
Abstract
Background
Jasmonate (JA) is the important phytohormone to regulate plant growth and adaption to stress signals. MYC2, an bHLH transcription factor, is the master regulator of JA signaling. Although MYC2 in maize has been identified, its function remains to be clarified.
Results
To understand the function and regulatory mechanism of MYC2 in maize, the joint analysis of DAP-seq and RNA-seq is conducted to identify the binding sites and target genes of ZmMYC2. A total of 3183 genes are detected both in DAP-seq and RNA-seq data, potentially as the directly regulating genes of ZmMYC2. These genes are involved in various biological processes including plant growth and stress response. Besides the classic cis-elements like the G-box and E-box that are bound by MYC2, some new motifs are also revealed to be recognized by ZmMYC2, such as nGCATGCAnn, AAAAAAAA, CACGTGCGTGCG. The binding sites of many ZmMYC2 regulating genes are identified by IGV-sRNA.
Conclusions
All together, abundant target genes of ZmMYC2 are characterized with their binding sites, providing the basis to construct the regulatory network of ZmMYC2 and better understanding for JA signaling in maize.
Similar content being viewed by others


Background
To respond the changeable environment, plants evolve complex mechanisms to integrate exogenous environmental stimuli and endogenous plant hormone signaling to coordinate growth and stress response. Jasmonate (JA) is an important phytohormone to widely participate into plant growth, immunity and defense through reprogramming gene expression [1,2,3]. The biosynthesis of JA is initiated from α-linolenic acid with sequential oxidation and catalysis by LOX, AOS, AOC and other enzymes to form jasmonic acid, finally to generate bioactive JA-Ile by JAR1 [4]. When JA level is elevated in plants, JA-Ile is perceived and bound by the receptor complex consisting of the F-box protein COI1 and the JA signaling repressor JAZ, resulting in degradation of JAZ by the 26S proteasome and releasing of MYC2 to activate downstream gene expression [5,6,7].
MYC2 is the key transcription factor to regulate JA signaling genes with conserved functions in plants [8]. MYC2 belongs to the basic-helix-loop-helix (bHLH) family, widely involved in plant defense, growth and development, metabolism and signaling transduction. At the C-terminus, MYC2 has the bHLH domain consisting of the basic region with 10–15 aa and the α-HLH-α motif with 40 aa, essential for DNA binding. At the N-terminus, MYC2 has one trans-activation domain for recruiting MED25 and the JID domain for interacting with JAZ, which is required for fine-tuning downstream gene expression [5, 9, 10]. Under the resting state of JA signaling, JAZ interacts with MYC2 and inhibit regulation of downstream target genes. Once JA signaling is activated, JAZ is degraded and MYC2 is released to target downstream genes [10].
MYC2 mediates the core transcriptional regulation of JA signaling to play diverse roles in stress response, specialized metabolism and growth in many plants [11, 12]. MYC2 is involved in many biological processes, such as root growth [13], seed protein accumulation [14], light signaling [15,16,17], phytohormone signaling crosstalk [18,19,20,21]. MYC2 is also involved in many defensive processes against herbivory and pathogen infection [10, 22], as well as drought response [23,24,25] and salinity response [26,27,28]. In maize, ZmMYC2 has been characterized [29]. It regulates maize terpenoid phytoalexin metabolism through involving in JA and ethylene signaling [30]. ZmMYC2 was also reported to bind the promoters of benzoxazinoid and volatile terpenoid biosynthetic genes to regulate corresponding metabolites accumulation in response to herbivory [31]. The homologous ZmMYC7 was identified to regulate ZmERF17 to promote maize resistance to Fusarium graminearum [32].
To give the better understanding for ZmMYC2 function and regulatory mechanism, the joint analysis through DAP-seq and RNA-seq was conducted to identify ZmMYC2 target genes and binding sites on maize chromosome. Abundant genes were detected in this study to be regulated by ZmMYC2 directly or indirectly. A number of cis-elements were also identified as the binding sites of ZmMYC2 for transcriptional regulation. These data in this study could be used for construction of ZmMYC2 regulatory network and further function characterization.
Results
RNA-seq analysis of ZmMYC2 regulating genes
Previously we identified ZmMYC2 as the core transcription factor of maize JA signaling [29]. To further characterize ZmMYC2 function and explore the regulating genes, we used maize leaf protoplasts as the convenient and fast platform to transiently overexpress ZmMYC2. The successful transformation and high expression were verified by GFP observation and qPCR validation (Fig S1A-C). The protoplasts with ZmMYC2 overexpression were collected for RNA extraction and RNA-seq analysis. Clear clustering of detecting genes was observed for RNA-seq data as indicated in Fig S1C (Table S1). After overexpression of ZmMYC2 in maize protoplasts, 2616 genes were detected with upregulation and 3104 genes for downregulation (Fig. 1A and Table S2). GO analysis indicated that these DEGs were enriched in various processes such as cell wall, defense, antioxidant activity (Fig. 1B and Table S3). Further KEGG analysis revealed that these DEGs were enriched in many pathways such as plant hormone signal transduction, phenylpropanoid biosynthesis, glutathione metabolism, as well as MAPK cascade, starch and sugar metabolism (Fig. 1C and Table S4). These results suggest that ZmMYC2 exhibits diverse functions.

RNA-seq analysis of ZmMYC2 transient overexpression in maize protoplasts. (A) volcano plot of DEGs in ZmMYC2 transient overexpression maize protoplasts and the control. Log2Foldchange indicates the fold change of DEGs with upregulation or downregulation. (B) GO enrichment analysis of DEGs. BP, biological process. CC, cellular component. MF, molecular function. (C) KEGG enrichment analysis of DEGs in different pathways with upregulation (Up term) and downregulation (Down term). The specific enriched pathways only with upregulated DEGs were also shown (Specific term)
Target regulating genes of ZmMYC2
We have identified a lot of potential target genes with binding sites using DAP-seq [29]. To explore whether these genes could be regulated by ZmMYC2, we combined the data from DAP-seq and RNA-seq to find the direct target regulating genes of ZmMYC2. From both sequencing data, 3183 genes (Target regulating genes, TRGs) were identified to be bound and regulated by ZmMYC2 (Fig. 2A and Table S5), among which 1741 genes were upregulated and 1442 genes were downregulated by ZmMYC2 (Fig. 2B). In RNA-seq data, expression of other 2537 genes (Indirect regulating genes, IRGs) were observed to be affected by transient overexpression of ZmMYC2 in maize protoplasts, however, they were not detected in DAP-seq data with direct binding, indicating indirect regulation on these genes by ZmMYC2. Among IRG, 1363 genes were upregulated and 1174 genes were downregulated by ZmMYC2 (Fig. 2B). There were also 21, 069 genes to be detected in DAP-seq data with binding by ZmMYC2 but not identified as the DEGs in RNA-seq data (Table S6).
