. Then 30 μL Proteinase K (20 mg/mL) and 200 μL 10% sodium dodecyl sulphate were added to the tubes, followed by vortex for 3 min and incubation for 2 h at 55 °C. Subsequently, 200 μL 5 M sodium chloride and 400 μL CTAB/NaCl were added, and the mixture was vortexed for 5 min and incubated for 30 min at 65 °C. The resulting lysate was extracted with phenol: chloroform: isoamyl alcohol (volume 25:24:1). The DNA was precipitated by adding 0.1 volume of 3 M Na-acetate (pH 4.8) and 0.6 volume of isopropanol. The DNA pellets were air-dried under − 20 °C temperature for 2 h, resuspended in 100 μL ddH2O, and stored at − 20 °C.
Finally, amplicon sequencing was conducted with the extracted DNA using an Illumina MiSeq platform following the protocols described by previous study [74]. The gene-specific primers 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) were developed based on the V4 region of the bacterial 16S rRNA gene [91]. DNA amplification was carried out by polymerase chain reaction (PCR) with the Thermo Scientific Phusion High-Fidelity PCR Master Mix (New England Biolabs, UK), and the extracted DNA solution was diluted to 1 ng/μL for amplification with the specific primers. The amplification was carried out in a 30 μL reaction volume consisting of 15 μL of 2 × Phusion Master Mix (New England Biolabs, UK), 10 μL DNA template, 3 μL of each primer (2 μM), and 2 μL molecular water. Reaction was performed with 1 min at 98 °C, 30 cycles of 10 s at 98 °C, 30 s at 50 °C, 30 s at 72 °C, and a final extension at 72 °C for 5 min. Amplicons from three reactions for each sample were excised from gels, pooled, and purified using GeneJET Gel Extraction Kit (Thermo Scientific). The sequencing was performed on the Illumina MiSeq platform at the Novogene Bioinformatics Technology (Beijing, China).
Data processing, assembly, and annotation
Bacterial raw reads were produced by the Illumina MiSeq platform, the raw sequences were assembled for each sample, and low-quality sequences were filtered using QIIME [92]. The OTUs (Operational Taxonomic Units) table for each sample were clustered at the 97% similarity following the Uparse (http://drive5.com/uparse/), and the OTUs were classified and annotated based on the clustering results using the RDP database (http://rdp.cme.msu.edu) offering aligned and annotated for bacterial 16S rRNA sequences [93].
Statistical analysis
The relative abundances of the top 10 most abundant phyla in each sample were analyzed. To compare the similarity of composition in bacterial community, a NMDS (non-metric multidimensional scaling) analysis and a test of significance among sample types of bacterial community were performed. Variations of species composition among samples were tested by ANOSIM (An Analysis of Similarities) at species level and calculated using 999 permutations in vegan package. To assess the indicator bacterial group specialized in three sample types, LEfSe (Linear discriminant analysis effect size) analysis was performed in python 2.7 environment. Shared and unique OTUs among the three sample types were used to generate a Venn diagram in VennDiagram package. Statistical analysis on α diversity index by OTUs richness was performed with vegan package in R [94], the diversity indices of different sample types were compared with ANOVA (one-way analysis of variance), and the mean of diversity indices was tested by the LSD (Least Significant Difference) test in agricolae package [95]. The functional profiles of the bacteria were obtained by PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) analysis [96], based on the results from the normalize_by_copy_number.py analysis, and then the taxonomic file was uploaded to perform the functional prediction online (http://huttenhower.sph.harvard.edu/galaxy/). A co-occurrence network analysis at genus level was performed to explore the linkage of the different bacterial community in igraph package [97]. We calculated the spearman correlation matrix and filtered the correlation coefficient, which were both lower than ±0.8 and not at the significant level. Then the co-occurrence network was plotted. In order to describe the structure of the network, the average path length, diameter, clustering coefficient, module, co-occurrence index (C score), robustness, and counting up the node degrees were measured in igraph and bipartite packages [98]. Besides, the networks in collected samples and null model (r2dtable) were compared to test the network distribution.
