
Abstract
Background
The skin commensal Staphylococcus haemolyticus is an emerging nosocomial pathogen. Despite its clinical relevance, published information about S. haemolyticus virulence factors is scarce. In this study, the adhesive and biofilm forming properties of ten clinical and ten commensal S. haemolyticus strains were examined using standard adhesion and biofilm assays. One of the clinical strains was used to identify expressed surface proteins using bacterial surface shaving. Protein abundance was examined by a comparative analysis between bacterial protein expression after human keratinocyte (HaCaT) colonization and growth in cell culture media supplemented with serum. Relative protein quantification was performed by labeling peptides with tandem mass tags (TMT) prior to Mass Spectrometry analysis. Surface proteins can be used as novel targets for antimicrobial treatment and in diagnostics.
Results
Adherence to fibronectin, collagen and plastic was low in all tested strains, but with significantly higher adhesion to fibronectin (p = 0.041) and collagen (p = 0.001) in the commensal strains. There was a trend towards higher degree of biofilm formation in the clinical strains (p = 0.059).
By using surface shaving, 325 proteins were detected, of which 65 were classified as surface proteins. Analyses showed that the abundance of nineteen (5.8%) proteins were significantly changed following HaCaT colonization. The bacterial Toll/interleukin-1 like (TIRs) domain containing protein (p = 0.04), the transglycosylase SceD (p = 0.01), and the bifunctional autolysin Atl (p = 0.04) showed a 1.4, 1.6- and 1.5-fold increased abundance. The staphylococcal secretory antigen (SsaA) (p = 0.04) was significantly downregulated (− 1.5 fold change) following HaCaT colonization.
Among the 65 surface proteins the elastin binding protein (Ebps), LPXAG and LPXSG domain containing proteins and five LPXTG domain containing proteins were identified; three Sdr-like proteins, the extracellular matrix binding protein Embp and a SasH-like protein.
Conclusions
This study has provided novel knowledge about expression of S. haemolyticus surface proteins after direct contact with eukaryotic cells and in media supplemented with serum. We have identified surface proteins and immune evasive proteins previously only functionally described in other staphylococcal species. The identification of expressed proteins after host-microbe interaction offers a tool for the discovery and design of novel targets for antimicrobial treatment.
Similar content being viewed by others

Background
Staphylococcus haemolyticus is a coagulase-negative staphylococcus (CoNS) and a member of the skin microbiome. It is an increasing cause of nosocomial infections associated with indwelling medical devices, particularly affecting immunocompromised patients and premature babies [1,2,3]. A distinct characteristic of clinical S. haemolyticus strains is the ability to acquire resistance to several classes of antimicrobial agents [2]. The ability to colonize and form biofilms is regarded as the most important virulence trait for CoNS [4]. Adhesion is the first step to form biofilm on surfaces [5] and staphylococci express several adhesive surface molecules that interact with eukaryotic host cell receptors, abiotic surfaces or soluble macromolecules. The number of adhesive surface proteins varies among different staphylococcal species. In Staphylococcus aureus, 24 different cell wall anchored proteins have been identified, while CoNS express a smaller number [6]. Cell wall anchored (CWA) proteins are covalently attached to the peptidoglycan layer. The most prevalent CWA proteins are the microbial surface component recognizing adhesive matrix molecule (MSCRAMM) family. All CWA proteins contain an LPXTG motif (Leu-Pro-X-Thr-Gly; where X can be any amino acid) that anchor the protein to the cell wall [6]. The Sdr protein subfamily of MSCRAMMs contains a serine-aspartate repeat region [1, 6] and a signal peptide with an YSIRK motif. In S. aureus the majority (13/21) surface proteins harbors the YSIRK/GS signal sequence, allowing delivery of surface proteins to unique locations in the cell wall [7]. Sdr-like genes have previously been described in S. haemolyticus [8].
Another family of the CWA proteins is the Serine Rich Repeats Proteins family. Like the Sdr proteins, they have a serine repeat region, but with alanine, valine or threonine instead of aspartate [9]. Bacterial surface proteins can act as new targets in treatment and prevention of infections in multiresistant bacteria. One method to examine bacterial surface proteins is by surface shaving. Surface-shaving is a technique where peptides from bacterial surface proteins are cleaved off when protease treatment is applied followed by a Liquid Chromatography tandem Mass Spectrometry (LC-MS/MS) analysis [10]. The Lipid-based Protein Immobilization (LPI™) technology enables surface shaving of intact bacterial cells in a flow cell, and thus promotes detection of proteins expressed in the surface proteome over the highly abundant cytosolic proteins. The flow cell channels, binds intact cells by a passive process. As the surface is similar in each channel, the same number of cells are bound. Thus, combining the surface shaving approach with protocols for relative quantification, such as tandem mass tags (TMT), makes studies of low abundant virulence factors possible [11,12,13,14,15,16,17].
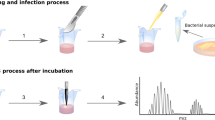
Several studies on surface proteins and their relevance in host-pathogen interactions and virulence have been performed after bacterial growth in standard laboratory medium [18,19,20,21,22]. In order to mimic a more biological relevant host-microbe interaction, we developed a novel method to investigate expressed surface proteins of a clinical S. haemolyticus isolate after colonization of human keratinocytes (HaCaT) before bacterial surface shaving was performed (Fig. 1). To our knowledge surface protein shaving of bacteria subsequent to colonization of mammalian skin cells has never been described before.

Bacterial surface protein shaving, graphical abstract. Comparison of S. haemolyticus surface protein expression after HaCaT colonization (top) and the control group (bottom). Bacterial surface proteins (multicolored) are degraded by the protease Trypsin (scissors)
In this study, we aimed to investigate the adhesive and biofilm forming abilities of ten commensal and ten clinical strains. We have previously shown that there are specific genetic signatures associated with clinical S. haemolyticus strains compared to commensal strains [23], thus we wanted to investigate if any functional differences in adhesive properties between commensal and clinical isolates could be observed. Furthermore, the expression of surface-associated proteins of one clinical S. haemolyticus strain was investigated by mass spectrometry and proteomics. The LPI surface shaving approach and relative quantification proteomics using TMT labels was employed to identify possible novel targets for treatment, prevention and biofilm formation.
Results
We wanted to examine if commensal and clinical strains had different ability to interact and adhere to selected host proteins. The adhesive ability of ten commensal and ten clinical strains to both uncoated plastic and plastic coated with fibronectin and collagen was examined to determine if binding to fibronectin or collagen would enhance binding to plastic, as we observed that binding to plastic in its native form was generally low. Further the biofilm forming capacity was examined. Eventually, one isolate was selected for bacterial surface shaving.
Adhesion to plastic and host matrix proteins
Both clinical and commensal strains adhered to plastic but no significant difference was observed between the two groups. Fibronectin and collagen binding were low for all strains, but still significantly higher for the commensal strains compared to clinical strains, p = 0.041 and p = 0.001 respectively (Fig. 2a-c).

Adhesion and biofilm assays of S. haemolyticus. Columns with black bars are clinical isolates and white bars are commensal isolates. Sample no. 6 was chosen for bacterial surface shaving (marked with asterisk). a-c Solid phase host matrix binding assay; a) Adhesion to fibronectin; b) Adhesion to plastic; c) Adhesion to collagen; d) Semi-quantitative determination of biofilm formation; e) Adhesion to human keratinocytes
Semi-quantitative determination of biofilm formation
The biofilm-forming ability of the strains was determined using a semi-quantitative assay. All strains formed biofilms and a trend towards higher biofilm formation was observed for the clinical strains (p = 0.059) where 5/10 clinical strains formed substantial amounts of biofilm in this assay (OD570 > 3) compared to 0/10 commensal strains (Fig. 2d).
