
Abstract
Background
The detection of signatures of selection in genomic regions provides insights into the evolutionary process, enabling discoveries regarding complex phenotypic traits. In this research, we focused on identifying genomic regions affected by different selection pressures, mainly highlighting the recent positive selection, as well as understanding the candidate genes and functional pathways associated with the signatures of selection in the Mangalarga Marchador genome. Besides, we seek to direct the discussion about genes and traits of importance in this breed, especially traits related to the type and quality of gait, temperament, conformation, and locomotor system.
Results
Three different methods were used to search for signals of selection: Tajima’s D (TD), the integrated haplotype score (iHS), and runs of homozygosity (ROH). The samples were composed of males (n = 62) and females (n = 130) that were initially chosen considering well-defined phenotypes for gait: picada (n = 86) and batida (n = 106). All horses were genotyped using a 670 k Axiom® Equine Genotyping Array (Axiom MNEC670). In total, 27, 104 (chosen), and 38 candidate genes were observed within the signatures of selection identified in TD, iHS, and ROH analyses, respectively. The genes are acting in essential biological processes. The enrichment analysis highlighted the following functions: anterior/posterior pattern for the set of genes (GLI3, HOXC9, HOXC6, HOXC5, HOXC4, HOXC13, HOXC11, and HOXC10); limb morphogenesis, skeletal system, proximal/distal pattern formation, JUN kinase activity (CCL19 and MAP3K6); and muscle stretch response (MAPK14). Other candidate genes were associated with energy metabolism, bronchodilator response, NADH regeneration, reproduction, keratinization, and the immunological system.
Conclusions
Our findings revealed evidence of signatures of selection in the MM breed that encompass genes acting on athletic performance, limb development, and energy to muscle activity, with the particular involvement of the HOX family genes. The genome of MM is marked by recent positive selection. However, Tajima’s D and iHS results point also to the presence of balancing selection in specific regions of the genome.
Similar content being viewed by others


Background
The “batida” and “picada” gait types are the main trait of the Mangalarga Marchador horse (MM), representing the unique natural movement allowed in intermediate speeds [1]. The main difference between batida and picada gaits is how the movement is executed, being the diagonal support more frequent than the triple support in the batida gait. In the picada gait, the lateral and triple supports overlap, providing a softer execution to the movement. This difference in movement characterizes the main phenotypic segregation in MM horses [2, 3], and because of this, gait is prioritized in studies involving this breed.
Andersson et al. [4] described the influence of DMRT3 gene and transcription factors, involved in the coordination of limb movement, in gaitedness across horse breeds. Promerová et al. [5] explained in detail the genetic mechanisms behind gait, including allelic patterns associated with equine locomotion across breeds. The frequency of DMRT3 allele A (mutant) was almost 100% in gaited horses, so the AA homozygous condition was believed to be associated with gait. However, post-investigations of the allelic patterns have shown that breeds without the gait phenotype could also have the mutant allele (A), as well as gaited horses could have the wild type allele (C) [5]. Although DMRT3 appears to be important for gaits in certain breeds, other genes are certainly involved in the expression of this trait.
Selection signatures studies represent a strategy for elucidating not only the complexity over the artificial/natural selection imposed on gait segregations, but also the complexity present in other economically important traits. It is of major interest to better understand the genetic aspects involved with complex phenotypes for the genetic improvement of MM horses. Investigations about hitchhiking effects on genomic regions and recent adaptive fixations were first conducted by Lewontin & Krakauer [6]. Current studies bring the concept of selection signatures, which are particular patterns of DNA identified in genomic regions with mutation and/or which have been under natural/artificial selection pressures in the population [7,8,9]. The exploitation of selection signatures aids in identifying regions in the genome under selective pressure that may harbor genes and variants that modulate important phenotypes in horses [10, 11].
Over the past few years, the interest in the detection of selection signatures in horses and other species has resulted in the increased number of publications on this topic, being the selection signatures described as results of domestication and selection processes that aimed to increase herd performance and productivity [12, 13]. There are several approaches to identifying signatures of selection [14,15,16,17,18,19,20,21,22]. Weigand & Leese [23] gathered several of the approaches in a review study, addressing the particularities of each approach in a non-model species perspective. In this study, we used three different approaches to search for signatures of selection in the genome of MM: Tajima’s D (TD) [20], the integrated haplotype score (iHS) [22], and runs of homozygosity (ROH) [21]. The choice of these three methods was made taking into account the genetic structure of our dataset, as horses of both gait modalities were not assigned into subpopulations. Therefore, we used standard within-population approaches to scan for signatures of selection in the MM breed, especially to detect recent signatures. In addition, a detailed discussion on signatures of selection that overlap with candidate genes and gene pathways previously described in the literature were provided, focusing more on candidates related to traits of importance in this breed, especially those related to the type and quality of gait, temperament, conformation, and locomotor system (muscular and skeletal structure).
Results
Genetic structure and linkage disequilibrium (LD)
Prior information related to the gait groups of each individual, batida and picada, was considered in the PCA analysis to investigate whether individuals who belong to the same group would cluster together. The top five eigenvectors explained 54.98% of the cumulative variance, with 40.33% assigned to cluster 1 for PCA 1 x PCA 2 (Fig. 1).

Principal Component Analysis (PCA) based on genotype data for the top five eigenvectors in Mangalarga Marchador horse, considering individuals with the batida and picada gait types. The core PCAs were highlighted in cluster 1
Only one cluster persisted in the dataset, meaning that all the individuals are genetically related when genomic information is considered. Some individuals were assigned distant from the center of cluster 1, implying that they are less genetically related to the others. The dispersion of the dataset and segregations (substructures) was attributed to the importance of sires from different families in the breed formation when the most significant number of clusters was assessed. Although animals with different gaits present distinct phenotypes, they are not discriminated by their genotypes which means they are not genetically distant. Therefore, one population, including all animals in the dataset, was taken into consideration for genomic scans of selection signatures.