We conducted GO analysis for regulating genes of ZmMYC2 and compared the top 15 GO terms of TRGs and IRGs (Fig. 2C). TRGs are mainly enriched in hormone response, stress response, metabolism and enzymatic activity, indicating that ZmMYC2 directly targets and regulates these genes to play roles in such processes. IRGs are mainly enriched in ribosome, photosynthesis and structure molecule activity, suggesting the indirect role of ZmMYC2 in these processes through regulating IRGs.

Joint analysis of RNA-seq and DAP-seq for ZmMYC2. (A) Venn plot of shared and specific genes by joint analysis of RNA-seq and DAP-seq for ZmMYC2. (B) Volcano plots for target regulating genes (TRGs) and indirect regulating genes (IRGs) of ZmMYC2. (C) Predominant GO terms of TRGs and IRGs through GO enrichment analysis
ZmMYC2 is involved in plant growth and stress response
To further analyze the function of ZmMYC2 in regulation of TRGs, we downloaded transcriptomic data related to maize growth and stress response from Maize eFP database for association analysis with our data [33]. 1429 TRGs were screened to be involved in growth and stress response (Table S7), including 303 genes for cold response, 430 for heat, 205 for salinity and 151 for drought by PEG treatment (Fig. 3A and Table S8). In the mean time, 197 and 407 TRGs were observed to respond to C. zeina and C. graminicola infection, respectively (Fig. 3B and Table S9). 389 and 422 TRGs were involved in root and apical meristem development, respectively (Fig. 3C and Table S10). All these genes were clustered into four categories (C1-4), among which C1 and C2 exhibited upregulation by ZmMYC2 but increased or decreased expression in the corresponding treatments, respectively. Genes in C3 and C4 were repressed by transient overexpression of ZmMYC2 but also showed increased or decreased expression in the corresponding treatments, respectively. All four category genes were detected in abiotic stress responses but C1 and C3 were mainly observed in pathogen infection responses with C1 as the predominant cluster (Fig. 3A-B), suggesting the positive role of ZmMYC2 in pathogen defense. In root and apical meristem development, C1 and C3 were mainly observed but with C3 as the predominant cluster (Fig. 3C), implicating negative roles of ZmMYC2 in growth.

Cluster analysis for gene expression analysis of ZmMYC2 target regulating genes. Gene expression of ZmMYC2 target regulating genes (TRGs) were compared among the RNA-seq data in this study and the transcriptomic data on Maize eFP including root and shoot apical meristem (SAM) growth, treatments of PEG, salt, heat and cold, infection of Colletotrichum graminicola and Cercospora zeina. (A) Cluster analysis of ZmMYC2 TRGs with expression comparison among RNA-seq data in this study and the data with abiotic stress treatments in Maize eFP, biotic stress treatments (B), root and SAM growth (C). C1-4, clustered genes. C1 and C2 exhibited upregulation by ZmMYC2 but upregulation (C1) or downregulation (C2) in the corresponding treatments. C3 and C4 were repressed by ZmMYC2 but showed increasing (C3) or decreasing (C4) expression in the corresponding treatments
ZmMYC2 regulates plant hormone related genes
MYC2 plays the key role in plant hormone signaling crosstalk [10, 34]. Further analysis indicated that ZmMYC2 targeted many genes related to plant hormone biosynthesis and signal transduction (Fig. 4 and Table S11). Among these genes, four types of plant hormones were revealed to be regulated by ZmMYC2 through targeting their biosynthesis genes including JA, abscisic acid (ABA), Auxin and brassinolide (BR) (Fig. 4A-B and F-G). In addition, ZmMYC2 widely regulated plant hormone signaling genes with mainly targeting JAZ, PP2C, SnRK2, ARF, Aux/IAA, BSK and TGA. In these hormone related genes, some have been characterized functionally in maize growth or stress response. For instance, JA related genes, ZmLOX10 and ZmJAZ9 were involved in insect resistance, and ZmJAZ8 played roles in defense to C. graminicola infection [31, 35, 36]. Auxin biosynthesis genes, ZmYucca6 and ZmYucca9 were reported to participate into root geotropism and lodging resistance [37]. Auxin signaling genes, ZmIAA8, ZmGH3.2, ZmGH3.8 and ZmIAA8 were involved in drought resistance and seed development, and ZmGH3.2 and ZmGH3.8 with roles in seed aging or leaf senescence, respectively [38,39,40,41]. ABA synthesis gene ZmNCED3 was also identified in drought response and regulated by ZmMYC2 directly here (Fig. 4B) [42].

Plant hormone related genes regulated by ZmMYC2. Graphs of eight plant hormone synthesis and signling with key genes shown. (A-H), Jasmonate (JA), abscisic acid (ABA), cytokinin, gibberellin (GA), ethylene, auxin, brassinolide (BR) and salicylic acid (SA). Plant hormone names are labeled in orange. The key genes regulated by ZmMYC2 as TRGs are highlighted in blue. Circles close to genes indicate fold enrichment in DAP-seq and fold change in RNA-seq data for specific genes, respectively. Value changes are shown by color intensity. The genes with functional characterization were labeled in purple
ZmMYC2 regulates abundant transcription factors
Many transcription factors were also identified as target regulating genes of ZmMYC2 (Fig. 5A and Table S12). Among which, the top three transcription factor families are MYB, bHLH and AP2. We further listed these transcription factors by categories with fold change of gene expression in RNA-seq and enrichment in DAP-seq (Fig. 5B-H). Some of these transcription factors have been characterized functionally (Table 1). For example, overexpression of ZmWRKY114 impaired salinity tolerance [43]. ZmMYB42 negatively regulated lignin synthesis, which was activated by ZmMYB69 to inhibit lignin biosynthesis in maize stem [44, 45]. ZmNAC111 promoted stomotal closure to increase water utilization and drought resistance [46]. ZmARF34 interacted with ZmRUM1 to participate lateral root formation [47]. In these genes, some of them have been identified to respond to JA signal, indicating that ZmMYC2 could mediate such response. For other transcription factors, our results indicated that ZmMYC2 might work upstream to regulate these genes, which might be investigated in the future study.