Adhesion to human keratinocytes
The strains were screened for their ability to adhere to human keratinocytes. In three clinical and one commensal strain > 60% of the inoculum adhered to the keratinocytes, while seven strains showed an adhesion of ~ 10–20% of the inoculum, which was in the same range as the S. aureus (NCTC 8325–4) control strain (Fig. 2e). On average, the clinical strains adhered better to the keratinocytes compared to the commensal strains, although the findings were not statistically significant (p = 0.4). One strain, displaying high adhesion to HaCaT cells in addition to being a strong biofilm producer, was chosen for further analyzes.
Bacterial surface protein shaving
Expressed surface proteins of a clinical S. haemolyticus isolate either colonizing HaCaT cells or grown in cell culture medium supplemented with serum, was examined by surface shaving using a Lipid-based Protein Immobilization flow cell. Relative quantification of protein abundance was performed by labelling proteins with tandem mass tags (protein markers) prior to LC-MS/MS.
Protein identification and subcellular localization of S. haemolyticus proteins detected by surface shaving
Cell surface shaving of bacteria colonizing HaCaT cells or incubated in cell culture media supplemented with serum resulted in identification of 436 proteins by LC-MS/MS analysis. Only proteins with ≥ #2 peptide-spectrum matches (PSMs) were included for further analysis, resulting in 325 proteins (Supplementary Table 1 and 2).
Subcellular localization analysis of the 325 proteins in silico and functional annotation predicted 249/325 (76.6%) cytoplasmic proteins, 65/325 (20.0%) surface proteins (i.e. proteins predicted to originate from the cytoplasmic membrane, cell wall or extracellular origin), and 11/325 (3.4%) as undefined proteins.
Clusters of orthologous groups
The 65 identified surface proteins were distributed in Clusters of Orthologous Groups (COG). A higher percentage of proteins in COG groups M (cell wall/membrane/envelope biogenesis) and P (inorganic ion transport and metabolism) was found when we compared the COG distribution of the identified surface proteins (65) to the COG distribution of the total number of predicted proteins (2539) encoded in the S. haemolyticus genome (Fig. 3).

Clusters of Orthologous Groups of proteins (COG). Comparison of Clusters of Orthologous Groups of proteins (COG) between the total proteins of the strain (#2539) and surface proteins (#65) found after HaCaT colonization
S. haemolyticus surface proteins
Characteristic motifs of surface proteins such as signal peptides and LPXTG motifs were identified by bioinformatic tools. The covalently anchored cell wall proteins classified as MSCRAMMs are characterized by the C-terminal LPXTG sorting signal. A total of 19 proteins were predicted to have LPXTG motifs based on in silico analysis of the whole genome sequence of S. haemolyticus 53–38, of these seven were annotated as adhesion proteins, four were hypothetical proteins and two were DUF 402 and 368.
Of the 325 proteins identified after surface shaving, 65 were annotated as surface proteins (Table 1). Three of the LPXTG proteins identified as adhesins by the in silico analysis were expressed on the S. haemolyticus surface. Five LPXTG, one LPXSG and one LPXAG domain containing surface proteins were identified. Three Serine-Aspartate-Repeat (Sdr-like) proteins, the extracellular matrix binding protein (Embp), one Mannosylglucosyl-3-phosphoglycerate phosphatase (SasH-like), and two uncharacterized surface proteins were identified. Other well characterized proteins identified surface proteins were the lytic transglycosylase immunodominant staphylococcal antigen A (IsaA), the Immunodominant staphylococcal antigen B (IsaB) and the elastin binding protein (EbpS).
HaCaT colonisation causes changes in abundance of proteins
We wanted to explore if protein abundance differed when S. haemolyticus colonized HaCaT cells compared to when grown in cell culture media supplemented with serum. The large majority of proteins were found similarly abundant when comparing the two conditions, this included EbpS, IsaB and cytoplasmic proteins (Supplementary Table 1).
Only nineteen of 325 proteins (5.8%) showed a significant change in abundance (≥ ± 1.2 fold change) following HaCaT colonization (Table 2). The lytic transglycosylase Staphylococcus epidermidis D protein (SceD) (p = 0.01) and the autolysin Atl (p = 0.04) showed significantly increased abundance with a fold increase of 1.6 and 1.5 respectively when S. haemolyticus colonized keratinocytes. The Toll/interleukin-1 like (TIRs) domain protein (p = 0.04) also had an increase in abundance (1.4-fold) after HaCaT co-incubation, while the Staphylococcal secretory antigen (SsaA) was significantly (p = 0.04) less abundant following keratinocyte colonization, showing a 1.5-fold reduced abundance.
Moonlighting proteins identified by surface shaving
Several proteins that have previously been shown in other bacteria to have moonlighting functions - proteins dually engaged intracellularly and with important adhesive functions extracellularly - were found among the predicted cytoplasmic proteins. These are the moonlighting proteins glyceraldehyde-3- phosphate dehydrogenase (GAPDH), [24,25,26], enolase [27], aldolase (ALDA) [26], triose phosphate isomerase (TPI) [28], fructose-bisphosphate aldolase (FBA) [29], ornithine carbamoyl transferase (ARGF) [30], pyruvate kinase (PYK) [31], Inosine 5′-monophosphate dehydrogenase (IMPDH) [32], Clp [33], DNaK [34] and (Atl) [35].
Discussion
The ability to adhere to and colonize implanted biomaterials in addition to biofilm formation is considered the main virulence factors of S. haemolyticus and other coagulase-negative staphylococci.
[1,2,3]. Despite the clinical relevance of S. haemolyticus, published information about virulence factors is scarce compared to literature published on other staphylococcal species. We recently published a comparative analysis of clinical and commensal S. haemolyticus isolates [23]. We identified distinct differences in the population structure, where the clinical isolates clustered together separately from the commensal isolates. Clinical isolates were more antibiotic resistant and had different versions of genes encoding surface proteins [23]. In this study, adhesive properties and biofilm formation was compared between clinical and commensal isolates, while the expressed surface proteins were characterized in one clinical isolate after keratinocyte colonization or incubation in cell culture medium supplemented with serum.
Solid phase host matrix protein binding assay
We found that both fibronectin and collagen binding was low for all S. haemolyticus strains. However, fibronectin and collagen binding was significantly higher for commensal compared to the clinical strains. Fibronectin is a glycoprotein found in substantial amounts in blood and loose connective tissue [36] while collagen is an abundant class of proteins in humans, offering structural support to connective tissues and the extracellular matrix [37]. In S. aureus, fibronectin binding is described as a crucial step in host cell adhesion. Adhesion mainly involves binding by bacterial fibronectin binding proteins (FNBPs) to fibronectin which forms a bridge between (α5)β1 integrin on mammalian cells [38]. Low fibronectin binding in S. haemolyticus was previously shown when compared to S. aureus [39], while a varying capacity of fibronectin binding in clinical S. haemolyticus and other CoNS was demonstrated by Switalski et al. [40]. FnBPA and FnBPB involved in S. aureus fibronectin binding have not been identified in CoNS so far, but fibronectin binding by the extracellular matrix binding protein (Embp) has been shown in S. epidermidis. Expression of Embp in S. epidermidis was shown to be induced by supplementation of serum in the growth media [41]. Embp mediates adhesion to fibronectin and biofilm accumulation in S. epidermidis [42], and is present in 90% of clinical S. epidermidis strains [43]. Cell culture media supplemented with serum was also used in the adhesion assays in this study, where low binding was observed for all strains tested. We identified Embp on the surface of S. haemolyticus in the presence of serum. However, if Embp mediates fibronectin binding in S. haemolyticus, this did not result in good fibronectin binding in the adhesion assay in this study. Our findings reflect that the role of Embp in fibronectin binding of S. haemolyticus needs to be further investigated.
Cooperative binding of collagen in the presence of vitronectin has previously been demonstrated for S. haemolyticus [44]. Paulsson et al. used different bacterial growth media to induce optimal binding to both collagen and vitronectin. Thus, the type of media used in our experiments might not have been optimal for expression of proteins conferring collagen and fibronectin binding, which also could explain the low binding capacity observed in our experiments.