A decrease in LD was observed as the physical distance between the markers increased. The r2 values were below 0.20 at distances below 15 kb (Additional file 1: Fig. S1). Further detailed aspects of the population structure and LD have been reported in Santos et al. [24] using the same database with imputed data. As we chose not to conduct the study with imputed data from two different platforms, the analyses were conducted with only 192 animals genotyped on Axiom MNEC670. Slight changes were noticed between our results and those reported in Santos et al. [24], which were possibly attributed to the different approaches used in the studies, as well as the reduction in the number of animals. However, the conclusions regarding the genetic structure and DL remained the same.
Signatures of selection and candidate genes identification
High TD values were identified under balanced selection in a wide aspect, and the majority of the equine autosomes demonstrated at least one significant signal of selection (Fig. 2, Additional file 2: Data S1). In general, high proportion of SNPs was noticed under balance selection or sudden population contraction scenarios. Values of -log10(p-value) ≥ 2 from empirical p-values were considered to be significant signals.

Patterns of genome-wide polymorphisms for Tajima’s D statistics were calculated in 20 kb windows across the genome. The threshold (−log10(p-value) ≥ 2) was highlighted in dashed line. The negative tail represents signals of recent positive selection. The top ten regions with the most significant values for negative tail were marked with small black arrowheads
In total, 147 genomic regions with negative and positive tails were identified as significant selection signals in the TD test (P < 0.01). As some limitations or biases inherent to the Tajima’s D approach can persist when genotyping data is used, we considered only negative values, which correspond to recent positive selection signals. The recent positive selection signals were observed on the autosomes ECA 1, 6, 7, 8, 20, and 26 (Fig. 2). In total, the TD signals encompassed 27 candidate genes (Table 1). The balancing selection results from this test were not prioritized in our study due to its subjectivity and the limited amount of information necessary for a better understanding.
The iHS positive and negative values were considered in our study, capturing ancient and recent signatures of selection. In total, 292 genomic regions were observed as signatures of selection in the iHS test (Additional file 2: Data S2). They were distributed along the genome, except for chromosomes ECA 21, 22, 26, 28, and 31 (Fig. 3a).

Genome-wide distribution of selection signatures in the equine autosomal chromosomes. a) -log10(p-value) for the Integrated Haplotype Score (iHS) plotted against chromosome position, with the significant threshold highlighted with the dashed line (P < 0.001). b) Runs of Homozygosity (ROH) islands with dashed line represented by the significant ROH hotspot mean frequency threshold ≥0.50
In total, 251 genomic regions were consistent in the iHS positive tail, representing the ancestral allele state, while 41 regions were consistent in the negative tail, representing the derived allele state. Genomic annotations were verified for the significant iHS signals. In total, 332 candidate genes were found within the signatures of selection (Additional file 3: Table S1).
Due to the large number of significant signals found in the iHS test, we did not follow the commonly used method of choosing to display only the top regions. We consider three parameters to prioritize candidate genes in our list: (I) genes within highlighted genomic regions based on the extremes iHS and piHS values, (II) genes related to locomotion, athletic performance, growth, fertility, conformation, pigmentation, and metabolism, and (III) genes that were also found in the Tajima’s D and ROH approaches. Considering these criteria, the shortened list comprised of 104 chosen genes. The genomic regions and their respective genes were shown in Table 2.
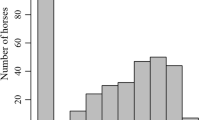
In the ROH analysis, 340 SNPs were observed within ROH island regions (mean hotspot) that were regions with frequencies ≥0.5 in the population (Additional file 2: Data S3). In total, 67,478 ROH segments were identified (Fig. 3b). The longest shared homozygous segment was detected in the ECA7, with length above 16 Mb. The number of ROH segments identified in ECA7 was 2846. Most of the ROH segments found in the MM genome corresponded to short segments with lengths around 1–2 Mb (Fig. 4). The ROH size is inversely correlated with age, where longer ROH is originated from recent common ancestors and shorter ROH is originated from distant common ancestors [19, 25, 26].

Shared homozygosity interval for the most representative chromosomes in the ROH approach. Green horizontal lines represent the length of ROH. Based on footprints, one can observe regions shared between individuals in the population
The same principle of gene annotation used for the iHS approach was used for ROH, adding 125 k upstream and downstream of the significant region. Most of the significant SNPs were located close to each other and, consequently, shared the same windows. Furthermore, windows found in ROH overlapped 38 genes (Table 3).
Nine genes were common between ROH and iHS tests. One gene is located on ECA1 (RASGRP1), and eight are located within ECA23 (C9orf24, CNTFR, DCTN3, DNAI1, ENHO, FAM219A, RPP25L, and SIGMAR1). No common genomic regions nor genes were found among TD and other statistics. Therefore, we sought to broaden the understanding of these genes through enrichment analysis and gene networks.
Enrichment analyses
Genes with biological processes relevant to horses were analyzed for pathways, molecular functions, and cellular components. The enrichment analyses were performed separately considering the gene lists derived from the three approaches, Tajima’s D, iHS, and ROH. To visualize the gene enrichment results, biological processes that are most relevant to the study were assessed (Additional file 3: Table S2). Most of the gene enrichment Gene Ontology (GO) terms for the biological process were attributed to cellular and metabolic processes (Fig. 5). Significant candidate genes with evidence of direct association with gait, locomotor system, energy, exercise, athletic performance, reproduction, and fertility were highlighted in Table 4. More details about the entire pool of identified genes can be accessed in Additional file 3: Table S2. The five main significant biological functions possibly associated with gait and locomotor system were represented in Fig. 6.

