
Abstract
Background
In recent years, animal welfare and health has become more and more important in pig breeding. So far, numerous parameters have been considered as important biomarkers, especially in the immune reaction and inflammation. Previous studies have shown moderate to high heritabilities in most of these traits. However, the genetic background of health and robustness of pigs needs to be extensively clarified. The objective of this study was to identify genomic regions with a biological relevance for the immunocompetence of piglets. Genome-wide Association Studies (GWAS) in 535 Landrace (LR) and 461 Large White (LW) piglets were performed, investigating 20 immune relevant traits. Besides the health indicators of the complete and differential blood count, eight different cytokines and haptoglobin were recorded in all piglets and their biological dams to capture mediating processes and acute phase reactions. Additionally, all animals were genotyped using the Illumina PorcineSNP60v2 BeadChip.
Results
In summary, GWAS detected 25 genome-wide and 452 chromosome-wide significant SNPs associated with 17 immune relevant traits in the two maternal pig lines LR and LW. Only small differences were observed considering the maternal immune records as covariate within the statistical model. Furthermore, the study identified across- and within-breed differences as well as relevant candidate genes. In LR more significant associations and related candidate genes were detected, compared with LW. The results detected in LR and LW are partly in accordance with previously identified quantitative trait loci (QTL) regions. In addition, promising novel genomic regions were identified which might be of interest for further detailed analysis. Especially putative pleiotropic regions on SSC5, SSC12, SSC15, SSC16 and SSC17 are of major interest with regard to the interacting structure of the immune system. The comparison with already identified QTL gives indications on interactions with traits affecting piglet survival and also production traits.
Conclusion
In conclusion, results suggest a polygenic and breed-specific background of immune relevant traits. The current study provides knowledge about regions with biological relevance for health and immune traits. Identified markers and putative pleiotropic regions provide first indications in the context of balancing a breeding-based modification of the porcine immune system.
Similar content being viewed by others

Background
Health and immune traits are characterising the immunocompetence of piglets [1–3]. Through the last years, the societal importance of health and animal welfare has increased [4]. The enhancement of immunocompetence is a potential determinant to prevent performance reduction through health impairment [5, 6]. In literature, several approaches to influence and improve the porcine immune response and immune system have been discussed (e.g., [2, 7, 8]). In this context, haematological parameters and cytokines might be considered as important markers of immune reaction [9, 10]. As the immune system is known as a highly interactive system [11], indicators triggering a health-promoting immunocompetence need to be investigated regarding negative impacts and interactions on economical and societal phenotypes [3, 5].
Piglets are extremely susceptible to infections in their first stage of life [12]. For this reason, innate immune defense mechanisms and colostrum intake are essential to newborn’s immunity [13]. Consequently, it is suggested that the immunocompetence of the biological dam is linked to the immune status of the piglet due to several components of colostrum [13–15].
Genetic background
Previous studies have estimated heritabilities under various conditions regarding e.g., breed, age (including developmental stage), number of animals and the experimental design (e.g., [1, 3, 5, 10, 16–20]). These studies have shown mostly moderate to high heritabilities for several components of the immune system. Overall, the innate and adaptive immune system indicate genetic foundation and slightly higher individual genetic variances in traits of the adaptive immune system [3, 21]. Especially immune cells, red blood cell (RBC) characteristics and cytokines suggest a large genetic component [3, 19]. Until now, a small number of studies investigated genetic parameters for cytokines in pigs (e.g., [3, 18]).
Heritability estimation in a Landrace and Large White population under non-challenging conditions has confirmed findings to a large extent [22, 23]. RBC and their characteristics showed heritabilities between 0.3 and 0.8 in Landrace and between 0.5 and 0.6 in Large White. In cytokines, heritability estimates were in a broad, breed-specific range from almost 0 to high values of 0.7 (Tumor necrosis factor (TNF)- α) in LR and 0.4 (Interleukin (IL)-8) in LW. Therefore, genetic foundation of immune relevant traits and responsiveness for selective breeding is assumed.
Although the genetic background of the porcine immune system still remains unclear, there are some studies trying to identify immune relevant quantitative trait loci (QTL). So far, the number of QTL associated with health and immune response traits in pigs increased from a limited number in 2007 [24] to a moderate number in 2020. According to the latest release of the AnimalQTLdb (Release 43, December 2020, https://www.animalgenome.org/cgi-bin/QTLdb/SS/summary) [25], 2,776 QTL representing associations with blood parameters, 3,231 QTL with immune capacity, and 609 QTL with disease susceptibility were published.