Adherence to plastic and semi-quantitative determination of biofilm formation
When we examined the ability to form biofilm we found trends towards more biofilm formation in the clinical strains compared to the commensal strains. However, all strains had the ability to form biofilm. In S. epidermidis, similar biofilm forming abilities were observed for both clinical and commensal strains, despite differences in population structure. Rather, different biofilm morphotypes and biofilm encoding genes were found among distinct genetic lineages indicating that biofilm formation is an important property of both commensal and clinical strains [45, 46].
We did not find any correlation between adherence to plastic and the degree of biofilm formation. As adherence is the first step in biofilm formation, one could expect an observed correlation between adhesion to plastic and biofilm formation. The discrepancy in these results can be explained by the use of different media when performing the two assays. It has previously been shown that the amount of biofilm varies depending on the media [47], making comparisons of results from different methods difficult.
Adhesion to human keratinocytes and bacterial surface protein shaving
We found a trend towards higher adhesion to keratinocytes for the clinical strains compared to the commensal strains. We selected one clinical strain with good adhesive and biofilm forming properties, and performed bacterial surface shaving. To date, most surface protein expression analyses are performed on bacteria incubated in bacterial growth medium [18,19,20,21,22]. As S. haemolyticus constitute a significant proportion of the skin microbiota of humans [1, 48, 49], we decided to choose a more biological relevant condition to study protein expression; incubation of S. haemolyticus with keratinocytes prior to bacterial surface shaving. Abundance of proteins following keratinocyte colonization was compared to protein abundance following growth in cell culture medium supplemented with bovine serum.
We identified 65 surface proteins in total, of which SceD and Atl were significantly more abundant when S. haemolyticus was colonizing keratinocytes. Transglycosylases cleave the β-1,4 glycosidic bond between N-acetylmuramic acid and N-acetylglucosamine residues of peptidoglycan, accompanied with formation of 1,6-anhydromuramic acid residues [50]. In S. aureus the transglycosylases SceD and IsaA are well described virulence factors involved in cell wall remodeling, contributing to resistance to antimicrobial peptides, adhesion and pathogenicity, shown in a murine septic arthritis model [51]. SceD has also been shown to have a pronounced upregulation upon nasal colonization of humans and rats [51, 52].
Biofilm formation is an important virulence factor in S. haemolyticus, and in this study we showed a trend towards stronger biofilm formation in clinical S. haemolyticus isolates. The bifunctional autolysin Atl was significantly more abundant in S. haemolyticus colonizing HaCaT cells. Atl homologs are described in several staphylococcal species [1]. In S. epidermidis and S. aureus, Atl is important for initial adhesion and biofilm formation [53], and has in S. epidermidis been demonstrated to mediate adhesion to vitronectin [54]. In S. aureus IsaA is involved in biofilm formation and isaA mutants form significantly less biofilm [55]. In this study we identified IsaA when S. haemolyticus was grown in the presence of serum. The S. haemolyticus biofilm is mainly composed of environmental DNA (eDNA) and proteins [47]. As Atl also mediates adhesion indirectly by hydrolysis of the bacterial cell wall causing the release of proteins and eDNA [1], it is likely that Atl and IsaA expression also in S. haemolyticus have similar functions as observed in S. epidermidis and S. aureus in both adhesion and biofilm formation.
In silico analysis of the genome sequence of the clinical S. haemolyticus isolate used for HaCaT colonization identified 19 LPXTG containing genes. Seven of these genes were annotated as genes encoding proteins involved in adhesion, while six had unknown function. These findings resemble what is found in S. aureus, where 21 LPXTG genes were predicted in silico, of which eleven had unknown function [56]. In this study, five LPXTG and two LPXSG, LPXAG containing proteins were identified after surface shaving. We identified three Sdr-like proteins which were expressed both when S. haemolyticus were co-incubated with HaCaT cells, and when grown in media containing serum. In S. aureus, transcription of SdrD and SdrG is increased in the presence of blood and serum [57, 58]. As both tested conditions contained media supplemented with serum, this could explain the expression of the Sdr-like proteins under both conditions.
In S. epidermidis, three Sdr proteins have been identified; SdrF, SdrG (Fbe) and SdrH. SdrF has been shown to mediate strong binding to keratins, keratinocytes and nasal epithelial cells [59]. In S. aureus, SdrD has been shown to mediate adhesion to keratinocytes through binding to desmoglein1, expressed in human epidermis [60]. The expression of Sdr-like proteins in S. haemolyticus after HaCaT colonization and grown in the presence of serum suggests that it might exert similar functions in keratinocyte binding, as found in S. epidermidis and S. aureus.
HaCaT colonization resulted in the significant upregulation of a TIR protein. TIR domain containing proteins have been shown in several pathogenic bacteria [61], but has not previously been described in S. haemolyticus. TirS in S. aureus increases survival in the host by blocking the cascade reaction leading to activation of the nuclear factor–ĸB (NF-ĸB), which regulates the expression of a pro-inflammatory immune response [62]. Bacterial circumvention of the host immune defense is an important mechanism in bacterial host colonization.
Cytoplasmic proteins
Many of the proteins identified in this experiment were predicted as cytoplasmic proteins. Detection of some cytoplasmic proteins are inevitable when performing surface shaving [10, 63]. The presence of predicted cytoplasmic proteins after bacterial surface shaving can be due to cellular lysis, moonlighting proteins or protein containing membrane-vesicles (MV) [10, 63, 64].
We recently showed that S. haemolyticus produces MVs [65]. The S. haemolyticus MV cargo mainly contained cytoplasmic proteins, amongst them several moonlighting proteins, which are proteins that express more than one function when transported to a different cellular location [24]. Release of MVs in incubation buffer after culturing and washing of cells might add to the identification of predicted cytoplasmic proteins [10].
Strengths and limitations of the study
The main advantage of the developed method is the direct contact between bacteria and mammalian cells before bacterial surface shaving, mimicking a more relevant host-microbe interaction compared to other protein expression systems. S. haemolyticus surface shaving subsequent to colonization of human keratinocytes has to our knowledge not been described before. By using the LPI™ approach for bacterial surface shaving, whole cells are immobilized by a passive process (personal communication Nanoxis Consulting AB) within a flow cell prior to digestion, allowing binding of intact cells only. In this study we only used one clinical isolate. In order to find surface proteins that are present only in clinical vs. commensal isolates, several isolates from different commensal and clinical lineages need to be compared.
The separation of bacteria from the mammalian cells by FACS is time consuming, leading to a low throughput of samples. The individual sorting of samples before being concentrated and subsequently subjected to surface shaving in individual LPI flow cell channels, might have led to slight variations in the concentration of cells or even slight differences in expression due to slight differences in handling time. However, we kept all samples on ice and in PBS throughout the experiment in order to minimize potential alteration of gene expression.
Conclusion
This is to our knowledge the first described study using surface shaving of expressed staphylococcal proteins after direct contact with eukaryotic cells and in cell culture media supplemented with serum. Gaining information about surface exposed proteins is important in order to better understand host-pathogen interactions, biofilm formation and for the discovery and design of novel targets for antimicrobial and anti-biofilm treatment. Thus, this method is transferable to other bacterial species and mammalian cell types. The method has provided novel knowledge about the S. haemolyticus surface proteins in a clinical isolate. We have identified surface proteins and immune evasive proteins previously only functionally described in other staphylococcal species. We have also identified hypothetical surface proteins, of which future analysis should be undertaken in order to describe function. Further functional assays should be performed to determine the importance of the different identified proteins in host microbe interactions and biofilm formation.
Methods
Bacterial strains and mammalian cell lines
Ten clinical and ten commensal S. haemolyticus strains were included in the study (Table 3). The clinical strains are a subset of a larger collection, isolated from blood, catheters and wounds [2]. The commensal strains are a subset of a collection of strains from the skin of healthy adults [49]. HaCaT cells were from a human keratinocyte cell line [66] (Cell Lines Service (CLS), Germany, no. 300493).