PANTHER GO-Slim pie chart analysis for biological processes for Tajima’s D, integrated haplotype score (iHS), and runs of homozygosity (ROH) approaches used to identify signatures of selection

Functional annotation for the top five significant biological functions possibly related to gait and locomotor system (P < 0.05)
Discussion
To the best of our knowledge, this is the first study to provide a whole scan for signatures of selection in the MM genome. Our findings shed light on the possible candidate genes/gene groups involved in the regions undergoing selection in this breed. The results and discussion found here can be useful for the comprehension of signatures of selection in other equine breeds.
Overall, quadrupeds use many footfall patterns during locomotion. The gaits are generally considered to be discrete patterns of footfalls and are divided into symmetrical and asymmetrical [27]. The allele A of the DMRT3 gene is only related to the picada gait in the MM, with two genotypes AA and CA, while the genotype CC is related to batida gait [28]; however, some studies have shown gait ability (the lateral gait pattern) is under the influence of a set of genes [5, 29]. Other discoveries suggested that alleles related to the type of gait were differently fixed within each gait type [5]. In Icelandic horses, no SNP demonstrated genome-wide significance for DMRT3, implying that the ability to pace goes beyond the presence of a single genetic variant [30]. Considering these facts, there is still a lack of information regarding the genetic architecture behind gait.
Selection in the MM breed is based exclusively on competitions where gaited performance records are evaluated relative to that of competitors, often being an empirical selection. Thus, we presumed that time under strong artificial selection is necessary to identify a possible gait type segregation to well-defined lineages. In this regard, it is essential to understand which genes in the MM population are most relevant to accomplish such goals. According to Arnason et al. [31], the thoroughbred carried out a long history of artificial selection for galloping speed while being ridden by a jockey, and it might be the same for MM. A well-defined breeding scheme can shift the allele frequencies of the desirable phenotype, and well-defined lineages could be achieved by selection. We next focused on exploring the signatures of positive recent selection found in the MM population and understanding the genes and pathways associated with these regions. As no significant region was associated with the DMRT3 gene in our analyses, we included the DMRT3 gene in a network analysis to get insights on the interactions between the observed candidate genes and DMRT3. The identification of genomic regions modified by positive selection has provided discoveries of adaptive directions in different species. Nowadays, the search for signatures of selection is one of the branches of the theoretical and applied evolutionary studies [7]. This study covered three distinct methods to scan for signatures of selection, which diverge according to their concepts and methodology. This implies that each method captures different patterns of genetic variation in different time scales. Due to the density of the genotyping panel used and the complexity of the biological information, these methods still present pitfalls and cannot exploit the genetic variation present within the entire genome. To overcome this limitation, it is essential that results from multiple methods, i. e., in our case, Tajima’s D, iHS and ROH, are used in a complementary way [32].
Common significant regions were found between iHS and ROH. Eight common candidate genes (C9orf24, CNTFR, DNAI1, ENHO, DCTN3, FAM219A, RPP25L, and SIGMAR1) were located on ECA23, and one common gene (RASGRP1) was located on ECA1. It is interesting that the genes mentioned above on ECA23 are located ~ 28 Mb away from DMRT3. The existence of LD in this region is possible biologically, and determining the exact gene(s) under selection can be challenging. Thus, we performed a network analysis including the DMRT3 gene, and only one occurrence of low co-expression was found between DCTN3 and DMRT3. Therefore, we excluded the possibility of any significant relationship between the eight candidate genes with DMRT3. However, the limitations of using non-model species may have interfered in our presumptions. Besides that, according to Ma et al. [33] and Ablondi et al. [34], during evolution, a series of unknown demographic events further increased the difficulty in detecting modified genomic regions due to different selective pressures. The use of next-generation sequencing (NGS) technologies can be promising for elucidating the relationships between loci in ECA23 because sequencing offers a higher genome coverage and more precision on the position of causative mutations and selection signatures [35]. Complementing the conclusions regarding common candidate regions observed among the used approaches, only a few common candidate genes were found for iHS and ROH. Each method presents specific features implying that non-overlapping regions of signatures of selection between different methods should be treated as complementary information to better understand the different patterns of variation in the MM genome.
The TD results suggest that the MM population is under strong balancing selection; however, many hitchhiking effects were highlighted in the statistics based on the extended haplotype homozygosity and footprints on homozygous regions. The pronounced balancing status in the studied population supported by the TD results was an interesting consequence, possibly explained by the nonexistence of any breeding program in the breed during the past years.
In a previous study developed by our group with the same database, we investigated the runs of homozygosity and populational inbreeding (Froh) in the MM breed [25]. A compressive discussion on the length of ROH segments is given in this previous study, which brings important information to understand the breed age and genetic variability. The MM breed is a relatively old breed compared to most modern breeds having originated in the mid-eighteenth century. The class with the highest percentage of ROH was 0–2 Mbps, with 92.78% of the observations, suggesting the majority of ROH segments was classified as short segments [25].
In the present study, we found only one long ROH segment (> 16 Mbps) located on ECA7 (Fig. 4). The number of short segments was more abundant, possibly due to events of recombination that occurred in the past and caused its reduction [19, 36], or due to the limitation of using genotyping data, thereby overestimating the number of short ROH [37]. Again, sequencing data can add value to ROH studies as it covers more genetic variability [38]. However, one of the strengths of ROH analysis is that long homozygous segments can be reliably identified, even from relatively modest marker densities [38].