Availability of data and materials
All datasets are presented in the main text and the additional file. Besides, the raw datasets and R code are available on Github digital repository (https://github.com/dongyiyi/Data-from-Co-occurrence-pattern-and-function-prediction-of-bacterial-community-in-Karst-cave). All raw data can be acquired from the corresponding author on request.
Abbreviations
- PICRUSt:
-
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States
- EDTA:
-
Ethylene Diamine Tetraacetic Acid
- CTAB:
-
Cetyltrimethylammonium bromide
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- NMDS:
-
Non-metric Multidimensional Scaling
- LEfSe:
-
Linear Discriminant Analysis Effect Size
- LSD:
-
Least Significant Difference
- OTU:
-
Operational Taxonomic Units
- rRNA:
-
Ribosomal RNA
- PCR:
-
Polymerase Chain Reaction
- ANOVA:
-
One-way Analysis of Variance
- ANOSIM:
-
An Analysis of Similarities
References
Rothschild LJ, Mancinelli RL. Life in extreme environments. Nature. 2001;409(6823):1092–101.
Seufferheld MJ, Alvarez HM, Farias ME. Role of polyphosphates in microbial adaptation to extreme environments. Appl Environ Microbiol. 2008;74(19):5867–74.
Engel AS. Microbial diversity of cave ecosystems. In: Geomicrobiology: Molecular and Environmental Perspective. Edited by Barton LL, Mandl M, Loy A. Dordrecht: Springer; 2010. p. 219–38.
Northup DE, Lavoie KH. Geomicrobiology of caves: a review. Geomicrobiol J. 2001;18(3):199–222.
Sarbu SM, Kane TC, Kinkle BK. A chemoautotrophically based cave ecosystem. Science. 1996;272(5270):1953–5.
Desai MS, Assig K, Dattagupta S. Nitrogen fixation in distinct microbial niches within a chemoautotrophy-driven cave ecosystem. ISME J. 2013;7(12):2411–23.
Simon KS. Cave ecosystems. In: Encyclopedia of caves. Edited by White WB, Culver DC, Pipan T. London: Elsevier Academic Press; 2019. p. 223–6.
Engel AS, Meisinger DB, Porter ML, Payn RA, Schmid M, Stern LA, Schleifer K, Lee NM. Linking phylogenetic and functional diversity to nutrient spiraling in microbial mats from lower Kane cave (USA). ISME J. 2010;4(1):98–110.
Adetutu EM, Ball AS. Microbial diversity and activity in caves. Microbiol Aust. 2014;35(4):192–4.
Barberán A, Bates ST, Casamayor EO, Fierer N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012;6(2):343–51.
Ortiz M, Neilson JW, Nelson WM, Legatzki A, Byrne A, Yu Y, Wing RA, Soderlund CA, Pryor BM, Pierson LS. Profiling bacterial diversity and taxonomic composition on speleothem surfaces in Kartchner caverns. AZ Microbial Ecol. 2013;65(2):371–83.
Adetutu EM, Thorpe K, Shahsavari E, Bourne S, Cao X, Mazaheri Nezhad Fard R, Kirby G, Ball AS. Bacterial community survey of sediments at Naracoorte Caves, Australia. Int J Speleol. 2012;41(2):2.
Marques EL, Silva GS, Dias JC, Gross E, Costa MS, Rezende RP. Cave drip water-related samples as a natural environment for aromatic hydrocarbon-degrading bacteria. Microorganisms. 2019;7(2):33.
Liu Q, Wang H, Zhao R, Qiu X, Gong L. Bacteria isolated from dripping water in the oligotrophic Heshang cave in Central China. J Earth Sci. 2010;21:325.
Liu SJ, Zhu HZ, Zhang ZF, Zhou N, Jiang CY, Wang BJ, Cai L. Diversity, distribution and co-occurrence patterns of bacterial communities in a karst cave system. Front Microbiol. 2019;10:1726.