Underlying studies focused on classical haematological parameters, characterised by measurements of the blood count. First QTL were detected using microsatellites (e.g., [26, 27]) or concentrated on particular genes (e.g., [28–30]). Other studies to detect immune relevant QTL vary widely in treatment, methods of phenotype determination and breed (e.g., [10, 31–37]). In addition, different ages and developmental stages are covered, limiting the comparability of the studies. Only a limited number of studies identified QTL associated with the acute phase protein haptoglobin [38] or cytokines. Studies investigating the cytokines Interferon (IFN)- γ and IL-10 used vaccination with Mycoplasma hyopneumoniae, tetanus toxoid, the porcine reproductive and respiratory syndrome virus (PRRSV) [39, 40] and the classical swine fever virus [41, 42]. Another study analysing cytokines, including IL-6 and IL-12, was restricted to exonic regions of a single gene [43].
Aim of the study
The aim of this study was to identify biological relevant markers associated with health and immune traits in piglets under non-challenge conditions in a Landrace (LR) and a Large White (LW) population. Besides, the genetic background of the piglets’ immune system and the role of the maternal immunocompetence is discussed.
Results
In total, 535 piglets of LR and 461 piglets of LW were phenotyped for the complete and differential blood count (15 traits), eight cytokines and haptoglobin in an experiment conducted under mostly practical, but high hygienic conditions and without challenging the animals. The number of samples per trait, least-squares means and standard errors are listed in Table 1. Slight differences in the number of records resulted from laboratory techniques. Differences between the breeds were tested using a generalized linear mixed model corrected for the environmental effects of breed, age and weight at sample collection and their interaction, a combined herd, year, season, sex effect and litter number (model 0). Due to the analytical method, batch was an additional fixed effect and biological dam an additional random effect in the analyses of cytokines. In general, there were significant differences in blood measurements between the breeds, especially in red blood cell characteristics and cytokines. Further differences were identified in the percentages of neutrophils and lymphocytes. The ratio of these closely antagonistic linked traits was above 1 (1.06) in LR but noticeable below 1 (0.76) in the LW population.
Genome-wide association studies
Different genetic backgrounds led to breed-specific investigations of LR and LW (see Fig. 1). For identifying associations between SNP genotypes and phenotypic information, model (0) without the fixed breed effect was extended by a fixed SNP effect and further on denoted as model (1). Genome-wide association studies (GWAS) based on model (1) identified a total of 25 genome-wide and 452 chromosome-wide significant SNPs (p ≤0.05, corrected for q-value) associated with 17 traits characterising the immunocompetence in unchallenged pigs (Table 2). As expected from more conservative correction methods to avoid false positive results, applying the Bonferroni threshold of 0.05 confirmed 12.8% (LR) and 10.7% (LW) of those markers. With the Bonferroni threshold for adjusted p-values of 0.1, the amount of validated SNPs was increased up to 17.9% (LR) and 13.2% (LW).

Genetic distances between the analysed animals from Landrace (green circle) and Large White (red triangle) displayed as the first two principal components (PC)
Most of the associations are variants within intergenic (LR: 119, LW: 94) or intronic (LR: 142, LW: 89) regions. A lower number of significant SNPs is located within untranslated (3-prime-UTR variant, 5-prime-UTR variant) or upstream and downstream regions of a gene. In addition, GWAS on immune relevant traits identified a few synonymous variants and a variant within a splice site (IL-12, LW) or a non-coding exon sequence (WBC, LR) as well as a missense variant (haemoglobin, LW). Each significant SNP explained between 1.7-5.2% and 2.2-5.5% of the phenotypic variance in LR and LW, respectively.
Tables 3 and 4 provide an overview of the Top 5 SNPs for each immune relevant trait analysed, explaining the largest proportion of the phenotypic variance. A complete list of significant SNPs is given in Additional files 1 and 2.
Due to the unclear influence of the dam on the immunocompetence of the piglets, model (1) was extended by the corresponding trait measured in the biological dam, included as a fixed covariate (model 1*). The effect was significant in twelve (LR) and ten (LW) traits out of 20. GWAS using model (1*) confirmed the majority (LR: 206, LW: 127) of the significant genomic regions from basic analysis using model (1). In some traits without any significant SNP (TNF- α in LR, lymphocytes and IL-4 in LW), model (1*) revealed single markers which were among the leading SNPs identified by model (1), but did not exceed the chromosome-wide threshold. Consequently, the benefit of model (1*) is limited. Conspicuous deviations from results based on model (1) were identified in IL-6 in LW. Markers associated with IL-6 diverged in the chromosomal region.
Due to the overall minor role of the additional covariate, only results of model (1) are discussed further.
Regarding the genomic background within the breeds, remarkably more significances were found within LR (n SNPs = 296) compared to LW (n SNPs = 197). Significant genomic regions of different traits did not overlap in a window of 70 kb. Window definition followed the twofold mean distance of markers identified by Ramos et al. [44].