Evidence for this long ROH segment on ECA7 has already been described in the literature [34]. We cannot fail to consider that equestrian sports prioritize high performance, therefore, conditioning to a positive selection based on directional selection [34, 39]. Another view suggested that strong bottlenecks occurred in this region during the breed formation. Ablondi et al. [34] found similar results for ECA7 in Swedish Warmblood horses and Exmoor ponies. Thus, we speculate that this candidate region of signature of selection in ECA7 is possibly a consequence of a previous bottleneck and not recent positive selection because of the similarity in the results found in distinct breeds. In this sense, our findings corroborate the argument reported in Ablondi et al. [34] for an intense bottleneck, but pointing to a common moment in the evolutionary process for some breeds.
Four genes (TRIR, TNPO2, WDR83, and WDR83OS) were highlighted within this longest shared homozygosity segment located on ECA7. These genes were identified under biological functions for localization (GO:0051179) and metabolic processes (GO:0008152). It has been shown that the TRIR gene has a significant role in cellular functions [40]. Other genes, TNPO2 and WDR83, were related to tumor development. One region on ECA1 encompassed the gene RASGRP1, which was found in common between ROH and iHS and played a key role in the development of T and B cells [41]. Studies have associated RASGRP with disease phenotypes in bovine animals [42, 43] and dogs [44].
In general, the genetic signals for the three statistics were most enriched in ontologies corresponding to “biological regulation,” “metabolic process,” and “cellular process.” In the Panther results for iHS and ROH candidate genes, the ontology “localization” was also very representative. Some highlighted candidate genes were associated with gait and locomotor system aspects, with eight of them regulating anterior/posterior pattern specification (Fig. 5).
The HOX genes encode homeodomain transcription factors in developing many embryonic structures in vertebrates and invertebrates [45]. According to Pineault & Wellik [46], as development progresses, tight spatial and temporal control of gene expression and cellular behavior sculpts the developing embryo, adding specific morphological and functional characteristics that determine the adult animal’s lifestyle and functionality.
The GLI3 gene was identified under the same HOX gene group to regulate anterior/posterior pattern specification. Exploring this information, we found that GLI3 is a transcriptional activator and a repressor of the sonic hedgehog pathway and plays a vital role in limb development. GLI3 has been described in the literature as an embryonic patterning of human limbs and other structures [47]. The relationship between the HOX genes and limb musculoskeletal development has been well described in the literature. Pineault & Wellik [46] suggested that the integration of the musculoskeletal system is regulated in part by HOX function in the stromal connective tissue and plays critical roles in skeletal patterning throughout the axial and appendicular skeleton. Evidence to support these genes as possibly regulating limb formation and other processes associated with the locomotor system was reported by Grilz-Seger et al. [48], who found several GO terms shared by more than one breed when studying a set of European and Near Eastern horse breeds; high significance levels were reached for the GO terms “anterior/posterior pattern specification” (GO:0009952), “embryonic skeletal system morphogenesis” (GO:0048704), and “sequence-specific DNA binding” (GO:0043565), mainly based upon the HOXB-cluster in the breeds Gidran, Lipizzan, Posavina, and Noriker.
Other significant signals in the present study were found for the CCL19 and MAP3K6 genes enriched for the activation of JUN kinase (JNK) activity. Exercise stimulates c-Jun NH2 Kinase Activity and c-Jun transcriptional activity in human skeletal muscle, showing that the JNK pathway may serve as a link between contractile activity and transcriptional responses in skeletal muscle [49]. Exercise causes selective changes over gene expression, leading to differentiation in skeletal muscle structure and function, which provides strong evidence that this regulation may be associated with gait type segregation in the skeletal muscle on limbs. The effect of activity during exercise in c-jun mRNA expression is via the phosphorylation of two serine residues through the JNKs in the c-Jun transactivation domain, leading to increased transcriptional activity [49].
It is well known in the modern horse that athletic performance has been the target of selection in recent years for many breeds. Increasingly, a perfect horse is being idealized in countless sporting modalities. Indeed, candidate genes were highlighted under important biological functions related to exercise physiology, energy mechanisms, catabolic processes, morphogenesis (bone, skeletal system, and cartilage), and fertility. However, these genes/functions do not act alone in the MM performance. As observed in the network analysis, gene functions are dependent, with the major part of them being regulated in sets.
The interpretation of the network analysis is that most candidate genes, either core genes or peripheral genes, are interconnected. Any peripheral gene is likely to affect the regulation or function of a hub gene. An explanation for the high interconnection in networks is that networks have structures consisting of distinct modules of connected nodes and frequent long-range connections. Under these conditions, any two nodes in the graph are usually connected by just a few steps [50].
Overall, the application of classical and recent techniques in genomics has successfully permitted the identification of several putative selection signatures in the MM population. Based on our discussions, gait may have a polygenic basis and is influenced by many genetic components. Further exploration would be strengthened by searching for signatures of selection by comparing the MM to a non-gaited breed. This method could then be compared to the regions found within the breed and would clarify whether these signatures are unique to the breed (or the gait) rather than being general signatures of selection in horses or if they could potentially detect new genetic bases of gait in the MM. Among the biological processes, genes of biological interest such as the HOX gene family were enriched in the ontology corresponding to “anterior/posterior pattern specification.” Biological processes related to limb morphogenesis, the skeletal system, proximal/distal pattern formation, JUN kinase activity (CCL19 and MAP3K6), and muscle stretch response (MAPK14), among others, were reported. Finally, identifying genes and pathways that drive phenotypes is still a challenge; here, we pinpoint some important genes and gene pathways involved in complex selective processes that could be useful in other studies and for the genetic improvement of this breed.