Xue L, Ren H, Li S, Leng X, Yao X. Soil bacterial community structure and co-occurrence pattern during vegetation restoration in karst rocky desertification area. Front Microbiol. 2017;8:2377.
Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10(8):538–50.
Yan MM, Chen SN, Huang TL, Li BQ, Li N, Liu KW, Zong RR, Miao YT, Huang X. Community compositions of phytoplankton and eukaryotes during the mixing periods of a drinking water reservoir: dynamics and interactions. Int J Environ Res Public Health. 2020;17(4):1128.
Baldassano SN, Bassett DS. Topological distortion and reorganized modular structure of gut microbial co-occurrence networks in inflammatory bowel disease. Sci Rep. 2016;6(1):1–14.
Milici M, Deng ZL, Tomasch J, Decelle J, Wos-Oxley ML, Wang H, Jáuregui R, Plumeier I, Giebel HA, Badewien TH. Co-occurrence analysis of microbial taxa in the Atlantic Ocean reveals high connectivity in the free-living bacterioplankton. Front Microbiol. 2016;7:649.
Kara EL, Hanson PC, Hu YH, Winslow L, McMahon KD. A decade of seasonal dynamics and co-occurrences within freshwater bacterioplankton communities from eutrophic Lake Mendota, WI, USA. ISME J. 2013;7(3):680–4.
Xiong K, Zhu W. Karst geomorphology and speleogenesis of the Zhijin cave area. Carsologica Sinica. 1994;13(3):281–92.
Pulido-Bosch A, Martin-Rosales W, López-Chicano M, Rodriguez-Navarro C, Vallejos A. Human impact in a tourist karstic cave (Aracena, Spain). Environ Geol. 1997;31(3–4):142–9.
Chelius MK, Beresford G, Horton H, Quirk M, Selby G, Simpson RT, Horrocks R, Moore JC. Impacts of alterations of organic inputs on the bacterial community within the sediments of wind cave, South Dakota, USA. Int J Speleol. 2009;38(1):1.
Ikner LA, Toomey RS, Nolan G, Neilson JW, Pryor BM, Maier RM. Culturable microbial diversity and the impact of tourism in Kartchner caverns, Arizona. Microbial Ecol. 2007;53(1):30–42.
Shapiro J, Pringle A. Anthropogenic influences on the diversity of fungi isolated from caves in Kentucky and Tennessee. Am Midl Nat. 2010;163(1):76–86.
Groth I, Vettermann R, Schuetze B, Schumann P, Sáiz-Jiménez C. Actinomycetes in karstic caves of northern Spain (Altamira and Tito Bustillo). J Microbiol Methods. 1999;36(1–2):115–22.
Pate D. Successful cave management strategies at Carlsbad Caverns National Park. In: 14th National Cave and Karst Management Symposium. Chattanooga: Southeastern Cave Conservancy Inc; 1999. p. 148–51.
Griffin DW, Gray MA, Lyles MB, Northup DE. The transport of nonindigenous microorganisms into caves by human visitation: a case study at Carlsbad caverns National Park. Geomicrobiol J. 2014;31(3):175–85.
Wang H, Luo SQ, Yang QD, Peng X. The changing rules and factors of CO2 concentration in Zhijin cave of Guizhou province in past 20 years. Hubei Agric Sci. 2014;53(6):1268–70 (Chinese with English abstract).
Luo SQ, Yi WY, Li P. Environmental monitoring and affecting factor analysis of Zhijin cave. Guizhou Sci. 2014;32(6):92–6 (Chinese with English abstract).
Dong YY, Ai YL, Huang Y, Wei WZ, Sun SY, Niu XJ, Zhang CB, Zhou ZF, Weng QB. The effects of human activities on the culturable bacteria diversity of the waters in Zhijindong cave. J Guizhou Normal Univ. 2017;4(35):64–70 (Chinese with English abstract).