Solid phase host matrix protein binding assay
The ability of S. haemolyticus to adhere to collagen, fibronectin and plastic was determined using a protocol based on Edwards et al. [67]. Bacterial cultures were grown for 10 h (Optical density (OD)600 0.7–1.0) in Dulbecco’s Modified Eagle’s Medium (DMEM) (Merck, Germany) with 10% heat inactivated Fetal Bovine serum (FBS) (Thermo Fisher Scientific, MA, USA), pelleted and re-suspended to a concentration of 108 colony forming units (CFU)/mL. Microtiterplates (96 well) pre-coated with collagen (Thermo Fisher Scientific, MA, USA) or fibronectin, 1 μg/well (R&D Systems, MN, USA) were blocked with 150 μl 3% Bovine Serum Albumin (BSA) (Merck, Germany) for 1 h at room temperature and then washed 2x with Phosphate Buffered Saline (PBS) (Merck, Germany). Inoculum was added to plastic (CAT.NO 163320, Thermo Fisher Scientific, MA, USA), collagen and fibronectin plates and incubated for 1 h at 37 °C followed by 1x wash with PBS. The plates were fixed at 55 °C for 1 h and stained with 0.25% crystal violet (Merck, Germany) for five minutes. Biomass of adherent bacteria was determined by solubilization of crystal violet with 150 μL 70% EtOH. Absorbance (Abs) was measured at 590 nm (Versamax, Molecular Devices, CA, USA). Values from bacterial binding to wells coated with BSA only were subtracted.
Semi-quantitative determination of biofilm formation
We performed semi-quantitative determination of biofilm production as described previously [47, 68]. Biofilm formation was induced in Tryptic Soy Broth (TSB) (BD, NJ, USA / Merck, Germany) with 1% glucose (Merck, Germany) in 96-well microtiter plates (Thermo Fisher Scientific, MA, USA). All strains were tested in eight wells with three parallel runs and controls were included on each plate. After 24 h, wells were washed, fixed and stained with 0.1% crystal violet (Merck, Germany). Crystal violet was dissolved from the biofilm with 70% ethanol for 10 min and Abs570 was determined (Versamax, Molecular Devices, CA, USA). We removed the highest and lowest outlier for each parallel and the remaining six values were averaged. Based on the distribution of the tested strains, strains with average OD values over 1 were considered strong biofilm-producers.
Adhesion to human keratinocytes
S. haemolyticus adhesion to human keratinocytes (HaCaT) was determined. HaCaT (2 × 105 cells/ml) were added to 24-well plates (Thermo Fisher Scientific, MA, USA) and allowed to attach for 16 h (37 °C, 5% CO2) in DMEM with 10% FBS. Bacterial cultures were grown at 37 °C to late exponential phase (OD600 0.7–1.0) in DMEM with 10% FBS, and then washed twice in Dulbecco’s Phosphate Buffered Saline (DPBS) (Merck, Germany). Approximately 2 × 106 CFU in DMEM with 10% FBS were added to each well of a cell culture plate to achieve a multiplicity of infection dose (MOI) of 10:1. The plates were centrifuged at 900xG (Eppendorf 5430R, Germany) for 10 min at 37 °C and incubated for 30 min. at 37 °C in 5% CO2 [69]. After incubation, the plates were thoroughly washed to remove all unbound bacterial cells. To enumerate the number of adhered bacteria, 0.25 mg/mL Trypsin-EDTA (Merck, Germany) and 0.1% mg/mL Triton X-100 (Merck, Germany) were added, and the suspension was pipetted in order to fully lyse the HaCaT cells. CFU/mL was determined by plating on blood agar plates (Thermo Fisher Scientific, MA, USA) and incubated at 37 °C overnight. Three biological replicates were performed.
Bacterial surface protein shaving
Preparation of bacteria for cell surface shaving
To explore the expression of surface proteins in S. haemolyticus when colonizing HaCaT cells, one clinical bacterial strain (53–38) with strong adhesive and biofilm-forming properties (Table 3) was co-incubated with HaCaT cells. We wanted to further explore this isolate as adhesion and biofilm formation is regarded as important virulence traits in the coagulase negative staphylococci. A bacterial control sample (same bacterial isolate) grown in cell culture media supplemented with serum but without HaCaT cultivation was included. Three biological replicates were performed for all samples and both conditions. The workflow of the bacterial surface shaving experiment is summarized in Fig. 1 and Supplementary Table 3.
HaCaT cells were seeded in 6-well plates, and bacterial cultures were grown to late exponential phase (OD600 0.6 ± 0.1) in DMEM with 10% FBS, washed twice in DPBS and resuspended in DMEM with 10% FBS and further handled as previously described for the HaCaT adhesion assay. A MOI of 100:1 was used and bacteria were centrifuged with HaCaT cells for 10 min, and further incubated for 50 min. After incubation, tissue culture plates were washed 4 times with DPBS to remove free-floating bacteria. Mechanical detachment of eukaryotic and bacterial cells from the tissue culture plates was performed with a cell scraper (VWR, PA, USA) followed by pipetting in DPBS. Cells were transferred to polystyrene tubes with a cell strainer cap (Thermo Fisher Scientific, MA, USA). Twelve wells from two tissue culture plates were used for each replicate.
The samples were prepared for Fluorescence-activated cell sorting (FACS), in order to separate bacteria from HaCaT cells, by labelling with the Vancomycin BODIPY™ FL Conjugate (Thermo Fisher Scientific, MA, USA) (0.6 μg/mL), targeting the Gram-positive bacterial cell wall [70].
The bacterial control samples that were not co-cultivated with HaCaT cells were grown to late exponential phase in DMEM with 10% FBS (OD600 0.6 ± 0.1) and resuspended in DPBS after centrifugation and washing and further stored on ice. Samples were then prepared for FACS by Vancomycin BODIPY™ labelling, in order to treat the bacterial control samples in a similar manner to the test samples.
Fluorescence-activated cell sorting system (FACS)
S. haemolyticus was sorted from HaCaT cells by using FACS Aria III (BD, NJ, USA) (Software BD FACSDiva 8.0.1), according to size and fluorescence. Based on the size of single bacteria (1 μm) and the fluorescent signal strength, the gating was set to sort single or doublets of bacteria. Fluorescent beads (Polystyrene Particle, Flow Cytometry grade PPS-6 K and Nano Blank Polystyrene NFPPS-52-4 K (Spherotech, IL, USA)) were used for calibration. Vancomycin BODIPY™ was excited with a 488 nm blue laser. A FITC-detector was used to read the emitted green, fluorescent light. Normal density filter 1.0 was used in front of the FSC detector. After FACS all samples were stored on ice.
Surface shaving - sample processing and generation of peptides by LPI™ HexaLane flow cell
In order to concentrate the bacterial samples after FACS (≈230 mL), samples were centrifuged twice, both steps at 10000xG for 30 min at 4 °C in swing bucket rotors (Beckman Coulter, CA, USA), resulting in samples containing approximately 2.8 × 107 CFU/mL. The concentrated samples were resuspended in ice cold PBS, kept on ice and immediately loaded into the LPI™ HexaLane Flow Cell (Nanoxis Consulting AB, Sweden), as seen in Fig. 4, step 1. To allow bacterial attachment, the flow cell was incubated for 35 min at room temperature. The cells attach to the gold coated channels in the Flow Cell by a passive process (personal communication Nanoxis Consulting AB). Unbound bacteria were removed by washing the channels with 200 μL PBS using a syringe pump (Harvard Apparatus, MA, USA) at a flow rate of 50 μL/min. Enzymatic digestion of bacterial surface proteins was performed by injecting 100 μL of trypsin (Promega, WI, USA) (40 μg/mL) into the LPI HexaLane Flow Cell channels and further incubated for 20 min at room temperature. After digestion, peptides were eluted in 200 μL PBS and the digestion was terminated by adding 4 μL formic acid (neat) (Merck, Germany). The peptide samples were centrifuged for 10 min at 10000xG, in order to remove any cell debris and the supernatants were subsequently dried using a SpeedVac (Eppendorf, Germany) and stored at − 20 °C.