Transcription factors regulated by ZmMYC2. (A) Bar plot of transcription factors regulated by ZmMYC2 with families and gene numbers. (B-H) Lollipop plot of seven transcription factor families regulated by ZmMYC2. Fold enrichment (green circle) by DAP-seq and log2fold change (red circle) by RNA-seq were shown for each genes. The genes highlighted in purple were functional characterized in previous studies
ZmMYC2 recognizes diverse cis-elements
ZmMYC2 belongs to the bHLH family that can recognizes and binds to cis-elements like the G-box, E-box and CAnnTG [65, 66]. DAP-seq analysis revealed that ZmMYC2 bound to abundant and diverse target motifs. The motifs with percentage of target sequences more than 10% were shown in Fig. 6, including the 8 bp motifs nCATGTGn (70.42%), AAAAAAAA (40.53%), CACGTGTT (29.71%); the 10 bp motifs nGCATGCAnn (32.93%), GTGTGCATAT (18.03%); the 12 bp motif CACGTGCGTGCG (33.79%). Among these motifs, the classic E-box and G-box sequence were also detected (Fig. 6A and C). Centrality analysis indicated that these motifs were located in the corresponding peaks detected by DAP-seq in the pattern of normal distribution and with peak value of ∼ 0.5, suggesting as the potential cis-elements of ZmMYC2. In addition, we also analyzed the binding sites of ZmMYC2 on some TRGs and found that most binding sites for these genes are located on promoters (Fig. 7). However, binding on the gene exon or intron or 3’ UTR was also observed, for example, ZmARF5, ZmPYL, ZmMYB69. All these results indicate that ZmMYC2 might conduct functions through acting on diverse cis-elements.

The main motifs bound by ZmMYC2. The main motifs were detected by DAP-seq to be bound by ZmMYC2 with length of 8 (A-C), 10 (D-E) and 12 (F). % of Target is percentage of target sequences with motif. The core sequences of E-box and G-box were labeled in red (A, C). Normal distribution of motifs is shown to reveal motif centrality. The x-axis indicates the relative position of motif in the peak. The y-axis indicates the count of motif on the different position of peak. Gaussian fitting curve is highlight in dark red

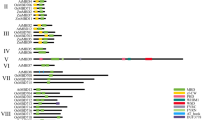
Motif distribution bound by ZmMYC2 on selected genes
Gene location on maize chromosomes are shown with gene names. The initial codon and transcription direction are indicted with the red arrow. The peaks indicate the binding of ZmMYC2 on genes by DAP-seq analysis with fold enrichment
Discussion
MYC2 as the master regulator of JA signaling plays important and diverse roles in plant growth and environmental responses. Here we explored the regulatory target genes of ZmMYC2 using the convenient protoplast transient expression system and revealed abundant TRGs and IRGs of ZmMYC2 with diverse target cis-elements in maize, providing the basis for further functional characterization of ZmMYC2 and target genes.
MYC2 has been reported to bind to the promoters of PLT1 and PLT2 and repress their gene expression, resulting in negative regulation on root stem cell and meristem activity [67]. ERF115 was involved in cell division in quiescent center and positively regulated by JA through MYC2, which promoted stem cell and regeneration after root damage [68]. Auxin synthesis genes Yucca6 and Yucca9 participated into root geotropism and lodging resistance in maize [69]. All these genes were found in our study to be the target genes of ZmMYC2, indicating involvement of ZmMYC2 in root development through regulating these genes. In addition, MYC2 also works in leaf senescence. MYC2 inhibited CAT2 gene expression, leading to H2O2 accumulation and upregulation of senescence associated genes SAG12, SAG13, SAG29, SAG113 and repression of photosynthesis related genes CAB1 and RBCS [70, 71]. JA induced senescence related gene Dof2.1 through MYC2 and Dof2.1 also activated MYC2 promoter, forming the positive feedback loop in leaf senescence [72]. During rape leaf senescence, MYC2 was activated by ABA and further elevated starch degrading genes AMY3, BAM1 and sugar transporting gene SUT1, SUT4 and SWEET11 [73]. In tomato, SlMYC2 increased expression of chlorophyll degradation gene SIPAO and promoted senescence [74]. In our results, we also found many senescence associated genes to be the target regulating genes of ZmMYC2 (Table S13), suggesting the function of ZmMYC2 in maize senescence regulation and providing basis for such investigation in the future.
MYC2 mediates plant hormone signaling crosstalk among JA, ABA, ethylene, GA and SA to widely involve in various biological processes [75]. In Arabidopsis, AtMYC2 bound to promoters of a number of defense related genes to positively regulate MKK4, RIN4, ICS1 and negatively regulate PEPR1, then playing dual roles in PTI and ETI through mediating SA bisoynthesis and signaling [76]. AtMYC2 also increased GA inactivation genes to inhibit endogenous GA level and plant growth through JA signaling [77]. MYC2 was also involved in drought response through activating RD22 and ADH, genes in ABA signaling pathway [78, 79]. Auxin synthesis was negatively regulated by MYC2 through inhibiting tryptophan biosynthesis, leading to repression of leaf vein development [80]. In this study, many hormone related genes were identified as the target regulating genes of ZmMYC2, indicating the important role of ZmMYC2 in hormone signaling crosstalk in maize, which could be characterized in the future investigation.
MYC2 mediates JA signaling to participate into biotic and abiotic stress responses. Overexpression of MYC2 in rice increased PR gene expression to confer resistance to bacterial blight [81]. In Arabidopsis, MYC2 interacted with EIN3 to regulate ERF gene expression and enhance PDF1.2 expression in response to pathogen infection [18]. Here we observed a number of PR genes to be upregulated by transient overexpression of ZmMYC2 in maize protoplasts, suggesting potential involvement of ZmMYC2 in disease resistance. In addition, MYC2 in apple bound to the G-box of MdCBF1 gene promoter to regulate cold tolerance [82]. In wheat, MYC2 regulated melatonin synthesis gene ASMT to improve drought resistance [83]. In Arabidopsis, MYC2 inhibited CAT2 gene expression and promoted H2O2 accumulation by JA and leaf senescence, resulting in lower salt tolerance [28, 70]. ZmMYC2 was also identified to regulate many abiotic stress response genes in this study (Table S13), implicating participation of ZmMYC2 in these processes.
MYC2 has been demonstrated to involve specialized metabolism regulation. At MYC2 directly bound to gene promoters of terpene synthases to increase terpene accumulation for defense to Bemisia tabaci [84]. In Tripterygium wilfordii, TwMYC2a and TwMYC2b negatively regulated gene expression of TwTPS27a/b and triptolide biosynthesis [85]. In maize, CUT&Tag-seq and RNA-seq analysis revealed that ZmMYC2 directly targeted and regulated ZmIGPS1/3 and BX10/11/12/14 in benzoxazinoid metabolism [31]. In this study, many benzoxazinoid related genes including ZmIGPS1, ZmBX4, ZmBX5, ZmBX6, ZmBX12 and ZmBX13 (Table S13), among which ZmIGPS1 and ZmBX12 have been demonstrated to be the target genes of ZmMYC2 [31], consistent with the previous study. In addition, ZmMYC2 was also reported to regulate terpenoid phytoalexin biosynthesis [30]. Here we detected ZmKSL2 (Zm00001d041082) as the target genes of ZmMYC2, which further validates our data accurancy. Furthermore, MYC3, the homolog of MYC2, dominantly affected flavonoid biosynthesis and cotton bollworm resistance [86]. Many flavonoid synthesis genes were also detected as the target regulating genes of ZmMYC2 (Table S13), suggesting the potential role of ZmMYC2 in maize flavonoid biosynthesis regulation.