Newman ME. Modularity and community structure in networks. Proc Natl Acad Sci. 2006;103(23):8577–82.
Ivanova V, Tomova I, Kamburov A, Tomova A, Vasileva-Tonkova E, Kambourova M. High phylogenetic diversity of bacteria in the area of prehistoric paintings in Magura Cave, Bulgaria. J Cave Karst Stud. 2013;75(3):218–28.
Gulecal-Pektas Y. Bacterial diversity and composition in Oylat cave (Turkey) with combined sanger/pyrosequencing approach. Pol J Microbiol. 2016;65:69–75.
Pašić L, Kovče B, Sket B, Herzog-Velikonja B. Diversity of microbial communities colonizing the walls of a Karstic cave in Slovenia. FEMS Microbiol Ecol. 2009;71(1):50–60.
Spain AM, Krumholz LR, Elshahed MS. Abundance, composition, diversity and novelty of soil Proteobacteria. ISME J. 2009;3(8):992–1000.
Martinez-Rodriguez A, Macedo-Raygoza G, Huerta-Robles AX, Reyes-Sepulveda I, Lozano-Lopez J, García-Ochoa EY, Fierro-Kong L, Medeiros MH, Di Mascio P, White JF. Agave seed endophytes: ecology and impacts on root architecture, nutrient acquisition, and cold stress tolerance. In: Seed Endophytes. Cham: Springer; 2019. p. 139–70.
Naumoff DG, Dedysh SN. Lateral gene transfer between the Bacteroidetes and Acidobacteria: the case of α-L-rhamnosidases. FEBS Lett. 2012;586(21):3843–51.
Bastida F, Selevsek N, Torres IF, Hernández T, García C. Soil restoration with organic amendments: linking cellular functionality and ecosystem processes. Sci Rep. 2015;5:15550.
Smit E, Leeflang P, Gommans S, van den Broek J, van Mil S, Wernars K. Diversity and seasonal fluctuations of the dominant members of the bacterial soil community in a wheat field as determined by cultivation and molecular methods. Appl Environ Microbiol. 2001;67(5):2284–91.
Nemergut DR, Cleveland CC, Wieder WR, Washenberger CL, Townsend AR. Plot-scale manipulations of organic matter inputs to soils correlate with shifts in microbial community composition in a lowland tropical rain forest. Soil Biol Biochem. 2010;42(12):2153–60.
Barron SK, Murdock CA, Blair BG, Meade ME, Barger TW. Analysis of bacterial diversity in soils from blowing spring cave (Lauderdale County, AL). J Ala Acad Sci. 2010;81:1–10.
Hügler M, Menendez C, Schägger H, Fuchs G. Malonyl-coenzyme a reductase from Chloroflexus aurantiacus, a key enzyme of the 3-hydroxypropionate cycle for autotrophic CO2 fixation. J Bacteriol. 2002;184(9):2404–10.
Lavoie KH, Winter AS, Read KJ, Hughes EM, Spilde MN, Northup DE. Comparison of bacterial communities from lava cave microbial mats to overlying surface soils from Lava Beds National Monument, USA. PloS One. 2017;12(2):1–27.
Karner MB, DeLong EF, Karl DM. Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature. 2001;409(6819):507–10.
Berg IA, Kockelkorn D, Buckel W, Fuchs G. A 3-hydroxypropionate/4-hydroxybutyrate autotrophic carbon dioxide assimilation pathway in Archaea. Science. 2007;318(5857):1782–6.
Pratscher J, Dumont MG, Conrad R. Ammonia oxidation coupled to CO2 fixation by archaea and bacteria in an agricultural soil. Proc Natl Acad Sci. 2011;108(10):4170–5.
Montano ET, Henderson LO. Studies of antibiotic production by cave bacteria. In Cave Microbiomes: A Novel Resource for Drug Discovery. Volume 1. Edited by Cheeptham N. New York: Springer; 2013. p. 109–30.