Methods
Sample collection, gait patterns, and DNA extraction
Blood sample were collected from competing horses during the 36th Brazilian National Exhibition of the Mangalarga Marchador breed, and also from horses raised in stud-farms located in the States of São Paulo and Minas Gerais. The dataset was composed of males (n = 62) and females (n = 130) that were initially chosen considering well-defined phenotypes for gait: picada (n = 86) and batida (n = 106). Also, animals from unrelated lineages were prioritized, avoiding the inclusion of full-sibs. Jugular blood samples (5 mL) were collected from each animal and mixed with 7.5 mg EDTA. We extracted genomic DNA from each sample using an Illustra Blood Genomic PrepMini Spin Kit (GE Healthcare, USA), according to the manufacturer’s instructions. The DNA was quantified using a Qubit® 3.0 Fluorometer (Invitrogen, USA), and quality assessment of DNA was achieved using the NanoDrop™ Lite Spectrophotometer (NanoDrop Lite, Thermo Scientific, USA), and 0.8% agarose gel electrophoresis. The final dilutions per sample were ~ 10 ng/μL.
Genotype, quality control, filter and phase genotypes
All horses were genotyped with the 670 k Axiom® Equine Genotyping Array (Axiom MNEC670). Quality control (QC) evaluations were performed using the Axiom™ Analysis Suite Software, version 4, with the default parameters for diploid organisms. QC was performed at sample level considering the following criteria: Dish QC (DQC) ≥ 0.82, call rate ≥ 97, percent of passing samples ≥95, average call rate for passing samples ≥98.5; and at SNP level using the threshold for call rate ≥ 97, with twenty-six other parameters that can be consulted in more detail (Additional file 4: Methods S1). The coordinates of the genotyping data were remapped to reference assembly of the equine genome EquCab3.0 [51], excluding non-autosomal chromosomes. The raw reports with the EquCab3.0 SNP coordinates for the MNEc670k array, used in our analysis, are available at https://www.animalgenome.org/repository/pub/UMN2018.1003/. Coordinates between the two assemblies was accessed using NCBI (https://www.ncbi.nlm.nih.gov/genome/tools/remap). The final genotyping file contained information from 545,219 SNPs, located within the 32 chromosomes, including chromosome X (Additional file 1: Fig. S2).
Additional QC analyses at SNP level were performed in VCFtools and R software in accordance with each method, being imposed a QC for Hardy-Weinberg <1e-8 for the three signature of selection statistics, minor allele frequency (MAF) < 0.01 for TD and iHS, MAF < 0.005 for ROH. SNPs were excluded based on these thresholds. SNP in the same position were removed. Thereby, two datasets were available due to the different QC applied for MAF; with a total of 422,656 SNP available in the dataset for TD and iHS analyses (MAF < 0.01), and 444,929 SNP available in the dataset used in ROH analysis (MAF < 0.005). We adopted an extreme lower MAF parameter for ROH to follow the recommendations of previous studies, which described possible underestimation problems when MAF is used [52]. Genotype phasing was performed in Beagle v.5.0, which provides faster and accurate algorithms [53], and the phased data was used in the TD and iHS analyses.
Population structure and linkage disequilibrium analyses
The principal component analysis (PCA) was performed in Plink 1.9 [21] using linkage disequilibrium. A pruning parameter was applied to remove correlations between SNP and keep approximately independent SNPs; the parameter --indep-pairwise was used. The relatedness between individuals was used for the computation of genome-wide IBD estimates. Before computing PCAs in the R software, close related individuals were excluded based on the high-values for pairwise PI_HAT statistic sum.
The linkage disequilibrium (LD) level was calculated for the entire panel using the phased data. To conduct the LD decay analysis, the PopLDdecay pipeline was used with default pruning [54]. The density was reduced to 347,935 SNPs after the LD pruning. Graphs and complementary analyses for the plot were conducted using the R packages pegas [55], ape [56], and ggplot2 [57].
Genome-wide scan for signals of positive selection
We used three distinct approaches to capture the evolutionary aspects of the selection in the MM. Each approach has some strengths and disadvantages, and the combination and reproducibility of the results add greater accuracy to the analyses.
Tajima’s D (TD)
Sliding windows of 20 kb across all autosomal regions were used in the TD analysis. The analysis was performed in the VCFtools (http://vcftools.sourceforge.net/), using the command option “--TajimaD”. Windows containing missing variants were ignored. Windows were sorted in ascending order of the TD values, using empirical p-values [58] of less than 0.01, before constructing the graphs.
Integrated haplotype score (iHS)
The R package rehh v.3.01 [22, 59] was used in the iHS analysis. Due to the absence of representative studies in horses and most non-model species for the designation of alleles as ‘ancestral’ or ‘derived’, iHS analysis was conducted using unpolarized alleles, which is a new feature of the latest version of the rehh package. This version allows the function to be defined as “FALSE”, which is ideal for the study of domestic animals as well as non-model organisms. The iHH (integrated EHH) values were computed for the major (most frequent) and minor (second-most frequent) alleles. Values of iHS ≥ 2 or ≤ 2 are already considered as significant signals of selection using the default settings [59] because they reflect on a p-value < 0.01. However, an ideal value for iHS or piHS (p-value for iHS) is not well defined in the literature. We used iHS values ≥3.5 or ≤ 3.5, for which piHS ≥3 was considered statistically significant, rejecting the null hypothesis at a level of significance equal to 0.1% (p-value< 0.001). The piHS values are products of iHS transformation to assign a p-value, being piHS = [−log10[1–2|ΦiHS-0.5|], wherein Φ iHS is the Gaussian cumulative distribution function of iHS.
Runs of homozygosity (ROH)
The analysis was conducted with Plink 2.0, using the following parameters “--homozyg --density 50 --gap 1000 --kb 250 --snp 50, --window-het 2, --window-missing 2, --window-snp 50, --window-threshold 0.05)” [60]. Binary runs of homozygosity were generated with the R script developed by Boison (https://github.com/soloboan/ROHs). SNPs with an ROH proportion lower than 0.01 were discarded. The signatures of selection for ROH, i.e., ROH islands, were defined as ROH regions (mean hotspot) with frequencies ≥0.5 in the population.