The use of LPI™ methodology together with TMT labelling when performing surface shaving. Three lanes were filled with bacterial cells after exposure to HaCaT cells (a) and three lanes were filled with bacterial cells only exposed to media (b). After surface shaving, the eluted peptides were tagged with TMT labels, pooled and subsequently analyzed by LC-MS/MS.
Protein identification and relative quantitation
The proteomic analysis was performed at The Proteomics Core Facility at Sahlgrenska Academy, Gothenburg University. Digested peptides were dissolved in 100 μL triethylammonium bicarbonate (TEAB) (350 mM, Thermo Fisher Scientific, MA, USA) and labelled using TMT 10-plex isobaric mass tagging reagents (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions. The TMT-set were fractionated into twelve fractions using Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s protocol, but with a modified gradient (Supplementary Table 4).
The fractions were analyzed on a QExactive HF mass spectrometer (MS) interfaced with Easy-nLC1200 liquid chromatography system (LC-MS/MS) (Thermo Fisher Scientific, MA, USA). Peptides were trapped on an Acclaim Pepmap 100 C18 trap column (100 μm × 2 cm, particle size 5 μm, Thermo Fisher Scientific, MA, USA) and separated on an in-house packed analytical column (75 μm × 300 mm, particle size 3 μm, Reprosil-Pur C18, Dr. Maisch, Germany) using a gradient from 7 to 35% B over 70 min followed by an increase to 100% B for 5 min at a flow of 300 nL/min. Solvent A was 0.2% formic acid and solvent B was 80% acetonitrile, 0.2% formic acid. The instrument operated in data-dependent mode where the precursor ion mass spectra were acquired at a resolution of 60,000, the 10 most intense ions were isolated in a 0.8 Da isolation window and fragmented using collision energy HCD settings at either 28 or 50. MS2 spectra were recorded at a resolution of 60,000, charge states 2 to 4 were selected for fragmentation and dynamic exclusion was set to 20 s with 10 ppm tolerance.
MS raw data files for the TMT set were merged for identification and relative quantification using Proteome Discoverer version 1.4 (Thermo Fisher Scientific, MA, USA). S. haemolyticus 53–38 with European Nucleotide Archive (ENA) accession number GCA_001226325.1 (Illumina sequence) and ENA accession number PRJEB36042 (PacBio sequence) [2] were aligned using BWA-MEM [71] and further used as reference proteome (2539 coding sequences). Structural and functional annotations were performed using Prokka [72]. Mascot 2.5 (Matrix Science Ltd., UK) was used as a search engine with precursor mass tolerance of 5 ppm and fragment mass tolerance of 200 mmu. Tryptic peptides were accepted with one missed cleavage and variable modifications of methionine oxidation, cysteine alkylation and fixed modifications of N-terminal TMT-label and lysine TMT-label were selected. Fixed Value of 13 was used for identification and the quantified proteins were filtered at 1% False Discovery Rate (FDR) resulting in a mascot score of at least 20. No missing values were present in the data set at Threshold of 2000. Proteins were grouped by sharing the same sequences to minimize redundancy. The resulting ratios were normalized in the Proteome Discoverer 1.4 and the sum of the samples cultivated with HaCaT was used as denominator. Only unique peptides were used for comparison between groups.
The mass spectrometry proteomics data has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD014450.
Statistical analyses
For the results from biofilm-, solid phase host matrix protein and HaCaT adhesion assays the data were analyzed using IBM SPSS software, version 25.0. The non-parametric Mann-Whitney U-test was used to compare two groups, a p value < 0.05 was considered statistically significant.
As the technical variation for the identified proteins was assumed to be 20%, only proteins displaying a higher degree of fold change (FC) than ±1.2 were considered as biologically significant regarding increased or reduced abundance of proteins. The most changed abundance of proteins had a threshold of at least ±1.5. Welch’s t-test was performed (3 parallels vs. 3 parallels) and only proteins passing filter p < 0.05 were considered statistically significant.
Bioinformatic analyses
LPXTG motifs were predicted in silico from the whole genome sequence of S. haemolyticus 53–38 using a manual sequence search. Prediction of the subcellular localization of proteins was done using PSORTb v.3.0 algorithms [73], CELLO v.2.5 [74] and LocateP v.2.0 [75]. Positive prediction of subcellular localization was determined by a two out of three or greater concurrent results between the databases. Surface proteins were defined as proteins predicted from cytoplasmic membrane, cell wall or extracellular origin.
Functional annotation of proteins was done with the EggNOG v.5.0 database with HMMER and Diamond mapping mode; i.e. functional description, seed orthologues, predicted name, KEGG KO and categorization of proteins into Clusters of Orthologous Groups of proteins (COG) [76], PHMMER v.3.3 [77, 78] and protein BLAST [79].
Moonlighting proteins were identified by using the MoonProt database and by manual searches based on published literature [80, 81].
Availability of data and materials
The proteomic raw data supporting the conclusions of this article is available in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD014450 [82]. The whole genome sequence of the strain used for surface shaving is depoasited in the European Nucleotide Archive with the unique identifier ERS066380 [83] and PRJEB36042 [84]. The dataset supporting the conclusions of this article is included within the article (and its Additional files S1–4).
Abbreviations
- Abs:
-
Absorbance
- ALDA:
-
Aldolase
- ARGF:
-
Ornithine carbamoyl transferase
- Atl:
-
Autolysin
- BSA:
-
Bovine Serum Albumin
- CFU:
-
Colony Forming Units
- CLS:
-
Cell Lines Service
- COG:
-
Clusters of Orthologous Groups
- CoNS:
-
Coagulase Negative Staphylococci
- CWA:
-
Cell Wall Anchored
- DMEM:
-
Dulbecco’s Modified Eagle’s Medium
- DPBS:
-
Dulbecco’s Phosphate Buffered Saline
- DUF:
-
Domain of Unknown Function
- Ebps:
-
Elastin binding protein
- eDNA:
-
Environmental DNA
- Embp:
-
Extracellular matrix binding protein
- ENA:
-
European Nucleotide Archive
- FACS:
-
Fluorescence-activated cell sorting
- FC:
-
Fold Change
- FBA:
-
Fructose-Bisphosphate Aldolase
- FBS:
-
Fetal Bovine Serum
- FDR:
-
False Discovery Rate
- FNBPs:
-
Bacterial Fibronectin binding proteins
- GAPDH:
-
Glyceraldehyde-3- phosphate dehydrogenase
- HaCaT:
-
Human Keratinocytes
- IgG:
-
Immunoglobulin G
- IMPDH:
-
Inosine 5′-monophosphate dehydrogenase
- IsaA:
-
Immunodominant staphylococcal antigen A
- IsaB:
-
Immunodominant staphylococcal antigen B
- LC-MS/MS:
-
Liquid Chromatography tandem Mass Spectrometry
- LPI:
-
Lipid-based Protein Immobilization
- LPXTG:
-
Leu-Pro-X-Thr-Gly; where X can be any amino acid
- MOI:
-
Multiplicity Of Infection
- MS:
-
Mass Spectrometry
- MSCRAMM:
-
Microbial Surface Component Recognizing Adhesive Matrix Molecule
- MV:
-
Membrane Vesicle
- NF-ĸB:
-
Nuclear Factor–ĸB
- OD:
-
Optical Density
- PBS:
-
Phosphate Buffered Saline
- PRIDE:
-
PRoteomics IDEntifications database
- PSM:
-
Peptide Spectrum match
- PYK:
-
PYruvate Kinase
- SasH:
-
Mannosylglucosyl-3-phosphoglycerate phosphatase
- SceD:
-
Lytic transglycosylase Staphylococcus epidermidis D protein
- Sdr:
-
Serine Aspartate repeat containing protein
- SsaA:
-
Staphylococcal secretory antigen
- TEAB:
-
Triethylammonium Bicarbonate
- TIR:
-
Toll/interleukin-1 receptor
- TMT:
-
Tandem Mass Tags
- TPI:
-
Triose Phosphate Isomerase
- TSB:
-
Tryptic Soy Broth
References
Becker K, Heilmann C, Peters G. Coagulase-negative staphylococci. Clin Microbiol Rev. 2014;27(4):870–926. https://doi.org/10.1128/cmr.00109-13.