MYC2 is usually reported to bind cis-elements like the G-box (CACGTG) and the E-box (ACGT or CANNTG) to regulate downstream target gene expression [65, 66]. For instance, MYC2 bound the G-box on target gene promoter to activate JA related gene expression and positively regulated JA signaling transduction and corresponding defense response [87, 88]. In Artemisia annua, AaMYC2 bound to the G-box like of CYP71AV1 and DBR2 promoters, two key genes of artemisinin biosynthesis, to elevate their expression [89]. SmMYC2 bound to the E-box on SmCYP98A14 gene promoter to regulate gene expression and tanshinone biosynthesis [66, 90]. In addition to G-box and E-box, we also found many new cis-elements to be recognized by ZmMYC2, which should be characterized in the future investigation about their functions and mechanisms.
We also found that ZmMYC2 regulated a lot of genes with various function annotations. Fox example, many glycosyl transferase encoding genes were observed as the TRGs of ZmMYC2 (Table S14). Rice glycosyl transferase UGT2 is regulated by bZIP23 to elevate salt tolerance [91]. UDP-glucose transferase EDR1 is the pivotal factor for endosperm development of rice [92]. In maize, the UDP-glucose transferase encoding gene sk1 is involved in pistil protection through blocking JA accumulation [93]. In addition, many glycoside hydrolase encoding genes were also regulated directly by ZmMYC2, which might be involved in specialized metabolism. In tartary buckwheat, FtGH1 (β-glucosidase) converted rutin to quercetin, playing the key role in rutin hydrolysis [94]. Glutathione S-transferase is a super family with the main functions of detoxification and specialized metabolism. The glutathione S-transferase encoding genes were also regulated by ZmMYC2. In tea tree, CsJAZ1-CsMYC2.2 mediated JA signaling to regulate CsGSTU45 for H2O2 accumulation, affecting resistance to Colletotrichum camelliae infection [95]. SWEET protein is the sugar transporter. In barley, SWEET11b is responsible for transportation of sugar and cytokinin [96]. SWEET17 in Arabidopsis regulated lateral root growth and drought resistance [97]. Many SWEET genes were also found to be TRGs of ZmMYC2, indicating participation of ZmMYC2 in regulating these processes.
Moreover, many genes encoding cytochrome P450, pentatricopeptide repeat (PRR) and protein kinase were identified to be regulated by ZmMYC2 with promoter binding (Table S14). These genes are widely involved in plant growth, development and stress response, suggesting the various functions of ZmMYC2. OsNBL3, the mitochondrion localized PRR, played a role in intron splicing of nad5 to increase stress response in rice [98]. Maize PRR genes regulated kernel size and yield determination [99, 100]. Protein kinases like LRR receptor-Like protein kinase, serine/threonine-protein kinase and calcium-dependent protein kinase function in most biological processes of plant life [101,102,103]. Although we observed regulation of these genes by ZmMYC2 here, the regulatory function and mechanism should be validated in the future investigation to be linked with specific biological process.
Conclusions
MYC2 not only plays as the master regulator of JA signaling, but also functions in many other biological processes in plants. Here we combined RNA-seq and DAP-seq data to explore the target regulating genes of ZmMYC2. A lot of genes were identified to regulated by ZmMYC2 through direct binding with various function annotation. Meanwhile, many new cis-elements were identified to be recognized and bound by ZmMYC2 besides the classic G-box and E-box. This investigation indicates the broad functions of ZmMYC2 through targeting diverse pathway genes including multiple plant hormone signaling genes and many transcription factor to participate various processes in plant growth and environmental responses (Fig. 8). Our study provides the important data basis for further function and mechanism characterization to clarify the complex signaling transduction and regulation in maize.

The proposed regulating model of ZmMYC2
ZmMYC2 targets to many plant hormone signaling genes and transcription factor encoding genes, which confers the broad function of ZmMYC2 in plant growth and stress responses through regulating these genes in various pathways
Materials and methods
Plant materials
Seeds of maize inbred line Mo17 were obtained from Maize Research Institute in Sichuan Agricultural University and germinated and grown in the growth chamber with the condition of 16 h light and 8 h dark at 28 °C. The maize seedling with two leaves were transferred into the darkness for 5 d until etiolation, ready for protoplast preparation.
Protoplast preparation and transfection
Maize leaf protoplast isolation was conducted as described previously [104]. ZmMYC2 was ligated into pBI221 and transfected into maize protoplasts as described previously [30], the empty pBI221 vector was used as the control. Specifically, 20 μg plamids were mixed with 200 μL protoplasts and 220 μL transfection buffer (40% PEG4000, 0.2 M mannitol, 0.1 M CaCl2) and kept in the darkness for 15 min. 1 mL W5 solution (154 mM NaCl, 125 mM CaCl2, 5 mM KCl and 2 mM MES) was added to terminate transfection. Protoplasts were collected with gentle centrifuge and resuspended into 500 μL W5 solution and incubated in the dark at 25 °C for 16–24 h, ready for protoplast collection and RNA extraction.
RNA extraction and RNA-seq analysis
RNA extraction from maize protoplasts was performed as described previously [64]. The HiPure HP Plant RNA Mini Kit (Magen Biotech, Guangzhou, China) was used to extract RNA from maize protoplasts following the manufacture protocol. RNA-seq analysis was conducted by Novogene (https://www.novogene.com/). cDNA library sequencing and data filtering were conducted on an Illumina NovaSeq 6000. Specifically, transcriptome libraries were prepared using a TruSeqTM RNA sample preparation kit from Illumina (San Diego, CA, USA). Paired-end libraries were sequenced by Illumina NovaSeq6000 sequencing. Deferentially expressed genes (DEGs) were identified using the DESeq2 software [105]. GO and KEGG enrichment were analyzed by GSEA (https://www.gsea-msigdb.org/gsea/index.jsp) [105, 106].
Quantitative PCR analysis
cDNA was prepared using the total RNA extracted above with the reverse transcriptase kit (Vazyme Biotech, Nanjing, China). Quantitative PCR (qPCR) analysis was conducted using the SYBR GREEN qPCR Master MIX (Vazyme Biotech) on the Bio-Rad CFX96 as described previously [64]. The maize gene ZmEf1a was used as the endogenous control according to previous study [64].
DAP-seq analysis
The maize gDNA was extracted from the two-week-old maize Mo17 leaves with the CTAB reagent. The gDNA was broken into fragments with 200–500 bp by ultrasonication for further binding. The recombinant ZmMYC2 protein was prepared and used to incubate with the fragmented gDNA as described previously [29]. The binding gDNA of ZmMYC2 was extracted with the mixture of chloroform and isopentanol (24:1, v/v) and re-purified for sequencing. DAP-seq analysis was conducted and reported in our previous study [29]. Peak analysis of promoter was carried out by IGV-sRNA [107, 108].