With exception of lymphocytes, mean corpuscular volume (MCV) and mean corpuscular haemoglobin (MCH), significant SNPs were identified for all traits analysed. In most cases, SNPs were only found within the LR or LW. For five traits (monocytes, haptoglobin, haematocrit, mean corpuscular haemoglobin concentration (MCHC), IL-10), detected markers were relevant in both breeds, but not in overlapping chromosomal regions. Moreover, WBC, neutrophils, IL-1 β, IL-4, and IL-8 were found to be only associated within LR. In contrast, haematopoietic traits, such as platelets, RBC, and haemoglobin as well as the cytokines IFN- γ, IL-12, IL-6 and TNF- α only showed associated SNPs within LW.
Identified SNPs were located across all autosomes with the exception of SSC8. But there were chromosome-specific clusters for certain trait complexes. For example, SSC12 showed associations with cell parameters such as WBC, neutrophils, monocytes, and RBC, whereas on SSC16 only associations with traits characterising RBC in LW were observed.
The largest number of significant markers was found for cytokines (Table 2) with an important region located on SSC17 in LR. In a narrow region between 37 and 39 to 40 Mb on SSC17, significantly associated SNPs for IL-1 β, IL-10 and IL-4 in LR were identified. These SNPs explained up to 3.7% of the phenotypic variance and associations with IL-10 were additionally confirmed with the more conservative Bonferroni correction at 5%.
In addition, 23 SNPs for IL-8 in LR and two SNPs for TNF- α in LW exceeded the genome-wide significance level according to q-value and partly the Bonferroni correction and explained up to 5.1% of the phenotypic variance.
Blood cell characteristics accounted for the second largest proportion of chromosome-wide significant SNPs in LR. Monocyte percentage, as the only subset of WBC showing significant SNPs in LR and LW, revealed SNPs on SSC12 and SSC18, respectively.
Putative pleiotropic SNPs were detected on SSC15 (ALGA0085557, 56.0 Mb) and SSC16 (ASGA0074790, 78.0 Mb) in LW as well as a genomic region on SSC17 in LR, affecting several cytokines.
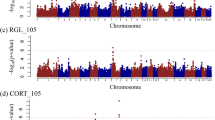
As has been shown, there were no overlapping significant SNPs for traits within LR and LW breed. However, SSC12 seems to be important for both breeds (see Fig. 2). Within a region of around 3.9 Mb, 16 SNPs were found significantly associated with nonidentical immune relevant traits in LR (neutrophils, monocytes) and LW (RBC). A similar observation was made on SSC5 (see Fig. 3). Within an interval of approximately the same size, a significant genomic region was found for MCHC in LR and haemoglobin in LW. In addition, several of the putative pleiotropic SNPs indicated significance under the more conservative Bonferroni correction.

Manhattan plots focusing on a putative pleiotropic region on SSC12 showing results in a neutrophils in Landrace, b monocytes in Landrace and c red blood cells in Large White

Manhattan plots focusing on a putative pleiotropic region on SSC5 showing results in a MCHC in Landrace and b haemoglobin in Large White
Discussion
Animal welfare and health are of social and ethical concerns in pig production and science [4]. In recent years, heritability of immune relevant traits has been confirmed (e.g., [1, 3, 5, 10, 16–20, 22, 23]). In this regard, the interacting and networking structure of immune traits, and basically the genetic background of health and robustness of animals needs to be refined (e.g., [6, 20, 36]). Therefore, this study identified biological relevant SNPs associated with traits characterising the immunocompetence of piglets.
Blood counts recorded in this study reflect the situation under high hygienic conditions in nucleus herds. Results indicate first insights into the genetic background of a LR and LW population in a non-challenging experimental design. Hence, these results may hardly be interpreted as reference values for pigs in commercial fattening systems, but they can be used as indicators for the genetic variation of the immune system.
In order to evaluate and characterise the immune status, reference values are needed. Such thresholds were determined by, for example, Seutter [45]. However, due to accelerated genetic progress accompanied by substantial changes in breeding objectives in the recent years, an update of physiological reference values is indispensable.
At the time point of blood sampling, mean concentrations in the here studied LR and LW populations differed, suggesting a diverging ability for immunocompetence at the critical time point around weaning. LW showed a clear lymphocytic blood count (N:L ratio = 0.76), whereas LR tended towards a granulocytic blood count (N:L ratio = 1.06). This led to the assumption of a more specific and higher responding immunocompetence of LW in the first weeks of age. On the other side, the neutrophil-to-lymphocyte ratio is influenced by non-genetic factors like stress [46] or age at blood sampling. However, taking into account that piglets of both breeds were raised under the same high hygienic and husbandry conditions, substantially deviating stress conditions were unlikely.
The impact of age at blood sampling has been discussed in contrasting ways. For one thing, neutrophils have been described as age-independent at an age of around seven weeks [16], whereas another study showed an effect in piglets up to an age of twelve weeks [45]. In our study, blood samples were collected from LR and LW piglets at an average age of 42 (SD = 3.3) to 44 (SD = 5.8) days. Estimated regression coefficients for age in neutrophils were different in LR (0.018% per day) and LW (0.669% per day). Minor importance of age in LR and higher importance in LW conflict with the conclusions of Seutter [45] and Henryon et al. [16] mentioned above. Contradictions might be due to genetic changes in modern breeds within the last decades.