Gene annotation and enrichment analysis
Gene annotation was carried out with the genomic regions identified as signatures of selection, considering the three methods separately. Window sizes were set at 125 kb upstream and downstream of each significant region/SNP. The window size was defined based on LD information and approximate values described in the literature. Genes within these windows were identified based on the most recent assembly of the equine genome sequence (EquCab3.0) using the BioMart R package [61]. Enrichment analysis was carried out on the PANTHER Classification System (www.pantherdb.org) to provide an accurate inference of biological processes, molecular functions, and cellular component analysis of the candidate genes. The enrichment analysis results were plotted using the ggplot2 R package [57] for better visualization. The p-values were adjusted to Benjamini–Hochberg (BH) (P < 0.05), which implements methods to analyze and visualize the functional profiles of genes and gene clusters [62]. We also used network analysis as a complementary approach to study the genes and how they are possibly functionally related (further details were provided in the Additional file 1: Gene network analysis).
Availability of data and materials
The data generated during this study are included in this published article and its supplementary information files. The data that support the findings of this study are available on request from the email address: rogerio.curi@unesp.br. The upload of this information was not possible due to privacy or ethical restrictions.
References
Andrade LS. A herança genética da marcha: matéria técnica; 2011.
ABCCMM. Associação Brasileira de Criadores de Cavalos da Raça Mangalarga. 2018.
USMMA. U.S. Mangalarga Marchador Association. 2019.
Andersson LS, Larhammar M, Memic F, Wootz H, Schwochow D, Rubin CJ, et al. Mutations in DMRT3 affect locomotion in horses and spinal circuit function in mice. Nature. 2012;488(7413):642–6. https://doi.org/10.1038/nature11399.
Promerová M, Andersson LS, Juras R, Penedo MCT, Reissmann M, Tozaki T, et al. Worldwide frequency distribution of the “gait keeper” mutation in the DMRT3 gene. Anim Genet. 2014;45(2):274–82. https://doi.org/10.1111/age.12120.
Lewontin RC, Krakauer J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics. 1973;74(1):175–95. https://doi.org/10.1093/genetics/74.1.175.
Nielsen R, Hellmann I, Hubisz M, Bustamante C, Clark AG. Recent and ongoing selection in the human genome. Nat Rev Genet. 2007;8(11):857–68. https://doi.org/10.1038/nrg2187.
Bertolini F, Servin B, Talenti A, Rochat E, Kim ES, Oget C, et al. Signatures of selection and environmental adaptation across the goat genome post-domestication. Genetics Proc Natl Acad Sci USA. 2008;105:17659–64.
Bamshad M, Wooding SP. Signatures of natural selection in the human genome. Nat Rev Genet. 2003;4(2):99–111. https://doi.org/10.1038/nrg999.
Avila F, Mickelson JR, Schaefer RJ, McCue ME. Genome-wide signatures of selection reveal genes associated with performance in American Quarter Horse subpopulations. Front Genet. 2018;9:1–13.
Srikanth K, Kim NY, Park WC, Kim JM, Kim KD, Lee KT, et al. Comprehensive genome and transcriptome analyses reveal genetic relationship, selection signature, and transcriptome landscape of small-sized Korean native Jeju horse. Sci Rep. 2019;9:1–16.
Qanbari S, Simianer H. Mapping signatures of positive selection in the genome of livestock. Livest Sci. 2014;166:133–43. https://doi.org/10.1016/j.livsci.2014.05.003.
de Simoni Gouveia JJ, Paiva SR, McManus CM, Caetano AR, Kijas JW, Facó O, et al. Genome-wide search for signatures of selection in three major Brazilian locally adapted sheep breeds. Livest Sci. 2017;197:36–45. https://doi.org/10.1016/j.livsci.2017.01.006.
Nielsen R. Molecular signatures of natural selection. Annu Rev Genet. 2005;39(1):197–218. https://doi.org/10.1146/annurev.genet.39.073003.112420.
Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419(6909):832–7. https://doi.org/10.1038/nature01140.
Purfield DC, McParland S, Wall E, Berry DP. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS One. 2017;12(5):1–23. https://doi.org/10.1371/journal.pone.0176780.
Fariello MI, Boitard S, Naya H, SanCristobal M, Servin B. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics. 2013;193(3):929–41. https://doi.org/10.1534/genetics.112.147231.
Pérez O’Brien AM, Utsunomiya YT, Mészáros G, Bickhart DM, Liu GE, Van Tassell CP, et al. Assessing signatures of selection through variation in linkage disequilibrium between taurine and indicine cattle. Genet Sel Evol. 2014;46(1):1–14. https://doi.org/10.1186/1297-9686-46-19.
Carlson CS, Thomas DJ, Eberle MA, Swanson JE, Livingston RJ, Rieder MJ, et al. Genomic regions exhibiting positive selection identified from dense genotype data. Genome Res. 2005;15(11):1553–65. https://doi.org/10.1101/gr.4326505.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–95. https://doi.org/10.1093/genetics/123.3.585.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. https://doi.org/10.1086/519795.
Gautier M, Vitalis R. Rehh an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics. 2012;28(8):1176–7. https://doi.org/10.1093/bioinformatics/bts115.