Centrality analysis of motifs
The main motifs detected by DAP-seq were located in the peaks by the Homer software to acquire the motif position and peak length (http://homer.ucsd.edu/homer/introduction/update.html). The relative position of motif in peaks was shown in the histogram by the ggplot2 software (http://www.sthda.com/english/wiki/ggplot2-barplots-quick-start-guide-r-software-and-data-visualization). All motif and peak information were included the raw data of DAP-seq.
Gene expression analysis
Maize gene expression data under different growth periods and stress treatment including PEG, Salt, Heat, Cold, infection of Colletotrichum graminicola and Cercospora zeina were obtained on the website of Maize eFP Browser (https://bar.utoronto.ca/) [109]. RNA-seq data analysis was conducted on ChiPlot (https://www.chiplot.online/#) and Bioinformatics (https://bioinformatics.com.cn/) [107, 108].
Data availability
DAP-seq and RNA-seq raw data were deposited into Genome Sequence Archive of CNCB-NGDC (https://ngdc.cncb.ac.cn/) with the accession ID of PRJCA022022 and PRJCA022023, respectively.
Change history
17 May 2024
A Correction to this paper has been published: https://doi.org/10.1186/s12864-024-10386-z
References
Browse J. Jasmonate passes muster: a receptor and targets for the defense hormone. Annu Rev Plant Biol. 2009;60:183–205.
Wasternack C, Strnad M. Jasmonates are signals in the biosynthesis of secondary metabolites pathways, transcription factors and applied aspects. New Biotechnol. 2019;48:1–11.
Zhang L, Zhang F, Melotto M, Yao J, He SY. Jasmonate signaling and manipulation by pathogens and insects. EXBOTJ. 2017;68:1371–85.
Huang H, Liu B, Liu L, Song S. Jasmonate action in plant growth and development. J Exp Bot. 2017;68:1349–59.
Chini A, Fonseca S, Fernández G, Adie B, Chico JM, Lorenzo O, et al. The JAZ family of repressors is the missing link in jasmonate signalling. Nature. 2007;448:666–71.
Thines B, Katsir L, Melotto M, Niu Y, Mandaokar A, Liu G, et al. JAZ repressor proteins are targets of the SCFCOI1 complex during jasmonate signalling. Nature. 2007;448:661–5.
Yan J, Zhang C, Gu M, Bai Z, Zhang W, Qi T, et al. The Arabidopsis CORONATINE INSENSITIVE1 protein is a Jasmonate receptor. Plant Cell. 2009;21:2220–36.
Peñuelas M, Monte I, Schweizer F, Vallat A, Reymond P, García-Casado G, et al. Jasmonate-related MYC transcription factors are functionally conserved in Marchantia polymorpha. Plant Cell. 2019;31:2491–509.
Çevik V, Kidd BN, Zhang P, Hill C, Kiddle S, Denby KJ, et al. MEDIATOR25 acts as an Integrative Hub for the regulation of Jasmonate-responsive gene expression in Arabidopsis. Plant Physiol. 2012;160:541–55.
Kazan K, Manners JM. MYC2: the master in action. Mol Plant. 2013;6:686–703.
Wasternack C, Jasmonates. An update on biosynthesis, signal transduction and action in plant stress response, growth and development. Ann Botany. 2007;100:681–97.
Ali S, Baek K-H. Jasmonic acid signaling pathway in response to abiotic stresses in plants. Int J Mol Sci. 2020:19.
Gasperini D, Chételat A, Acosta IF, Goossens J, Pauwels L, Goossens A, et al. Multilayered organization of jasmonate signalling in the regulation of root growth. PLoS Genet. 2015;11:e1005300.
Gao C, Qi S, Liu K, Li D, Jin C, Li Z, et al. MYC2, MYC3, and MYC4 function redundantly in seed storage protein accumulation in Arabidopsis. Plant Physiol Biochem. 2016;108:63–70.
Maurya JP, Sethi V, Gangappa SN, Gupta N, Chattopadhyay S. Interaction of MYC2 and GBF1 results in functional antagonism in blue light-mediated Arabidopsis seedling development. Plant J. 2015;83:439–50.
Zhang Q, Xie Z, Zhang R, Xu P, Liu H, Yang H, et al. Blue light regulates secondary cell wall thickening via MYC2/MYC4 activation of the NST1-directed transcriptional network in Arabidopsis. Plant Cell. 2018;30:2512–28.
Luo F, Zhang Q, Xin H, Liu H, Yang H, Doblin MS, et al. A phytochrome B-PIF4-MYC2/MYC4 module inhibits secondary cell wall thickening in response to shaded light. Plant Commun. 2022;3:100416.
Song S, Huang H, Gao H, Wang J, Wu D, Liu X, et al. Interaction between MYC2 and ETHYLENE INSENSITIVE3 modulates antagonism between Jasmonate and ethylene signaling in Arabidopsis. Plant Cell. 2014;26:263–79.
Zhang X, Zhu Z, An F, Hao D, Li P, Song J, et al. Jasmonate-activated MYC2 represses ETHYLENE INSENSITIVE3 activity to antagonize ethylene-promoted apical hook formation in Arabidopsis. Plant Cell. 2014;26:1105–17.
Hong G-J, Xue X-Y, Mao Y-B, Wang L-J, Chen X-Y. Arabidopsis MYC2 interacts with DELLA proteins in regulating sesquiterpene synthase gene expression. Plant Cell. 2012;24:2635–48.
Chen R, Jiang H, Li L, Zhai Q, Qi L, Zhou W, et al. The Arabidopsis mediator subunit MED25 differentially regulates Jasmonate and abscisic acid signaling through interacting with the MYC2 and ABI5 transcription factors. Plant Cell. 2012;24:2898–916.
Du M, Zhao J, Tzeng DTW, Liu Y, Deng L, Yang T, et al. MYC2 orchestrates a hierarchical transcriptional cascade that regulates jasmonate-mediated plant immunity in tomato. Plant Cell. 2017;29:1883–906.
Xu B-Q, Wang J-J, Peng Y, Huang H, Sun L-L, Yang R, et al. SlMYC2 mediates stomatal movement in response to drought stress by repressing SlCHS1 expression. Front Plant Sci. 2022;13:952758.
Zhao W, Huang H, Wang J, Wang X, Xu B, Yao X, et al. Jasmonic acid enhances osmotic stress responses by MYC2-mediated inhibition of protein phosphatase 2C1 and response regulators 26 transcription factor in tomato. Plant J. 2023;113:546–61.
Xing Q, Liao J, Cao S, Li M, Lv T, Qi H. CmLOX10 positively regulates drought tolerance through jasmonic acid -mediated stomatal closure in oriental melon. Sci Rep. 2020;10:17452.