Cavanagh JP, Hjerde E, Holden MT, Kahlke T, Klingenberg C, Flaegstad T, et al. Whole-genome sequencing reveals clonal expansion of multiresistant Staphylococcus haemolyticus in European hospitals. J Antimicrob Chemother. 2014;69(11):2920–7. https://doi.org/10.1093/jac/dku271.
Czekaj T, Ciszewski M, Szewczyk EM. Staphylococcus haemolyticus - an emerging threat in the twilight of the antibiotics age. Microbiology. 2015;161(11):2061–8. https://doi.org/10.1099/mic.0.000178.
Heilmann C, Ziebuhr W, Becker K. Are coagulase-negative staphylococci virulent? Clin Microbiol Infect. 2018. https://doi.org/10.1016/j.cmi.2018.11.012.
Kline KA, Falker S, Dahlberg S, Normark S, Henriques-Normark B. Bacterial adhesins in host-microbe interactions. Cell Host Microbe. 2009;5(6):580–92. https://doi.org/10.1016/j.chom.2009.05.011.
Foster TJ, Geoghegan JA, Ganesh VK, Hook M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat Rev Microbiol. 2014;12(1):49–62. https://doi.org/10.1038/nrmicro3161.
DeDent A, Bae T, Missiakas DM, Schneewind O. Signal peptides direct surface proteins to two distinct envelope locations of Staphylococcus aureus. EMBO J. 2008;27(20):2656–68. https://doi.org/10.1038/emboj.2008.185.
Takeuchi F, Watanabe S, Baba T, Yuzawa H, Ito T, Morimoto Y, et al. Whole-genome sequencing of Staphylococcus haemolyticus uncovers the extreme plasticity of its genome and the evolution of human-colonizing staphylococcal species. J Bacteriol. 2005;187(21):7292–308. https://doi.org/10.1128/jb.187.21.7292-7308.2005.
Arora S, Uhlemann AC, Lowy FD, Hook M. A novel MSCRAMM subfamily in coagulase negative staphylococcal species. Front Microbiol. 2016;7:540. https://doi.org/10.3389/fmicb.2016.00540.
Olaya-Abril A, Jimenez-Munguia I, Gomez-Gascon L, Rodriguez-Ortega MJ. Surfomics: shaving live organisms for a fast proteomic identification of surface proteins. J Proteome. 2014;97:164–76. https://doi.org/10.1016/j.jprot.2013.03.035.
Karlsson R, Karlsson A, Backman O, Johansson BR, Hulth S. Identification of key proteins involved in the anammox reaction. FEMS Microbiol Lett. 2009;297(1):87–94. https://doi.org/10.1111/j.1574-6968.2009.01677.x.
Karlsson R, Davidson M, Svensson-Stadler L, Karlsson A, Olesen K, Carlsohn E, et al. Strain-level typing and identification of bacteria using mass spectrometry-based proteomics. J Proteome Res. 2012;11(5):2710–20. https://doi.org/10.1021/pr2010633.
Karlsson R, Karlsson A, Backman O, Johansson BR, Hulth S. Subcellular localization of an ATPase in anammox bacteria using proteomics and immunogold electron microscopy. FEMS Microbiol Lett. 2014;354(1):10–8. https://doi.org/10.1111/1574-6968.12425.
Karlsson R, Thorell K, Hosseini S, Kenny D, Sihlbom C, Sjoling A, et al. Comparative analysis of two Helicobacter pylori strains using genomics and mass spectrometry-based proteomics. Front Microbiol. 2016;7:1757. https://doi.org/10.3389/fmicb.2016.01757.
Jansson ET, Trkulja CL, Olofsson J, Millingen M, Wikstrom J, Jesorka A, et al. Microfluidic flow cell for sequential digestion of immobilized proteoliposomes. Anal Chem. 2012;84(13):5582–8. https://doi.org/10.1021/ac300519q.
Gonzales-Siles L, Karlsson R, Kenny D, Karlsson A, Sjoling A. Proteomic analysis of enterotoxigenic Escherichia coli (ETEC) in neutral and alkaline conditions. BMC Microbiol. 2017;17(1):11. https://doi.org/10.1186/s12866-016-0914-1.
Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75(8):1895–904. https://doi.org/10.1021/ac0262560.
Marin E, Haesaert A, Padilla L, Adan J, Hernaez ML, Monteoliva L, et al. Unraveling Gardnerella vaginalis surface proteins using cell shaving proteomics. Front Microbiol. 2018;9:975. https://doi.org/10.3389/fmicb.2018.00975.
Solis N, Cain JA, Cordwell SJ. Comparative analysis of Staphylococcus epidermidis strains utilizing quantitative and cell surface shaving proteomics. J Proteome. 2016;130:190–9. https://doi.org/10.1016/j.jprot.2015.09.011.
Bowden MG, Chen W, Singvall J, Xu Y, Peacock SJ, Valtulina V, et al. Identification and preliminary characterization of cell-wall-anchored proteins of Staphylococcus epidermidis. Microbiology. 2005;151(Pt 5):1453–64. https://doi.org/10.1099/mic.0.27534-0.
Gomez-Gascon L, Luque I, Olaya-Abril A, Jimenez-Munguia I, Orbegozo-Medina RA, Peralbo E, et al. Exploring the pan-surfome of Streptococcus suis: looking for common protein antigens. J Proteome. 2012;75(18):5654–66. https://doi.org/10.1016/j.jprot.2012.07.025.
Rodriguez-Ortega MJ, Norais N, Bensi G, Liberatori S, Capo S, Mora M, et al. Characterization and identification of vaccine candidate proteins through analysis of the group a Streptococcus surface proteome. Nat Biotechnol. 2006;24(2):191–7. https://doi.org/10.1038/nbt1179.
Pain M, Hjerde E, Klingenberg C, Cavanagh JP. Comparative genomic analysis of Staphylococcus haemolyticus reveals key to hospital adaptation and pathogenicity. Front Microbiol. 2019;10(2096). https://doi.org/10.3389/fmicb.2019.02096.
Kainulainen V, Korhonen TK. Dancing to another tune-adhesive moonlighting proteins in bacteria. Biology. 2014;3(1):178–204. https://doi.org/10.3390/biology3010178.
Glenting J, Beck HC, Vrang A, Riemann H, Ravn P, Hansen AM, et al. Anchorless surface associated glycolytic enzymes from Lactobacillus plantarum 299v bind to epithelial cells and extracellular matrix proteins. Microbiol Res. 2013;168(5):245–53. https://doi.org/10.1016/j.micres.2013.01.003.
Henderson B, Martin A. Bacterial virulence in the moonlight: multitasking bacterial moonlighting proteins are virulence determinants in infectious disease. Infect Immun. 2011;79(9):3476–91. https://doi.org/10.1128/iai.00179-11.
Antikainen J, Kuparinen V, Lahteenmaki K, Korhonen TK. Enolases from gram-positive bacterial pathogens and commensal lactobacilli share functional similarity in virulence-associated traits. FEMS Immunol Med Microbiol. 2007;51(3):526–34. https://doi.org/10.1111/j.1574-695X.2007.00330.x.
Yamaguchi M, Ikeda R, Nishimura M, Kawamoto S. Localization by scanning immunoelectron microscopy of triosephosphate isomerase, the molecules responsible for contact-mediated killing of Cryptococcus, on the surface of Staphylococcus. Microbiol Immunol. 2010;54(6):368–70. https://doi.org/10.1111/j.1348-0421.2010.00225.x.
Blau K, Portnoi M, Shagan M, Kaganovich A, Rom S, Kafka D, et al. Flamingo cadherin: a putative host receptor for Streptococcus pneumoniae. J Infect Dis. 2007;195(12):1828–37. https://doi.org/10.1086/518038.