Adetutu EM, Thorpe K, Bourne S, Cao X, Shahsavari E, Kirby G, Ball AS. Phylogenetic diversity of fungal communities in areas accessible and not accessible to tourists in Naracoorte caves. Mycologia. 2011;103(5):959–68.
Timonen S, Hurek T. Characterization of culturable bacterial populations associating with Pinus sylvestris-Suillus bovinus mycorrhizospheres. Can J Microbiol. 2006;52(8):769–78.
Nishiyama M, Sugita R, Otsuka S, Senoo K. Community structure of bacteria on different types of mineral particles in a sandy soil. Soil Sci Plant Nutr. 2012;58(5):562–7.
Gudiña EJ, Pereira JF, Rodrigues LR, Coutinho JA, Teixeira JA. Isolation and study of microorganisms from oil samples for application in microbial enhanced oil recovery. Int Biodeterior Biodegradation. 2012;68:56–64.
Darsa K, Thatheyus AJ, Ramya D. Biodegradation of petroleum compound using the bacterium Bacillus subtilis. Sci Internat. 2014;2(1):20–5.
Jeong SW, Kim J. Psychrobacillus soli sp. nov., capable of degrading oil, isolated from oil-contaminated soil. Int J Syst Evol Microbiol. 2015;65(9):3046–52.
Zhang YJ, Tang SK, Shi R, Klenk HP, Chen C, Yang LL, Zhou Y, Li WJ. Salinisphaera halophila sp. nov., a moderately halophilic bacterium isolated from brine of a salt well. Int J Syst Evol Microbiol. 2012;62(9):2174–9.
Shimane Y, Tsuruwaka Y, Miyazaki M, Mori K, Minegishi H, Echigo A, Ohta Y, Maruyama T, Grant WD, Hatada Y. Salinisphaera japonica sp. nov., a moderately halophilic bacterium isolated from the surface of a deep-sea fish, Malacocottus gibber, and emended description of the genus Salinisphaera. Int J Syst Evol Microbiol. 2013;63(6):2180–5.
Crespo-Medina M, Chatziefthimiou A, Cruz-Matos R, Pérez-Rodríguez I, Barkay T, Lutz RA, Starovoytov V, Vetriani C. Salinisphaera hydrothermalis sp. nov., a mesophilic, halotolerant, facultatively autotrophic, thiosulfate-oxidizing gammaproteobacterium from deep-sea hydrothermal vents, and emended description of the genus Salinisphaera. Int J Syst Evol Microbiol. 2009;59(Pt 6):1497.
Juni E, Bøvre K. Moraxellaceae. In: Bergey's Manual of Systematics of Archaea and Bacteria; 2015. p. 1–11.
Wu Y, Tan L, Liu W, Wang B, Wang J, Cai Y, Lin X. Profiling bacterial diversity in a limestone cave of the western loess plateau of China. Front Microbiol. 2015;6:244.
Leuko S, Koskinen K, Sanna L, D’Angeli IM, De Waele J, Marcia P, Moissl-Eichinger C, Rettberg P. The influence of human exploration on the microbial community structure and ammonia oxidizing potential of the Su Bentu limestone cave in Sardinia, Italy. PloS one. 2017;12(7):1–22.
Lavoie KH, Northup DE. Bacteria as indicators of human impact in caves. In: Proceedings of the 17th National Cave and Karst Management Symposium; 2006. p. 40–7.
Barton HA, Northup DE. Geomicrobiology in cave environments: past, current and future perspectives. J Cave Karst Stud. 2007;69(1):163–78.
Fortin D, Langley S. Formation and occurrence of biogenic iron-rich minerals. Earth Sci Rev. 2005;72(1–2):1–19.
Tang K, Baskaran V, Nemati M. Bacteria of the Sulphur cycle: an overview of microbiology, biokinetics and their role in petroleum and mining industries. Biochem Eng J. 2009;44(1):73–94.
He Z, Gentry TJ, Schadt CW, Wu L, Liebich J, Chong SC, Huang Z, Wu W, Gu B, Jardine P. GeoChip: a comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J. 2007;1(1):67–77.