Deng H, Li Q, Cao R, Ren Y, Wang G, Guo H, et al. Overexpression of SmMYC2 enhances salt resistance in Arabidopsis thaliana and salvia miltiorrhiza hairy roots. J Plant Physiol. 2023;280:153862.
Liu S, Zhang P, Li C, Xia G. The moss jasmonate ZIM-domain protein PnJAZ1 confers salinity tolerance via crosstalk with the abscisic acid signalling pathway. Plant Sci. 2019;280:1–11.
Song R-F, Li T-T, Liu W-C. Jasmonic acid impairs Arabidopsis seedling salt stress tolerance through MYC2-mediated repression of CAT2 expression. Front Plant Sci. 2021;12:730228.
Fu J, Liu L, Liu Q, Shen Q, Wang C, Yang P, et al. ZmMYC2 exhibits diverse functions and enhances JA signaling in transgenic Arabidopsis. Plant Cell Rep. 2020;39:273–88.
Fu J, Wang L, Pei W, Yan J, He L, Ma B, et al. ZmEREB92 interacts with ZmMYC2 to activate maize terpenoid phytoalexin biosynthesis upon Fusarium graminearum infection through jasmonic acid/ethylene signaling. New Phytol. 2023;237:1302–19.
Ma C, Li R, Sun Y, Zhang M, Li S, Xu Y, et al. ZmMYC2s play important roles in maize responses to simulated herbivory and jasmonate. J Integr Plant Biol. 2023;65:1041–58.
Cao H, Zhang K, Li W, Pang X, Liu P, Si H, et al. ZmMYC7 directly regulates ZmERF147 to increase maize resistance to Fusarium Graminearum. Crop J. 2023;11:79–88.
Hoopes GM, Hamilton JP, Wood JC, Esteban E, Pasha A, Vaillancourt B, et al. An updated gene atlas for maize reveals organ-specific and stress-induced genes. Plant J. 2019;97:1154–67.
Luo L, Wang Y, Qiu L, Han X, Zhu Y, Liu L, et al. MYC2: a master switch for plant physiological processes and specialized metabolite synthesis. Int J Mol Sci. 2023;24:3511.
Huang P-C, Grunseich JM, Berg-Falloure KM, Tolley JP, Koiwa H, Bernal JS, et al. Maize OPR2 and LOX10 mediate defense against fall armyworm and western corn rootworm by tissue-specific regulation of Jasmonic acid and ketol metabolism. Genes. 2023;14:1732.
Agostini RB, Ariel F, Rius SP, Vargas WA, Campos-Bermudez VA. Trichoderma root colonization in maize triggers epigenetic changes in genes related to the jasmonic and salicylic acid pathways that prime defenses against Colletotrichum graminicola leaf infection. J Exp Bot. 2023;74:2016–28.
Zheng Z, Wang B, Zhuo C, Xie Y, Zhang X, Liu Y, et al. Local auxin biosynthesis regulates brace root angle and lodging resistance in maize. New Phytol. 2023;238:142–54.
Wang Y, Nie L, Ma J, Zhou B, Han X, Cheng J, et al. Transcriptomic variations and network hubs controlling seed size and weight during maize seed development. Front Plant Sci. 2022;13:828923.
Ma H, Liu C, Li Z, Ran Q, Xie G, Wang B, et al. ZmbZIP4 contributes to stress resistance in maize by regulating ABA synthesis and root development. Plant Physiol. 2018;178:753–70.
Feng X, Liu L, Li Z, Sun F, Wu X, Hao D, et al. Potential interaction between autophagy and auxin during maize leaf senescence. J Exp Bot. 2021;72:3554–68.
Han Q, Chen K, Yan D, Hao G, Qi J, Wang C, et al. ZmDREB2A regulates ZmGH3.2 and ZmRAFS, shifting metabolism towards seed aging tolerance over seedling growth. Plant J. 2020;104:268–82.
Mao Y, Xu J, Wang Q, Li G, Tang X, Liu T, et al. A natural antisense transcript acts as a negative regulator for the maize drought stress response gene ZmNAC48. J Exp Bot. 2021;72:2790–806.
Bo C, Chen H, Luo G, Li W, Zhang X, Ma Q, et al. Maize WRKY114 gene negatively regulates salt-stress tolerance in transgenic rice. Plant Cell Rep. 2020;39:135–48.
Fornalé S, Sonbol F-M, Maes T, Capellades M, Puigdomènech P, Rigau J, et al. Down-regulation of the maize and Arabidopsis thaliana caffeic acid O-methyl-transferase genes by two new maize R2R3-MYB transcription factors. Plant Mol Biol. 2006;62:809–23.
Qiang Z, Sun H, Ge F, Li W, Li C, Wang S, et al. The transcription factor ZmMYB69 represses lignin biosynthesis by activating ZmMYB31/42 expression in maize. Plant Physiol. 2022;189:1916–9.
Liu S, Liu X, Zhang X, Chang S, Ma C, Qin F. Co-expression of ZmVPP1 with ZmNAC111 confers robust drought resistance in maize. Genes. 2023;14:8.
Zhang Y, Marcon C, Tai H, von Behrens I, Ludwig Y, Hey S, et al. Conserved and unique features of the homeologous maize Aux/IAA proteins ROOTLESS WITH UNDETECTABLE MERISTEM 1 and RUM1-like 1. J Exp Bot. 2016;67:1137–47.
Yang F, Shi Y, Zhao M, Cheng B, Li X. ZmIAA5 regulates maize root growth and development by interacting with ZmARF5 under the specific binding of ZmTCP15/16/17. PeerJ. 2022;10:e13710.
Hou F, Liu K, Zhang N, Zou C, Yuan G, Gao S, et al. Association mapping uncovers maize ZmbZIP107 regulating root system architecture and lead absorption under lead stress. Front Plant Sci. 2022;13:1015151.
He R-Y, Zheng J-J, Chen Y, Pan Z-Y, Yang T, Zhou Y, et al. QTL-seq and transcriptomic integrative analyses reveal two positively regulated genes that control the low-temperature germination ability of MTP–maize introgression lines. Theor Appl Genet. 2023;136:116.
Li Z, Srivastava R, Tang J, Zheng Z, Howell SH. Cis-effects condition the induction of a major unfolded protein response factor, ZmbZIP60, in response to heat stress in maize. Front Plant Sci. 2018;9:833.
Feng X, Xiong J, Zhang W, Guan H, Zheng D, Xiong H, et al. ZmLBD5, a class-II LBD gene, negatively regulates drought tolerance by impairing abscisic acid synthesis. Plant J. 2022;112:1364–76.
Agarwal T, Grotewold E, Doseff AI, Gray J. MYB31/MYB42 syntelogs exhibit divergent regulation of phenylpropanoid genes in maize, sorghum and rice. Sci Rep. 2016;6:28502.