Hussain M, Peters G, Chhatwal GS, Herrmann M. A lithium chloride-extracted, broad-spectrum-adhesive 42-kilodalton protein of Staphylococcus epidermidis is ornithine carbamoyltransferase. Infect Immun. 1999;67(12):6688–90.
Katakura Y, Sano R, Hashimoto T, Ninomiya K, Shioya S. Lactic acid bacteria display on the cell surface cytosolic proteins that recognize yeast mannan. Appl Microbiol Biotechnol. 2010;86(1):319–26. https://doi.org/10.1007/s00253-009-2295-y.
Molkanen T, Tyynela J, Helin J, Kalkkinen N, Kuusela P. Enhanced activation of bound plasminogen on Staphylococcus aureus by staphylokinase. FEBS Lett. 2002;517(1–3):72–8. https://doi.org/10.1016/s0014-5793(02)02580-2.
Wang W, Jeffery CJ. An analysis of surface proteomics results reveals novel candidates for intracellular/surface moonlighting proteins in bacteria. Mol BioSyst. 2016;12(5):1420–31. https://doi.org/10.1039/c5mb00550g.
Amblee V, Jeffery CJ. Physical features of intracellular proteins that moonlight on the cell surface. PLoS One. 2015;10(6):e0130575. https://doi.org/10.1371/journal.pone.0130575.
Allignet J, England P, Old I, El Solh N. Several regions of the repeat domain of the Staphylococcus caprae autolysin, AtlC, are involved in fibronectin binding. FEMS Microbiol Lett. 2002;213(2):193–7. https://doi.org/10.1111/j.1574-6968.2002.tb11305.x.
Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115(Pt 20):3861–3. https://doi.org/10.1242/jcs.00059.
Madani A, Garakani K, Mofrad MRK. Molecular mechanics of Staphylococcus aureus adhesin, CNA, and the inhibition of bacterial adhesion by stretching collagen. PLoS One. 2017;12(6):e0179601. https://doi.org/10.1371/journal.pone.0179601.
Grundmeier M, Hussain M, Becker P, Heilmann C, Peters G, Sinha B. Truncation of fibronectin-binding proteins in Staphylococcus aureus strain Newman leads to deficient adherence and host cell invasion due to loss of the cell wall anchor function. Infect Immun. 2004;72(12):7155–63. https://doi.org/10.1128/iai.72.12.7155-7163.2004.
Maali Y, Martins-Simoes P, Valour F, Bouvard D, Rasigade JP, Bes M, et al. Pathophysiological mechanisms of Staphylococcus non-aureus bone and joint infection: Interspecies homogeneity and specific behavior of S pseudintermedius. Front Microbiol. 2016;7:1063. https://doi.org/10.3389/fmicb.2016.01063.
Switalski LM, Ryden C, Rubin K, Ljungh A, Hook M, Wadstrom T. Binding of fibronectin to Staphylococcus strains. Infect Immun. 1983;42(2):628–33. https://doi.org/10.1128/IAI.42.2.628-633.1983.
Christner M, Heinze C, Busch M, Franke G, Hentschke M, Bayard Duhring S, et al. sarA negatively regulates Staphylococcus epidermidis biofilm formation by modulating expression of 1 MDa extracellular matrix binding protein and autolysis-dependent release of eDNA. Mol Microbiol. 2012;86(2):394–410. https://doi.org/10.1111/j.1365-2958.2012.08203.x.
Christner M, Franke GC, Schommer NN, Wendt U, Wegert K, Pehle P, et al. The giant extracellular matrix-binding protein of Staphylococcus epidermidis mediates biofilm accumulation and attachment to fibronectin. Mol Microbiol. 2010;75(1):187–207. https://doi.org/10.1111/j.1365-2958.2009.06981.x.
Buttner H, Mack D, Rohde H. Structural basis of Staphylococcus epidermidis biofilm formation: mechanisms and molecular interactions. Front Cell Infect Microbiol. 2015;5:14. https://doi.org/10.3389/fcimb.2015.00014.
Paulsson M, Wadstrom T. Vitronectin and type-I collagen binding by Staphylococcus aureus and coagulase-negative staphylococci. FEMS Microbiol Immunol. 1990;2(1):55–62. https://doi.org/10.1111/j.1574-6968.1990.tb03479.x.
Harris LG, Murray S, Pascoe B, Bray J, Meric G, Mageiros L, et al. Biofilm morphotypes and population structure among Staphylococcus epidermidis from commensal and clinical samples. PLoS One. 2016;11(3):e0151240. https://doi.org/10.1371/journal.pone.0151240.
Freitas AI, Lopes N, Oliveira F, Bras S, Franca A, Vasconcelos C, et al. Comparative analysis between biofilm formation and gene expression in Staphylococcus epidermidis isolates. Future Microbiol. 2018;13:415–27. https://doi.org/10.2217/fmb-2017-0140.
Fredheim EG, Klingenberg C, Rohde H, Frankenberger S, Gaustad P, Flaegstad T, et al. Biofilm formation by Staphylococcus haemolyticus. J Clin Microbiol. 2009;47(4):1172–80. https://doi.org/10.1128/jcm.01891-08.
Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011;9(4):244–53. https://doi.org/10.1038/nrmicro2537.
Cavanagh JP, Wolden R, Heise P, Esaiassen E, Klingenberg C, Aarag Fredheim EG. Antimicrobial susceptibility and body site distribution of community isolates of coagulase-negative staphylococci. APMIS. 2016;124(11):973–8. https://doi.org/10.1111/apm.12591.
Holtje JV, Mirelman D, Sharon N, Schwarz U. Novel type of murein transglycosylase in Escherichia coli. J Bacteriol. 1975;124(3):1067–76. https://doi.org/10.1128/JB.124.3.1067-1076.1975.
Stapleton MR, Horsburgh MJ, Hayhurst EJ, Wright L, Jonsson IM, Tarkowski A, et al. Characterization of IsaA and SceD, two putative lytic transglycosylases of Staphylococcus aureus. J Bacteriol. 2007;189(20):7316–25. https://doi.org/10.1128/jb.00734-07.
Burian M, Wolz C, Goerke C. Regulatory adaptation of Staphylococcus aureus during nasal colonization of humans. PLoS One. 2010;5(4):e10040. https://doi.org/10.1371/journal.pone.0010040.
Biswas R, Voggu L, Simon UK, Hentschel P, Thumm G, Gotz F. Activity of the major staphylococcal autolysin Atl. FEMS Microbiol Lett. 2006;259(2):260–8. https://doi.org/10.1111/j.1574-6968.2006.00281.x.
Heilmann C, Hussain M, Peters G, Gotz F. Evidence for autolysin-mediated primary attachment of Staphylococcus epidermidis to a polystyrene surface. Mol Microbiol. 1997;24(5):1013–24.
Lopes AA, Yoshii Y, Yamada S, Nagakura M, Kinjo Y, Mizunoe Y, et al. Roles of lytic transglycosylases in biofilm formation and beta-lactam resistance in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2019. https://doi.org/10.1128/aac.01277-19.
Casas V, Vadillo S, San Juan C, Carrascal M, Abian J. The exposed proteomes of Brachyspira hyodysenteriae and B pilosicoli. Front Microbiol. 2016;7:1103. https://doi.org/10.3389/fmicb.2016.01103.
Sitkiewicz I, Babiak I, Hryniewicz W. Characterization of transcription within sdr region of Staphylococcus aureus. Antonie Van Leeuwenhoek. 2011;99(2):409–16. https://doi.org/10.1007/s10482-010-9476-7.
Askarian F, Uchiyama S, Valderrama JA, Ajayi C, JUE S, van Sorge NM, et al. Serine-aspartate repeat protein D increases Staphylococcus aureus virulence and survival in blood. Infect Immun. 2017;85(1). https://doi.org/10.1128/iai.00559-16.
Trivedi S, Uhlemann AC, Herman-Bausier P, Sullivan SB, Sowash MG, Flores EY, et al. The surface protein SdrF mediates Staphylococcus epidermidis adherence to keratin. J Infect Dis. 2017;215(12):1846–54. https://doi.org/10.1093/infdis/jix213.