Cañveras JC, Sanchez-Moral S, Soler V, Saiz-Jimenez C. Microorganisms and microbially induced fabrics in cave walls. Geomicrobiol J. 2001;18(3):223–40.
Parker CW, Wolf JA, Auler AS, Barton HA, Senko JM. Microbial reducibility of Fe (III) phases associated with the genesis of iron ore caves in the Iron quadrangle, Minas Gerais, Brazil. Minerals. 2013;3(4):395–411.
Jones DS, Macalady JL. The snotty and the stringy: Energy for subsurface life in caves. In: Their World: A Diversity of Microbial Environments. Volume 1. Edited by Hurst C. Cham: Springer; 2016. p. 203–24.
Lyu X, Shen C, Xie J, Fu Y, Jiang D, Hu Z, Tang L, Tang L, Ding F, Li K. A “footprint” of plant carbon fixation cycle functions during the development of a heterotrophic fungus. Sci Rep. 2015;5(1):1–13.
Berg IA, Kockelkorn D, Ramos-Vera WH, Say RF, Zarzycki J, Hügler M, Alber BE, Fuchs G. Autotrophic carbon fixation in archaea. Nat Rev Microbiol. 2010;8(6):447–60.
Huber H, Gallenberger M, Jahn U, Eylert E, Berg IA, Kockelkorn D, Eisenreich W, Fuchs G. A dicarboxylate/4-hydroxybutyrate autotrophic carbon assimilation cycle in the hyperthermophilic Archaeum Ignicoccus hospitalis. Proc Natl Acad Sci. 2008;105(22):7851–6.
Zhang LM, Offre PR, He JZ, Verhamme DT, Nicol GW, Prosser JI. Autotrophic ammonia oxidation by soil thaumarchaea. Proc Natl Acad Sci. 2010;107(40):17240–5.
Ortiz M, Legatzki A, Neilson JW, Fryslie B, Nelson WM, Wing RA, Soderlund CA, Pryor BM, Maier RM. Making a living while starving in the dark: Metagenomic insights into the energy dynamics of a carbonate cave. ISME J. 2014;8(2):478–91.
Mandal SD, Chatterjee R, Kumar NS. Dominant bacterial phyla in caves and their predicted functional roles in C and N cycle. BMC Microbiol. 2017;17(1):90.
Rondon MA, Lehmann J, Ramírez J, Hurtado M. Biological nitrogen fixation by common beans (Phaseolus vulgaris L.) increases with bio-char additions. Biol Fertil Soils. 2007;43(6):699–708.
Chistoserdova L, Kalyuzhnaya MG, Lidstrom ME. The expanding world of methylotrophic metabolism. Annu Rev Microbiol. 2009;63:477–99.
Sadoway T, Rule D, Watson K, Moote P, Soliman LC, Azad N, Donkor K, Horne D. Cure from the cave: volcanic cave actinomycetes and their potential in drug discovery. Int J Speleol. 2013;42(1):5.
Sharma S, Aneja MK, Mayer J, Munch JC, Schloter M. Diversity of transcripts of nitrite reductase genes (nirK and nirS) in rhizospheres of grain legumes. Appl Environ Microbiol. 2005;71(4):2001–7.
Kim BK, Jung MY, Yu DS, Park SJ, Oh TK, Rhee SK, Kim JF. Genome sequence of an ammonia-oxidizing soil archaeon, “Candidatus Nitrosoarchaeum koreensis” MY1. In Am Soc Microbiol. 2011;19(193):5539–40.
Falkowski PG, Fenchel T, Delong EF. The microbial engines that drive Earth's biogeochemical cycles. Science. 2008;320(5879):1034–39.
Royer EM, Melliar-Smith PM, Moser LE. An analysis of the optimum node density for ad hoc mobile networks. In: Proceedings of the IEEE International Conference on Communications (Cat No 01CH37240). Helsinki; 2001. p. 857–61.