Weigand H, Leese F. Detecting signatures of positive selection in non-model species using genomic data. Zool J Linnean Soc. 2018;184(2):528–83. https://doi.org/10.1093/zoolinnean/zly007.
dos Santos BA, Pereira GL, Bussiman F, Bussiman FD, Paschoal VR, de Souza Júnior SM, et al. Genomic analysis of the population structure in horses of the Brazilian Mangalarga Marchador breed. Livest Sci. 2019;229:49–55.
dos Santos WB, Schettini GP, Fonseca MG, Pereira GL, Chardulo LAL, et al. Fine-scale estimation of inbreeding rates, runs of homozygosity and genome-wide heterozygosity levels in the Mangalarga Marchador horse breed. J Anim Breed Genet. 2020;00(2):1–13. https://doi.org/10.1111/jbg.12508.
Gomez-Raya L, Rodríguez C, Barragán C, Silió L. Genomic inbreeding coefficients based on the distribution of the length of runs of homozygosity in a closed line of Iberian pigs. Genet Sel Evol. 2015;47(1):81. https://doi.org/10.1186/s12711-015-0153-1.
Robilliard JJ, Pfau T, Wilson AM. Gait characterization and classification in horses. J Exper Biol. 2007;210(2):187–97. https://doi.org/10.1242/jeb.02611.
Fonseca MG, Ferraz GC, Lage J, Pereira GL, et al. A genome-wide association study reveals differences in the genetic mechanism of control of the two gait patterns of the Brazilian Mangalarga Marchador breed. J Equine Vet Sci. 2017;53:64–7. https://doi.org/10.1016/j.jevs.2016.01.015.
Bussiman FO, dos Santos BA, Abreu Silva BC, Perez BC, Pereira GL, Chardulo LAL, et al. Allelic and genotypic frequencies of the DMRT3 gene in the Brazilian horse breed Mangalarga Marchador and their association with types of gait. Genet Mol Res. 2019;18:gmr18217.
Jäderkvist Fegraeus K, Hirschberg I, Árnason T, Andersson L, Velie BD, Andersson LS, et al. To pace or not to pace: a pilot study of four- and five-gaited Icelandic horses homozygous for the DMRT3 ‘gait keeper’ mutation. Anim Genet. 2017;48(6):694–7. https://doi.org/10.1111/age.12610.
Arnason T, Van Vleck LD. Genetic improvement of the Horse. In: Bowling AT, Ruvinsky A, editors. The Genetics of the Horse. Wallingford, Oxon: CABI Pub; 2000. p. 341.
Cadzow M, Boocock J, Nguyen HT, Wilcox P, Merriman TR, Black MA. A bioinformatics workflow for detecting signatures of selection in genomic data. Front Genet. 2014;5:1–8. https://doi.org/10.3389/fgene.2014.00293.
Ma Y, Ding X, Qanbari S, Weigend S, Zhang Q, Simianer H. Properties of different selection signature statistics and a new strategy for combining them. Heredity (Edinb). 2015;115(5):426–36. https://doi.org/10.1038/hdy.2015.42.
Ablondi M, Viklund Å, Lindgren G, Eriksson S, Mikko S. Signatures of selection in the genome of Swedish warmblood horses selected for sport performance. BMC Genomics. 2019;20(1):1–12. https://doi.org/10.1186/s12864-019-6079-1.
Boitard S, Schlötterer C, Nolte V, Pandey RV, Futschik A. Detecting selective sweeps from pooled next-generation sequencing samples. Mol Biol Evol. 2012;29(9):2177–86. https://doi.org/10.1093/molbev/mss090.
Stapley J, Feulner PGD, Johnston SE, Santure AW, Smadja CM. Variation in recombination frequency and distribution across eukaryotes: patterns and processes. Philos Trans R Soc B Biol Sci. 2017;372(1736):20160455. https://doi.org/10.1098/rstb.2016.0455.
Szmatoła T, Gurgul A, Jasielczuk I, Fu W, Ropka-Molik K. A detailed characteristics of bias associated with long runs of homozygosity identification based on medium density SNP microarrays. J Genomics. 2020;8:43–8. https://doi.org/10.7150/jgen.39147.
Ceballos FC, Joshi PK, Clark DW, Ramsay M, Wilson JF. Runs of homozygosity: windows into population history and trait architecture. Nat Rev Genet. 2018;19(4):220–34. https://doi.org/10.1038/nrg.2017.109.
Melo D, Marroig G. Directional selection can drive the evolution of modularity in complex traits. PNS. 2015;112(2):470–5. https://doi.org/10.1073/pnas.1322632112.
Park J, Lee J, Lee DH. Identification of protein phosphatase 4 inhibitory protein that plays an indispensable role in DNA damage response. Mol Cells. 2019;42(7):546–56. https://doi.org/10.14348/molcells.2019.0014.
Golinski ML, Vandhuick T, Derambure C, Fréret M, Lecuyer M, Guillou C, et al. Dysregulation of RasGRP1 in rheumatoid arthritis and modulation of RasGRP3 as a biomarker of TNFaα inhibitors. Arthritis Res Ther. 2015;17(1):1–12. https://doi.org/10.1186/s13075-015-0894-9.
Wu X, Guldbrandtsen B, Lund MS, Sahana G. Association analysis for feet and legs disorders with whole-genome sequence variants in 3 dairy cattle breeds. J Dairy Sci. 2016;99(9):7221–31. https://doi.org/10.3168/jds.2015-10705.
Kurz JP, Yang Z, Weiss RB, Wilson DJ, Rood KA, Liu GE, et al. A genome-wide association study for mastitis resistance in phenotypically well-characterized Holstein dairy cattle using a selective genotyping approach. Immunogenetics. 2019;71(1):35–47. https://doi.org/10.1007/s00251-018-1088-9.
Holden LA, Arumilli M, Hytönen MK, Hundi S, Salojärvi J, Brown KH, et al. Assembly and analysis of unmapped genome sequence reads reveal novel sequence and variation in dogs. Sci Rep. 2018;8:1–11.