Genome-wide association studies for immunocompetence
In total, GWAS revealed 477 SNPs associated with 17 immune relevant traits in LR and LW. With the exception of some traits, at least one SNP within most of the genomic regions of interest, identified according to q-value, was also significant after Bonferroni correction. Annotation to the reference genome Sscrofa 11.1 identified 113 genes in LR and 71 genes in LW. Results are partly in accordance with immune-related QTL published in previous studies (e.g., [10, 31, 33–35, 40, 42, 47]). The analyses showed clear breed differences. In LR (n SNPs = 280) more significant associated SNPs were found, compared to LW (n SNPs = 197). Moreover, all of the markers were breed-specific. This might be due to a strong impact of the breed on baseline values of the immune system [48]. Across-breed differences were probably caused by selection processes and the heterogeneity of the genetic structure within the breeds.
Due to the comprehensive results, the discussion focuses on the importance of the networking structure of immune relevant traits and trade-offs between the immune system of piglets and economically important fertility and production factors.
Analyses on WBC in LR identified significant associations on SSC4, SSC12 and SSC14. The genomic region on SSC14 (41.7 - 48.5 Mb) confirms a QTL for WBC in a German LR population [10] and overlaps with QTL for platelet count and plateletcrit [31]. In addition, a SNP for haemoglobin, haematocrit and RBC in a Duroc × Erhulian population has been revealed close to our result on SSC12 [34].
An intron variant (MARC0040388, 34.5 Mb) within the gene lactoperoxidase (LPO) on SSC12 showed chromosome-wide significance for neutrophils in LR. Pathway analysis indicates LPO to affect neutrophils in their phagocytolytic activity [49, 50]. A previous study in German LR has revealed a QTL for mean platelet volume in an enclosing region on SSC12 [10].
Our study revealed significant associations for monocytes in both breeds. In LR, the genomic regions for monocytes and neutrophils were located close together (<1 Mb) at 36 Mb to 37 Mb without any intersection. From the physiological point of view, relationships between neutrophils and monocytes are well known, especially in the innate immune system and the defense against pathogens [51]. Use of whole genome sequencing data might clarify the underlying genetic structure. In LW, results on SSC18 were found within QTL associated with mean platelet volume [10] and CD4+CD8- T cells and the ratio of CD4+ to CD8+ T cells [47].
Especially cytokines, as intercellular signalling molecules, are pleiotropic and interconnected in the mechanisms of the immune system [8, 40]. Nevertheless, literature on the genetic background in pigs is limited and underlying mechanisms of interaction in cytokines have not been investigated thoroughly. Therefore, this study reveals mostly novel insights into the genetic background affecting cytokine profiles in pigs under non-challenge conditions. The current study detected 23 genome-wide significant SNPs on SSC15 associated with IL-8 in LR. One of them is an intron variant (ASGA0070620, 120.4 Mb) within the gene transmembrane BAX inhibitor motif containing 1 (TMBIM1). TMBIM1 is involved in the neutrophil degranulation and thereby, for instance, in regulating the exocytosis of inflammatory mediators [49, 50, 52, 53]. In humans, IL-8 is well known to affect neutrophils, for instance through priming or adhesion [52].
GWAS on IL-12 concentration in LW piglets provided a moderate number of immune relevant candidate genes, inter alia RNA binding motif single stranded interacting protein 3 (RBMS3). Significant markers within RBMS3 were located within a QTL previously described to be associated with IL-10 level [42]. Significant markers for IL-12 in LW seem to be important in immune system processes as has been already shown by previous studies, reporting QTL associated with MCH and MCV in LR [10] and CD4+CD8+ percentage [47].
Furthermore, association analysis on TNF- α in LW detected significant SNPs on SSC1 and SSC9. An intron variant on SSC1 (ASGA0001781, 26.7 Mb) and an intergenic variant on SSC9 (ASGA0097568, 138.5 Mb) exceeded the genome-wide level.
The genetic component of the biological dam in the development of piglets’ immune system still remains unclear. The results indicate consequences on IL-6 in LW piglets depending on the corresponding cytokine status of the biological dam. Nevertheless, most of the genomic regions from basic analysis with model (1) were confirmed after the environmental correction. However, putative pleiotropic mechanisms in cytokines were supported in all statistical models.
Knowledge about pleiotropic mechanisms might help to clarify genetic relationships between the traits [54]. Indications on these putative pleiotropic regions were also observed within the here studied breeds LR and LW. In total, we identified two across-breed regions and three additional regions with putative pleiotropy in LR or LW including all trait complexes investigated in this study.