Poisot T, Gravel D. When is an ecological network complex? Connectance drives degree distribution and emerging network properties. PeerJ. 2014;2:e251.
Dunne JA, Williams RJ, Martinez ND. Food-web structure and network theory: the role of connectance and size. Proc Natl Acad Sci. 2002;99(20):12917–22.
Liu JH, Wang YX, Zhang XX, Wang ZG, Chen YG, Wen ML, Xu LH, Peng Q, Cui XL. Salinarimonas rosea gen. Nov., sp. nov., a new member of the α-2 subgroup of the Proteobacteria. Int J Syst Evol Microbiol. 2010;60(1):55–60.
de Issaly IS, Stoppani A. Carbohydrate metabolism in Pasteurella multocida. I. Mechanism of glucose oxidation. Proc Soc Exp Biol Med. 1964;115(2):466–71.
Hodgson DA. Primary metabolism and its control in streptomycetes: a most unusual group of bacteria. Adv Microb Physiol. 2000;42:47–238.
Zelezniak A, Andrejev S, Ponomarova O, Mende DR, Bork P, Patil KR. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc Natl Acad Sci. 2015;112(20):6449–54.
Morris BE, Henneberger R, Huber H, Moissl-Eichinger C. Microbial syntrophy: interaction for the common good. FEMS Microbiol Rev. 2013;37(3):384–406.
Hom EF, Murray AW. Niche engineering demonstrates a latent capacity for fungal-algal mutualism. Science. 2014;345(6192):94–8.
Scheuerl T, Hopkins M, Nowell RW, Rivett DW, Barraclough TG, Bell T. Bacterial adaptation is constrained in complex communities. Nat Commun. 2020;11(1):1–8.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci. 2011;108(Supplement 1):4516–22.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–7.
R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2018.
Steel RG, Torrie JH. Principles and procedures of statistics, a biometrical approach. New York: McGraw-Hill Kogakusha, Ltd.; 1980.
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Thurber RLV, Knight R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814.
Csardi G, Nepusz T. The igraph software package for complex network research. Int Complex Syst. 2006;1695(5):1–9.
Dormann CF, Fründ J, Blüthgen N, Gruber B. Indices, graphs and null models: analyzing bipartite ecological networks. Open Ecol J. 2009;2(1):7–24.
Acknowledgements
We acknowledged Prof. Zhongfa Zhou and his graduate students for their help to collect the samples. We also acknowledged the access provided by the administrative department of the Guizhou Zhijin cave Global Geopark, China.
Funding
This study was supported by the Joint Fund of the National Natural Science Foundation of China and the Karst Science Research Center of Guizhou Province (U1812401), Provincial Program on Platform and Talent Development of the Department of Science and Technology of Guizhou China (2019–5655, 2019–5617). The funders were not involved in the design of the study, collection, analysis, and interpretation of data, nor in preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
YD conducted the experimental works, interpreted the results, and wrote the manuscript. JG carried out bioinformatic, statistical analysis, and edited the manuscript. QW1, YA, YH, WW and SS performed the samples collection and processing. QW2 (corresponding author) was responsible for the primary conception of the study, supervised and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The permission to enter the study sites and to collect the samples was obtained from the Guizhou Zhijin cave Global Geopark, China.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Sample list and sequencing information. Table S4. Co-occurrence network descriptors for bacterial communities across the three sample types.
Additional file 2: Figure S1.
Venn diagram showing the exclusive and overlap of bacterial OTUs across the three sample types.
Additional file 3: Table S2.
List of the encoding genes and enzymes involved in carbon metabolism.
Additional file 4: Table S3.
List of the encoding genes and enzymes involved in nitrogen metabolism.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dong, Y., Gao, J., Wu, Q. et al. Co-occurrence pattern and function prediction of bacterial community in Karst cave. BMC Microbiol 20, 137 (2020). https://doi.org/10.1186/s12866-020-01806-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-020-01806-7