Li J, Guo G, Guo W, Guo G, Tong D, Ni Z, et al. miRNA164-directed cleavage of ZmNAC1 confers lateral root development in maize (Zea maysL). BMC Plant Biol. 2012;12:220.
Yang Z, Wang C, Qiu K, Chen H, Li Z, Li X, et al. The transcription factor ZmNAC126 accelerates leaf senescence downstream of the ethylene signalling pathway in maize. Plant Cell Environ. 2020;43:2287–300.
Chen Y, Li X, Xie X, Liu L, Fu J, Wang Q. Maize transcription factor ZmNAC2 enhances osmotic stress tolerance in transgenic Arabidopsis. J Plant Physiol. 2023;282:153948.
Cao H, Yang Z, Song S, Xue M, Liang G, Li N. Transcriptome analysis reveals genes potentially related to maize resistance to Rhizoctonia solani. Plant Physiol Biochem. 2022;193:78–89.
Shiriga K, Sharma R, Kumar K, Yadav SK, Hossain F, Thirunavukkarasu N. Genome-wide identification and expression pattern of drought-responsive members of the NAC family in maize. Meta Gene. 2014;2:407–17.
Sahito JH, Zhang X, Zhong H, He X, Zhen C, Ma P, et al. Identification, association of natural variation and expression analysis of ZmNAC9 gene response to low phosphorus in maize seedling stage. Plants. 2020;9:1447.
Fang X, Bo C, Wang M, Yuan H, Li W, Chen H, et al. Overexpression of the maize WRKY114 gene in transgenic rice reduce plant height by regulating the biosynthesis of GA. Plant Signal Behav. 2021;16:1967635.
Yu T, Lu X, Bai Y, Mei X, Guo Z, Liu C, et al. Overexpression of the maize transcription factor ZmVQ52 accelerates leaf senescence in Arabidopsis. PLoS ONE. 2019;14:e0221949.
Huo T, Wang C-T, Yu T-F, Wang D-M, Li M, Zhao D, et al. Overexpression of ZmWRKY65 transcription factor from maize confers stress resistances in transgenic Arabidopsis. Sci Rep. 2021;11:4024.
Fang X, Li W, Yuan H, Chen H, Bo C, Ma Q, et al. Mutation of ZmWRKY86 confers enhanced salt stress tolerance in maize. Plant Physiol Biochem. 2021;167:840–50.
Liu L, Zhang Y, Tang C, Shen Q, Fu J, Wang Q. Maize transcription factor ZmHsf28 positively regulates plant drought tolerance. Int J Mol Sci. 2023;24:8079.
Lian T, Xu Y, Li L, Su X-D. Crystal structure of tetrameric Arabidopsis MYC2 reveals the mechanism of enhanced interaction with DNA. Cell Rep. 2017;19:1334–42.
Ezer D, Shepherd SJK, Brestovitsky A, Dickinson P, Cortijo S, Charoensawan V, et al. The G-box transcriptional regulatory code in Arabidopsis. Plant Physiol. 2017;175:628–40.
Chen Q, Sun J, Zhai Q, Zhou W, Qi L, Xu L, et al. The basic helix-loop-helix transcription factor MYC2 directly represses PLETHORA expression during jasmonate-mediated modulation of the root stem cell niche in Arabidopsis. Plant Cell. 2011;23:3335–52.
Zhou W, Lozano-Torres JL, Blilou I, Zhang X, Zhai Q, Smant G, et al. A Jasmonate signaling network activates root stem cells and promotes regeneration. Cell. 2019;177:942–e95614.
Pérez-Alonso M-M, Sánchez-Parra B, Ortiz-García P, Santamaría ME, Díaz I, Pollmann S. Jasmonic acid-dependent MYC transcription factors bind to a tandem G-box motif in the YUCCA8 and YUCCA9 promoters to regulate biotic stress responses. Int J Mol Sci. 2021;22:9768.
Zhang Y, Ji T-T, Li T-T, Tian Y-Y, Wang L-F, Liu W-C. Jasmonic acid promotes leaf senescence through MYC2-mediated repression of CATALASE2 expression in Arabidopsis. Plant Sci. 2020;299:110604.
Qi T, Wang J, Huang H, Liu B, Gao H, Liu Y, et al. Regulation of Jasmonate-induced leaf senescence by antagonism between bHLH subgroup IIIe and IIId factors in Arabidopsis. Plant Cell. 2015;27:1634–49.
Zhuo M, Sakuraba Y, Yanagisawa SA, Jasmonate-Activated MYC. 2–Dof2.1–MYC2 transcriptional loop promotes leaf senescence in Arabidopsis. Plant Cell. 2020;32:242–62.
Lee B-R, Zaman R, La VH, Bae D-W, Kim T-H. Ethephon-induced ethylene enhances starch degradation and sucrose transport with an interactive abscisic acid-mediated manner in mature leaves of oilseed rape (Brassica napus L). Plants. 2021;10:1670.
Ding F, Wang C, Xu N, Zhang S, Wang M. SlMYC2 mediates jasmonate-induced tomato leaf senescence by promoting chlorophyll degradation and repressing carbon fixation. Plant Physiol Biochem. 2022;180:27–34.
Yang J. The crosstalks between Jasmonic acid and other plant hormone signaling highlight the involvement of Jasmonic acid as a core component in plant response to biotic and abiotic stresses. Front Plant Sci. 2019;10:12.
Gautam JK, Giri MK, Singh D, Chattopadhyay S, Nandi AK. MYC2 influences salicylic acid biosynthesis and defense against bacterial pathogens in Arabidopsis thaliana. Physiol Plant. 2021;173:2248–61.
Fukazawa J, Mori K, Ando H, Mori R, Kanno Y, Seo M, et al. Jasmonate inhibits plant growth and reduces gibberellin levels via microRNA5998 and transcription factor MYC2. Plant Physiol. 2023;193:2197–214.
Abe H, Urao T, Ito T, Seki M, Shinozaki K, Yamaguchi-Shinozaki K. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell. 2003;15:63–78.
Wei X, Mao L, Wei X, Xia M, Xu C. MYB41, MYB107, and MYC2 promote ABA-mediated primary fatty alcohol accumulation via activation of AchnFAR in wound suberization in kiwifruit. Hortic Res. 2020;7:86.
Huang C-F, Yu C-P, Wu Y-H, Lu M-YJ, Tu S-L, Wu S-H, et al. Elevated auxin biosynthesis and transport underlie high vein density in C4 leaves. Proc Natl Acad Sci U S A. 2017;114:E6884–91.
Uji Y, Taniguchi S, Tamaoki D, Shishido H, Akimitsu K, Gomi K. Overexpression of OsMYC2 results in the Up-regulation of early JA-rresponsive genes and bacterial blight resistance in rice. Plant Cell Physiol. 2016;57:1814–27.