Askarian F, Ajayi C, Hanssen AM, van Sorge NM, Pettersen I, Diep DB, et al. The interaction between Staphylococcus aureus SdrD and desmoglein 1 is important for adhesion to host cells. Sci Rep. 2016;6:22134. https://doi.org/10.1038/srep22134.
Rana RR, Zhang M, Spear AM, Atkins HS, Byrne B. Bacterial TIR-containing proteins and host innate immune system evasion. Med Microbiol Immunol. 2013;202(1):1–10. https://doi.org/10.1007/s00430-012-0253-2.
Askarian F, van Sorge NM, Sangvik M, Beasley FC, Henriksen JR, Sollid JU, et al. A Staphylococcus aureus TIR domain protein virulence factor blocks TLR2-mediated NF-kappaB signaling. J Innate Immun. 2014;6(4):485–98. https://doi.org/10.1159/000357618.
Solis N, Cordwell SJ. Cell shaving and false-positive control strategies coupled to novel statistical tools to profile gram-positive bacterial surface proteomes. Methods Mol Biol (Clifton, NJ). 2016;1440:47–55. https://doi.org/10.1007/978-1-4939-3676-2_4.
Olaya-Abril A, Prados-Rosales R, McConnell MJ, Martin-Pena R, Gonzalez-Reyes JA, Jimenez-Munguia I, et al. Characterization of protective extracellular membrane-derived vesicles produced by Streptococcus pneumoniae. J Proteome. 2014;106:46–60. https://doi.org/10.1016/j.jprot.2014.04.023.
Cavanagh JP, Pain M, Askarian F, Bruun JA, Urbarova I, Wai SN, et al. Comparative exoproteome profiling of an invasive and a commensal Staphylococcus haemolyticus isolate. J Proteome. 2019;197:106–14. https://doi.org/10.1016/j.jprot.2018.11.013.
Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106(3):761–71. https://doi.org/10.1083/jcb.106.3.761.
Edwards AM, Potts JR, Josefsson E, Massey RC. Staphylococcus aureus host cell invasion and virulence in sepsis is facilitated by the multiple repeats within FnBPA. PLoS Pathog. 2010;6(6):e1000964. https://doi.org/10.1371/journal.ppat.1000964.
Christensen GD, Simpson WA, Younger JJ, Baddour LM, Barrett FF, Melton DM, et al. Adherence of coagulase-negative staphylococci to plastic tissue culture plates: a quantitative model for the adherence of staphylococci to medical devices. J Clin Microbiol. 1985;22(6):996–1006. https://doi.org/10.1128/JCM.22.6.996-1006.1985.
Edwards AM, Massey RC. Invasion of human cells by a bacterial pathogen. J Vis Exp. 2011;49. https://doi.org/10.3791/2693.
Hildebrandt P, Surmann K, Salazar MG, Normann N, Volker U, Schmidt F. Alternative fluorescent labeling strategies for characterizing gram-positive pathogenic bacteria: flow cytometry supported counting, sorting, and proteome analysis of Staphylococcus aureus retrieved from infected host cells. Cytometry A. 2016;89(10):932–40. https://doi.org/10.1002/cyto.a.22981.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXivorg. 2013. https://doi.org/10.1007/s40242-013-2300-y.
Prokka ST. Rapid prokaryotic genome annotation. Bioinformatics (Oxford, England). 2014;30(14):2068–9. https://doi.org/10.1093/bioinformatics/btu153.
Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics (Oxford, England). 2010;26(13):1608–15. https://doi.org/10.1093/bioinformatics/btq249.
Yu CS, Chen YC, Lu CH, Hwang JK. Prediction of protein subcellular localization. Proteins. 2006;64(3):643–51. https://doi.org/10.1002/prot.21018.
Zhou M, Boekhorst J, Francke C, Siezen RJ. LocateP: genome-scale subcellular-location predictor for bacterial proteins. BMC Bioinformatics. 2008;9:173. https://doi.org/10.1186/1471-2105-9-173.
Huerta-Cepas J, Szklarczyk D, Heller D, Hernandez-Plaza A, Forslund SK, Cook H, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47(D1):D309–d14. https://doi.org/10.1093/nar/gky1085.
Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7(10):e1002195. https://doi.org/10.1371/journal.pcbi.1002195.
Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, Finn RD. HMMER web server: 2018 update. Nucleic Acids Res. 2018;46(W1):W200–w4. https://doi.org/10.1093/nar/gky448.
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic acids research. 2008;36(Web Server issue):W5–9.DOI: https://doi.org/10.1093/nar/gkn201.
Mani M, Chen C, Amblee V, Liu H, Mathur T, Zwicke G, et al. MoonProt: a database for proteins that are known to moonlight. Nucleic Acids Res. 2015;43(Database issue):D277–82. https://doi.org/10.1093/nar/gku954.
Chen C, Zabad S, Liu H, Wang W, Jeffery C. MoonProt 2.0: an expansion and update of the moonlighting proteins database. Nucleic Acids Res. 2018;46(D1):D640–d4. https://doi.org/10.1093/nar/gkx1043.
PRoteomics IDEntifications Database [Internet]. 2020. Available from: https://www.ebi.ac.uk/pride/archive.
European Nucleotide Archive, ERS066380 [Internet]. 2020. Available from: https://www.ebi.ac.uk/ena/data/search?query=ERS066380.
European Nucleotide Archive, [Internet]. 2020. Available from: https://www.ebi.ac.uk/.
Acknowledgements
We would like to acknowledge Annika Thorsell at The Proteomics Core Facility at Sahlgrenska Academy, Gothenburg University, for performing the proteomic analysis. We would also like to acknowledge Roy André Lyså at The Bio-imaging Platform at UiT, The Arctic University of Norway, for performing the cell sorting.
Authors` contributions
RW: Participated in experimental design and planning. Performed the surface shaving experiments, analyzed the results and wrote the first draft of the manuscript. MP: Participated in experimental design and planning. Performed the adhesion experiments, analyzed the results and participated in writing the first draft of the manuscript. RK: Participated in experimental design and performance of the cell surface shaving experiment. Performed the mass spectrometry analysis and read through the final version of the manuscript. AK: Participated in experimental design and performance of the cell surface shaving experiment. Performed the mass spectrometry analysis and read through the final version of the manuscript. EF: Participated in experimental design and planning. Performed the biofilm and adhesion experiments, analyzed the results and participated in writing the first draft of the manuscript. JPC: Conceptualized the experimental design. Participated in the surface shaving and adhesion experiments, analysis of results and in writing the manuscript. All authors have read and approved the manuscript.
Funding
The study was supported by grants from the Northern Norway Regional Health Authority (HNF1344–17). The publication charges for this article have been funded by a grant from the publication fund of UiT The Arctic University of Norway. The funders were not involved in the design, data collection or interpretation of the results obtained in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that the submitted work was not carried out in the presence of any personal, professional or financial relationships that could potentially be construed as a conflict of interest.
Authors AK and RK are affiliated to a company, Nanoxis Consulting AB. The Company did not have influence on the collection, analysis, or interpretation of data, the writing of the paper, or the decision to submit for publication.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
The cell surface shaving and LC-MS/MS analysis results identified 325 proteins with ≥ #2 peptide-spectrum matches (PSMs).
Additional file 2: Table S2.
FASTA sequences of the proteins from the cell surface shaving and LC-MS/MS analysis.
Additional file 3: Table S3
. Workflow for bacterial protein surface shaving samples. X = performed, − = not performed
Additional file 4: Table S4
. Manufacturer’s and modified gradient using the Pierce High pH Reversed-Phase Peptide Fractionation Kit.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wolden, R., Pain, M., Karlsson, R. et al. Identification of surface proteins in a clinical Staphylococcus haemolyticus isolate by bacterial surface shaving. BMC Microbiol 20, 80 (2020). https://doi.org/10.1186/s12866-020-01778-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-020-01778-8