Akam M. Hox genes and the evolution of diverse body plans. Philosoph Transact Biol Sci. 1995;349(1329):313–9. https://doi.org/10.1098/rstb.1995.0119.
Pineault KM, Wellik DM. Hox genes and limb musculoskeletal development. Curr Osteoporos Rep. 2014;12(4):420–7. https://doi.org/10.1007/s11914-014-0241-0.
Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/ posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100(4):423–34. https://doi.org/10.1016/S0092-8674(00)80678-9.
Grilz-Seger G, Neuditschko M, Ricard A, Velie B, Lindgren G, Mesarič M, et al. Genome-wide homozygosity patterns and evidence for selection in a set of European and near eastern horse breeds. Genes (Basel). 2019;10(7):491. https://doi.org/10.3390/genes10070491.
Aronson D, Boppart MD, Dufresne SD, Fielding RA, Goodyear LJ. Exercise stimulates c-Jun NH2 kinase activity and c-Jun transcriptional activity in human skeletal muscle. Biochem Biophys Res Commun. 1998;251(1):106–10. https://doi.org/10.1006/bbrc.1998.9435.
Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: from polygenic to omnigenic. Cell. 2017;169(7):1177–86. https://doi.org/10.1016/j.cell.2017.05.038.
Beeson SK, Schaefer RJ, Mason VC, McCue ME. Robust remapping of equine SNP array coordinates to EquCab3. Anim Genet. 2019;50(1):114–5. https://doi.org/10.1111/age.12745.
Ferenčaković M, Sölkner J, Curik I. Estimating autozygosity from high-throughput information: effects of SNP density and genotyping errors. Genet Sel Evol. 2013;45(1):1–9. https://doi.org/10.1186/1297-9686-45-42.
Pook T, Mayer M, Geibel J, Weigend S, Cavero D, Schoen CC, et al. Improving imputation quality in beagle for crop and livestock data. G3 genes, genomes. Genet. 2020;10(1):177–88. https://doi.org/10.1534/g3.119.400798.
Zhang C, Dong SS, Xu JY, He WM, Yang TL. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics. 2019;35(10):1786–8. https://doi.org/10.1093/bioinformatics/bty875.
Paradis E. Pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics. 2010;26(3):419–20. https://doi.org/10.1093/bioinformatics/btp696.
Paradis E, Schliep K. Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35(3):526–8. https://doi.org/10.1093/bioinformatics/bty633.
Wickham H. ggplot2: Elegant Graphics for Data Analysis; 2016.
Yu F, Keinan A, Chen H, Ferland RJ, Hill RS, Mignault AA, et al. Detecting natural selection by empirical comparison to random regions of the genome. Hum Mol Genet. 2009;18(24):4853–67. https://doi.org/10.1093/hmg/ddp457.
Gautier M, Klassmann A, Vitalis R. Rehh 2:0. A reimplementation of the R package rehh to detect positive selection from haplotype structure. Mol Ecol Res. 2017;17(1):78–90. https://doi.org/10.1111/1755-0998.12634.
Ceballos FC, Hazelhurst S, Ramsay M. Assessing runs of homozygosity: a comparison of SNP Array and whole genome sequence low coverage data. BMC Genomics. 2018;19(1):106. https://doi.org/10.1186/s12864-018-4489-0.
Smedley D, Haider S, Ballester B, Holland R, London D, Thorisson G, et al. BioMart - Biological queries made easy. BMC Genomics. 2009;10(1):1–12. https://doi.org/10.1186/1471-2164-10-22.
Hu JX, Zhao H, Zhou HH. False discovery rate control with groups. J Am Stat Assoc. 2010;105(491):1215–27. https://doi.org/10.1198/jasa.2010.tm09329.
Acknowledgements
We thank Luis Artur L. Chardulo and Jéssica M. Malheiros for the ideas and suggestions during the execution of this work.
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo – Fapesp (Grant 2016/19081–9). Wellington Bizarria dos Santos was the recipient of a Master’s fellowship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Capes (Finance code 001). Also financial supports from Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (141564/2020–20).
Author information
Authors and Affiliations
Contributions
RAC conceived and managed the project. FOB, GLP, WAB, ORMN and RAC collected and extracted the DNA samples. All authors contributed to design the study. WBS, GPS, AMM, JCCB, GCF and HNO conducted the data analysis, graphs and supplementary materials. WBS wrote the main manuscript text. WBS, GPS, AMM and RAC drafted the manuscript. All authors read, edited and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental procedures involving horses in this study were performed in accordance with the relevant guidelines of animal welfare. The project was approved by the Ethics Committee on Animal Use of the College of Veterinary and Animal Science (FMVZ), Unesp, Botucatu/SP (Approval No. 0029/2017).
Consent for publication
Not applicable.
Competing interests
The author(s) certify that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1
Genome-wide linkage disequilibrium (LD) decay plot for 192 Mangalarga Marchador based on 347,935 SNP markers. Fig. S2 Final density of 545,219 SNP in the Mangalarga Marchador horse genome after Axiom™ Analysis Suite pruning. Fig. S3 Interaction networks of candidate genes identified from signatures of selection. Different colored arrows indicate the types of evidence used in predicting the associations.
Additional file 2:
Data S1 TD output. Data S2 iHS output. Data S3 ROH output.
Additional file 3:
Table S1 All genomic annotations for iHS. Table S2 Gene enrichment results for TD, iHS, and ROH.
Additional file 4:
AxiomTM Analysis Suite final report.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Santos, W.B., Schettini, G.P., Maiorano, A.M. et al. Genome-wide scans for signatures of selection in Mangalarga Marchador horses using high-throughput SNP genotyping. BMC Genomics 22, 737 (2021). https://doi.org/10.1186/s12864-021-08053-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-08053-8