Wang Y, Xu H, Liu W, Wang N, Qu C, Jiang S, et al. Methyl jasmonate enhances apple’ cold tolerance through the JAZ–MYC2 pathway. Plant Cell Tiss Organ Cult. 2019;136:75–84.
Shamloo-Dashtpagerdi R, Lindlöf A, Nouripour-Sisakht J. Unraveling the regulatory role of MYC2 on ASMT gene expression in wheat: implications for melatonin biosynthesis and drought tolerance. Physiol Plant. 2023;175:e14015.
Zhao P, Zhang X, Gong Y, Wang D, Xu D, Wang N, et al. Red-light is an environmental effector for mutualism between begomovirus and its vector whitefly. PLoS Pathog. 2021;17:e1008770.
Huo Y, Zhang J, Zhang B, Chen L, Zhang X, Zhu C. MYC2 transcription factors TwMYC2a and TwMYC2b negatively regulate triptolide biosynthesis in tripterygium wilfordii hairy roots. Plants. 2021;10:679.
Wang D-D, Li P, Chen Q-Y, Chen X-Y, Yan Z-W, Wang M-Y, et al. Differential contributions of MYCs to insect defense reveals flavonoids alleviating growth inhibition caused by wounding in Arabidopsis. Front Plant Sci. 2021;12:700555.
You Y, Zhai Q, An C, Li C. LEUNIG_HOMOLOG mediates MYC2-dependent transcriptional activation in cooperation with the coactivators HAC1 and MED25. Plant Cell. 2019;31:2187–205.
Breeze E, Master, MYCs. MYC2, the Jasmonate signaling master switch. Plant Cell. 2019;31:9–10.
Kayani S-I, Shen Q, Ma Y, Fu X, Xie L, Zhong Y, et al. The YABBY family transcription factor AaYABBY5 directly targets cytochrome P450 monooxygenase (CYP71AV1) and double-bond reductase 2 (DBR2) involved in artemisinin biosynthesis in artemisia annua. Front Plant Sci. 2019;10:1084.
Du T, Niu J, Su J, Li S, Guo X, Li L, et al. SmbHLH37 functions antagonistically with SmMYC2 in regulating jasmonate-mediated biosynthesis of phenolic acids in salvia miltiorrhiza. Front Plant Sci. 2018;9:1720.
Wang T, Li X-K, Liu X, Yang X-Q, Li Y-J, Hou B-K. Rice glycosyltransferase gene UGT2 functions in salt stress tolerance under the regulation of bZIP23 transcription factor. Plant Cell Rep. 2023;42:17–28.
Wu Z, Zhang X, Chang G, Yang J, Wan J, Wang F, et al. Natural alleles of a uridine 5ʹ-diphospho-glucosyltransferase gene responsible for differential endosperm development between upland rice and paddy rice. J Integr Plant Biol. 2022;64:135–48.
Hayward AP, Moreno MA, Howard TP, Hague J, Nelson K, Heffelfinger C, et al. Control of sexuality by the sk1-encoded UDP-glycosyltransferase of maize. Sci Adv. 2016;2:e1600991.
Lai D, Zhang K, He Y, Fan Y, Li W, Shi Y, et al. Multi-omics identification of a key glycosyl hydrolase gene FtGH1 involved in rutin hydrolysis in Tartary buckwheat (Fagopyrum tataricum). Plant Biotechnol J. 2023. https://doi.org/10.1111/pbi.14259
Lv W, Jiang H, Cao Q, Ren H, Wang X, Wang Y. A tau class glutathione S-transferase in tea plant, CsGSTU45, facilitates tea plant susceptibility to colletotrichum camelliae infection mediated by jasmonate signaling pathway. Plant J. 2023. https://doi.org/10.1111/tpj.16567
Radchuk V, Belew ZM, Gündel A, Mayer S, Hilo A, Hensel G, et al. SWEET11b transports both sugar and cytokinin in developing barley grains. Plant Cell. 2023;35:2186–207.
Valifard M, Le Hir R, Müller J, Scheuring D, Neuhaus HE, Pommerrenig B. Vacuolar fructose transporter SWEET17 is critical for root development and drought tolerance. Plant Physiol. 2021;187:2716–30.
Qiu T, Zhao X, Feng H, Qi L, Yang J, Peng Y-L, et al. OsNBL3, a mitochondrion-localized pentatricopeptide repeat protein, is involved in splicing nad5 intron 4 and its disruption causes lesion mimic phenotype with enhanced resistance to biotic and abiotic stresses. Plant Biotechnol J. 2021;19:2277–90.
Long Y, Wang C, Liu C, Li H, Pu A, Dong Z, et al. Molecular mechanisms controlling grain size and weight and their biotechnological breeding applications in maize and other cereal crops. J Adv Res. 2023;23:00265–5.
Chen R, Wei Q, Liu Y, Li J, Du X, Chen Y, et al. The pentatricopeptide repeat protein EMP601 functions in maize seed development by affecting RNA editing of mitochondrial transcript ccmC. Crop J. 2023;11:1368–79.
Schulz P, Herde M, Romeis T. Calcium-dependent protein kinases: hubs in plant stress signaling and development. Plant Physiol. 2013;163:523–30.
Bender KW, Zipfel C. Paradigms of receptor kinase signaling in plants. Biochem J. 2023;480:835–54.
Son S, Park SR. The rice SnRK family: biological roles and cell signaling modules. Front Plant Sci. 2023;14:1285485.
Fu J, Liu Q, Wang C, Liang J, Liu L, Wang Q. ZmWRKY79 positively regulates maize phytoalexin biosynthetic gene expression and is involved in stress response. J Exp Bot. 2018;69:497–510.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13:1194–202.
Chen C, Wu Y, Li J, Wang X, Zeng Z, Xu J, et al. TBtools-II: a one for all, all for one bioinformatics platform for biological big-data mining. Mol Plant. 2023;16:1733–42.
Seeve CM, Cho I, Hearne LB, Srivastava GP, Joshi T, Smith DO, et al. Water-deficit‐induced changes in transcription factor expression in maize seedlings. Plant Cell Environ. 2017;40:686–701.
Funding
This work was supported by the NSFC (31971825 to Q.W., 32001446 to J.F.), the Talent Program of Scientific Innovation of Sichuan Province (2022JDRC0036).
Author information
Authors and Affiliations
Contributions
Q.W. and J.F. conceived the project. L.L., Y.Z., C.T. and J.W. performed the experiments and collected data. L.L., J.F. and Q.W. analyzed the data. L.L.and Q.W. wrote the paper.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Following publication of the original article, we have been notified that the y-axis labels in Figs. 1, 2, 3 and 8 had been mirrored and made unreadable.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, L., Zhang, Y., Tang, C. et al. Genome-wide identification of ZmMYC2 binding sites and target genes in maize. BMC Genomics 25, 397 (2024). https://doi.org/10.1186/s12864-024-10297-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10297-z