In LW, association studies identified a chromosome-wide significant SNP (ALGA0085557, 56.0 Mb) on SSC15 in haemoglobin and haematocrit. Located on SSC16, a SNP (ASGA0074790, 78.0 Mb) was found which was significantly associated with haemoglobin, haematocrit as well as RBC. Therefore, in red blood cell characteristics, pleiotropy in LW is assumed. These potentially pleiotropic SNPs underline the highly interacting mechanisms of the immune system. According to the biological context, this relationship is not unexpected. Yan et al. [35] reported consistently high genetic correlations between haemoglobin, haematocrit and RBC at day 18 (rg 0.77-0.85) and day 46 (rg 0.50-0.77).
In LR, a genomic region on SSC17 showed nine identical SNPs significantly associated with at least two of the cytokines IL-1 β, IL-4 and IL-10. One of these SNPs is a downstream gene variant of the gene reactive oxygen species modulator 1 (ROMO1). Among others, ROMO1 is classified in the biological process of a defense to a bacterium [55, 56]. Human monocytes and macrophages have shown an overexpression of the protein Romo1 in case of tumor disease. In this context, an inhibition of the immune response has been assumed [57].
Furthermore, we identified two regions on SSC5 and SSC12 with a size of around 3.6 and 3.9 Mb including three and 16 SNPs, respectively. Markers were associated with up to three different immune traits in LR and LW.
The adjoining SNPs on SSC5 were associated with MCHC in LR and haemoglobin in LW. Fibroblast growth factor 6 (FGF6), as a putative candidate gene within the described genomic region, is involved in signalling processes and thereby in cell growth and survival processes due to the Phosphatidylinositol 3-kinase cascade [49, 50, 58]. In addition, FGF6 was recently annotated to the current reference genome Sscrofa 11.1 [59]. Another of these markers (ASGA0025952, 65.8 Mb) is located within the gene A-kinase anchoring protein 3 (AKAP3). It is a missense variant probably causing an amino acid change. Biological pathways in humans indicate a link to signalling by regulating the mitogen-activated protein kinase (MAPK) pathway [49, 50].
A genomic region on SSC12 was detected around 50.1 to 54 Mb associated with different cell parameters of the immune system and the blood. SNPs revealed a significance for monocytes and neutrophils in LR and RBC in LW. A putative candidate gene is CST telomere replication complex component 1 (CTC1), directly associated with the percentage of neutrophils in LR on SSC12. Findings in humans outline CTC1 is involved in immune system processes and the development of the immune system [55, 56, 60].
Conserved functions might have an effect across species [24], and, thus, it is recommended to consider TMBIM1, RBMS3, ROMO1, FGF6, AKAP3 and CTC1 as putative candidate genes, although they are not yet fully described for pigs in detail.
Genetics of immunocompetence and economic traitsTaking the identified markers into consideration for further analyses, we need to check for undesirable relationships with traits involved in the survivability and diseases susceptibility but also regarding production traits and meat quality.
In this context, the current study identified markers associated with neutrophils in LR (SSC12), IL-8 concentration in LR (SSC15) as well as IL-10 and IL-12 concentration in LW (SSC1 and SSC13). Genomic regions were identified close to QTL for intramuscular fat content [61–63] or within a QTL previously reported for the genetic defect cryptorchidism [64].
Additionally, our findings for IL-8 in LR and MCHC in LW indicate a connection to QTL regions associated with feed conversion ratio and average daily gain in Duroc [65]. The genomic region on SSC14 identified in GWAS for WBC in LR was found close to a QTL previously described for ham weight in a LW population [66].
Results of this study indicate overlaps between regions significantly associated with immune relevant phenotypes and QTL for survivability of piglets and growing pigs. A putative relationship is supported by several studies which were summarized in the review of Heuß et al. [6]. In this regard, important results in our study were markers on SSC13 associated with the concentration of IL-12 in LW and markers associated with neutrophils in LR. These regions have been previously described to be associated with the number of stillborn [67, 68].
Furthermore, significant SNPs on SSC14, associated with WBC in LR and RBC in LW, have been previously described in the context of the reproductive traits litter size (WBC) [67] and mummified piglets (WBC, RBC) [68].
The relationship between the immune system and the survivability of piglets gains in importance. Besides, several putative pleiotropic regions have been discussed in “Genome-wide association studies for immunocompetence” section. In this context, the identified across-breed genomic region on SSC5 was additionally described to be associated with litter size in Duroc [69].
Conclusions
In summary, the study identified 477 SNP markers associated with immune relevant traits. Analyses were performed in piglets of a LR and LW population kept under high hygienic standards and without challenging the immune system. GWAS results pointed out clear differences between the breeds LR and LW. Comparison between previous studies and findings in this study indicate a complex genetic background of immune relevant traits and health indicators. Several interesting genomic regions suggest a pleiotropic background. Further investigations need to follow, considering relationships and antagonistic mechanisms between the immune system, survivability, performance traits and other economically important traits. In general, as health and immune traits are expected to become an essential part of balanced pig breeding, this study provides first insights into regions with special importance for the immune system of piglets.
Methods
Animals, sample collection, phenotyping
A total of 535 piglets (♂190/♀345) of LR and 461 piglets (♂170/♀291) of LW were analysed. Animals were a subset of the nucleus populations which aim to reflect the genetic variability of both populations with respect to their different breeding objectives. All piglets were born between 2015 and 2017 on five farms of the German breeding organization BHZP GmbH (Bundeshybridzuchtprogramm GmbH). All herds were subjected to high hygienic standards. The experimental design aimed to sample a triplet consisting of the mother and two full siblings born alive.
Piglets were routinely blood sampled in the week around weaning (⌀ 42 to 44 days), sows around piglets’ birth. Sampling was performed by collecting blood via the Vena jugularis.
Traits investigated are listed in Table 5. Traits of the complete and differential blood count were determined using the optoelectronic technology ADVIA® 2120. Band cells as well as basophils and eosinophils were excluded from further analyses due to low expression, resulting in a variation close to zero. Haptoglobin concentration was measured using a photometric technology. Cytokines were analysed with Millipore’s Milliplex Map Kit Porcine Cytokine/Chemokine Magnetic Bead Panel on a Luminex® 200, as well as the software xPonent 3.1. Milliplex Map Kit enabled simultaneous analysis of eight cytokines. Cytokine concentrations below the analytical threshold were replaced by one-half of the threshold [70, 71]. Measurements which did not fulfill the quality criteria determined by the laboratory technique were excluded from further analyses. Cytokines and haptoglobin measurements were log-transformed to assume a normal distribution and to take the skewness of distribution into account.
Descriptive statistics including least-squares means and breed differences are listed in Table 1. Model (0) was used to test for differences between the breeds. Correction was performed for the fixed environmental factors of breed (ΔBR), age (ΔA) and weight (ΔW) at sample collection and their interaction, a combined herd, year, season, sex effect (HYSS) and litter number (L). The intercept is represented by μ and regression coefficients are given as b1,b2,b3 and b4 and environmental residual effects as e. In the analyses of cytokines, batch (B) was an additional fixed effect and biological dam (u d) an additional random effect.
Model (0):
Note: Terms in [ ] are only used in the analysis of cytokines.
Genotyping
Blood and tissue samples were analysed using the PorcineSNP60v2 BeadChip (Illumina, San Diego, CA, USA).
Breed-specific quality control on autosomes was applied using the R-package GenABEL [72]. Samples and markers with a call rate <95% and markers with a low minor allele frequency (<1%) or high linkage disequilibrium (r2>0.8) in a region of 3 kb were excluded from further analysis, resulting in 41’872 (LR) and 42’388 (LW) markers in 534 (LR) and 461 (LW) piglets.
Genetic distances were visualised using multidimensional scaling based on a genomic relationship matrix calculated within the R-package GenABEL [72] (Fig. 1). Consequently, the two populations were analysed separately.
Statistical analyses and genome-wide association studies
In a first step, data were analysed with a generalized linear mixed model (model 1). Observations (y) were corrected for the fixed environmental effects age (ΔA) and weight (ΔW) at sample collection and their interaction, a fixed SNP effect, herd, year, season and sex, combined to a HYSS-effect (9 levels in LR, 13 levels in LW), and litter number (L, 1 - >4). The intercept is represented by μ and regression coefficients are given as b1,b2,b3 and b4. The environmental residual effects were denoted by e. In the analyses of cytokines, a fixed batch effect (B, 20 levels in LR, 19 levels in LW) and the random effect “biological dam of the piglet” (u d) were added to the model. Both additional effects have been consistently identified as relevant for cytokines.
Model (1):
Note: Terms in [ ] are only used in the analysis of cytokines.
In a second step, the impact of direct environmental maternal effects on the piglet’s immunocompetence were verified using model (1*). Accordingly, model (1) was extended by the corresponding blood parameter of the biological dam, modelled as a fixed covariate. Phenotypic information of all biological dams in LR (n = 262) and LW (n = 226) was available. In case of a significant effect of the covariate, the trait was considered for GWAS using model (1*).
Covariables age, weight and blood parameter of the biological dam, used in models (1) and (1*), were standardised to a mean value of zero.
The R-package GenABEL [72] with a generalized linear model approach was used to analyse all parameters except for the cytokines. In order to identify associations between SNP genotypes and phenotypic information, a fast score test was conducted. Because of limitations in the GenABEL-package, the analysis of cytokines was performed using ASReml® [73], which is more flexible in the inclusion of random effects. SNP effects were estimated one by one. P-values were assessed based on the effect and its standard error, assuming a normal distribution.
Lambda (λ) values were low to moderate in LR (0.99-1.87) and LW (0.95-1.67). Adjustment for biases through the population structure was performed according to the Genomic Control approach [74]. After correction, the λ values were in an acceptable range <1.08. In addition, correction for a modified false discovery rate [75, 76] of 5% was applied in order to take multiple testing into account using the R-package qvalue [76]. Q-values <5% were denoted as genome-wide/ chromosome-wide significant, based on the number of tests (total number of SNPs/ number of SNPs per chromosome). To test for a more stringent threshold in terms of multiple testing, additional validation through Bonferroni correction was performed using p.adjust R-command. Markers with adjusted p-values <0.05 and 0.1 were highlighted in Additional files 1 and 2.
The phenotypic variance explained by a single SNP was calculated according to the transformation of a student’s t-distribution into a z-distribution [77] using the following formula:
Annotation of significant SNPs
Markers were mapped to the porcine reference genome Sscrofa 11.1 and variants were identified according to Ensembl release 100 [78]. In addition, significant markers were examined regarding their biological function and position in putative candidate genes. Classification into known pathways was implemented using Reactome [49, 50]. Furthermore, a comparison with the literature deposited in the AnimalQTL database [25] was conducted.
Availability of data and materials
Data cannot be made publicly available, as they are owned by a third party, the BHZP GmbH. The datasets used and analysed during the current study are available from the corresponding author on reasonable request and with permission of the BHZP GmbH pig breeding company (henne@bhzp.de).
References
Edfors-Lilja I, Wattrang E, Magnusson U, Fossum C. Genetic variation in parameters reflecting immune competence of swine. Vet Immunol Immunopathol. 1994; 40(1):1–16.
Knap PW, Bishop SC. Relationships between genetic change and infectious disease in domestic livestock. Occ Publ Br Soc Anim Sci. 2000; 27:65–80.
Flori L, Gao Y, Laloë D, Lemonnier G, Leplat J-J, Teillaud A, Cossalter A-M, Laffitte J, Pinton P, de Vaureix C, et al.Immunity traits in pigs: Substantial senetic variation and limited covariation. PLoS One. 2011; 6(7):e22717.
Kanis E, Van den Belt H, Groen AF, Schakel J, De Greef KH. Breeding for improved welfare in pigs: a conceptual framework and its use in practice. Anim Sci. 2004; 78(2):315–29.
Clapperton M, Diack AB, Matika O, Glass EJ, Gladney CD, Mellencamp MA, Hoste A, Bishop SC. Traits associated with innate and adaptive immunity in pigs: heritability and associations with performance under different health status conditions. Genet Sel Evol. 2009; 41:54.
Heuß EM, Pröll-Cornelissen MJ, Neuhoff C, Tholen E, Große-Brinkhaus C. Invited review: Piglet survival: benefits of the immunocompetence. Animal. 2019; 13(10):2114–24.
Knap PW. Breeding robust pigs. Aust J Exp Agric. 2005; 45(8):763–73.
Pluske JR, Kim JC, Black JL. Manipulating the immune system for pigs to optimise performance. Anim Prod Sci. 2018; 58(4):666–80.
Fossum C. Cytokines as markers for infections and their effect on growth performance and well-being in the pig. Domest Anim Endocrinol. 1998; 15(5):439–44.
Ponsuksili S, Reyer H, Trakooljul N, Murani E, Wimmers K. Single- and Bayesian multi-marker genome-wide association for haematological parameters in pigs. PloS One. 2016; 11(7):e0159212.
Tizard IR. Veterinary Immunology-E-Book, 10th ed.St. Louis: Elsevier Health Sciences; 2017.
Matías J, Berzosa M, Pastor Y, Irache JM, Gamazo C. Maternal vaccination. immunization of sows during pregnancy against ETEC Infections. Vaccines (Basel). 2017; 5(4):48.
Salmon H, Berri M, Gerdts V, Meurens F. Humoral and cellular factors of maternal immunity in swine. Dev Comp Immunol. 2009; 33(3):384–93.
Tuboly S, Bernáth S, Glávits R, Medveczky I. Intestinal absorption of colostral lymphoid cells in newborn piglets. Vet Immunol Immunopathol. 1988; 20(1):75–85.
Collins AM. On-farm measures to monitor the health and immune status of pigs. Breeding Focus 2014 - Improving Resilience. University of New England: Animal Genetics and Breeding Unit; 2014, pp. 31–47.
Henryon M, Heegaard PMH, Nielsen J, Berg P, Juul-Madsen H. Immunological traits have the potential to improve selection of pigs for resistance to clinical and subclinical disease. Anim Sci. 2006; 82(5):597–606.
Clapperton M, Glass EJ, Bishop SC. Pig peripheral blood mononuclear leucocyte subsets are heritable and genetically correlated with performance. Animal. 2008; 2(11):1575–84.
Flori L, Gao Y, Oswald IP, Lefevre F, Bouffaud M, Mercat M-J, Bidanel J-P, Rogel-Gaillard C. Deciphering the genetic control of innate and adaptive immune responses in pig: a combined genetic and genomic study. BMC Proc. 2011; 5(Suppl 4):S32.
Mpetile Z, Young JM, Gabler NK, Dekkers JCM, Tuggle CK. Assessing peripheral blood cell profile of Yorkshire pigs divergently selected for residual feed intake. J Anim Sci. 2015; 93(3):892–9.
Ballester M, Ramayo-Caldas Y, González-Rodríguez O, Pascual M, Reixach J, Díaz M, Blanc F, López-Serrano S, Tibau J, Quintanilla R. Genetic parameters and associated genomic regions for global immunocompetence and other health-related traits in pigs. Sci Rep. 2020; 10:18462.
Mangino M, Roederer M, Beddall MH, Nestle FO, Spector TD. Innate and adaptive immune traits are differentially affected by genetic and environmental factors. Nat Commun. 2017; 8:13850.
Roth K, Dauben C, Heuß E, Pröll MJ, Henne H, Appel AK, Schellander K, Tholen E. Genomic analysis of immune traits of two maternal pig lines. In: 69th Annual Meeting of the European Federation of Animal Science, Dubrovnik, Croatia: 2018. p. 403.
Große-Brinkhaus C, Pröll-Cornelissen MJ, Roth K, Dauben C, Heuß EM, Appel AK, Henne H, Schellander K, Tholen E. Genetische Analysen von Immunmerkmalen. In: 11. Schweine-Workshop Uelzen 2019. DGfZ-Schriftenreihe Heft 77, Germany: 2019. p. 32–40.
Rothschild MF, Hu Z. -l., Jiang Z. Advances in QTL mapping in pigs. Int J Biol Sci. 2007; 3(3):192–7.
Hu Z-L, Park CA, Wu X-L, Reecy JM. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2013; 41(D1):871–9.
Edfors-Lilja I, Wattrang E, Marklund L, Moller M, Andersson-Eklund L, Andersson L, Fossum C. Mapping quantitative trait loci for immune capacity in the pig. J Immunol. 1998; 161(2):829–35.
Wattrang E, Almqvist M, Johansson A, Fossum C, Wallgren P, Pielberg G, Andersson L, Edfors-Lilja I. Confirmation of QTL on porcine chromosomes 1 and 8 influencing leukocyte numbers, haematological parameters and leukocyte function. Anim Genet. 2005; 36(4):337–45.
Bao WB, Ye L, Zhu J, Pan ZY, Zhu GQ, Huang XG, Wu SL. Polymorphism of M307 of the FUT1 Gene and Its Relationship with Some Immune Indexes in Sutai Pigs (Duroc x Meishan). Biochem Genet. 2011; 49(9-10):665–73.
Liu Y, Luo YR, Lu X, Qiu XT, Zhou JP, Gong YF, Ding XD, Zhang Q. Association analysis of polymorphisms of porcine LMP2 and LMP7 genes with haematological traits. Mol Biol Rep. 2011; 38(7):4455–60.
Sun N, Liu D, Chen H, Liu X, Meng F, Zhang X, Chen H, Xie S, Li X, Wu Z. Localization, expression change in PRRSV infection and association analysis of the porcine TAP1 gene. Int J Biol Sci. 2012; 8(1):49–58.
Wang JY, Luo YR, Fu WX, Lu X, Zhou JP, Ding XD, Liu JF, Zhang Q. Genome-wide association studies for hematological traits in swine. Anim Genet. 2013; 44(1):34–43.
Zhang Z, Hong Y, Gao J, Xiao S, Ma J, Zhang W, Ren J, Huang L. Genome-wide association study reveals constant and specific loci for hematological traits at three time stages in a White Duroc x Erhualian F2 resource population. PLoS One. 2013; 8(5):e63665.
Zhang F, Zhang Z, Yan X, Chen H, Zhang W, Hong Y, Huang L. Genome-wide association studies for hematological traits in Chinese Sutai pigs. BMC Genet. 2014; 15:41.
Zhang J, Chen JH, Liu XD, Wang HY, Liu XL, Li XY, Wu ZF, Zhu MJ, Zhao SH. Genomewide association studies for hematological traits and T lymphocyte subpopulations in a Duroc x Erhualian F2 resource population. J Anim Sci. 2016; 94(12):5028–41.
Yan G, Guo T, Xiao S, Zhang F, Xin W, Huang T, Xu W, Li Y, Zhang Z, Huang L. Imputation-based whole-genome sequence association study reveals constant and novel loci for hematological traits in a large-scale swine F2 resource population. Front Genet. 2018; 9:401.
Bovo S, Mazzoni G, Bertolini F, Schiavo G, Galimberti G, Gallo M, Dall’Olio S, Fontanesi L. Genome-wide association studies for 30 haematological and blood clinical-biochemical traits in Large White pigs reveal genomic regions affecting intermediate phenotypes. Sci Rep. 2019; 9:7003.
Bovo S, Ballan M, Schiavo G, Gallo M, Dall’Olio S, Fontanesi L. Haplotype-based genome-wide association studies reveal new loci for haematological and clinical–biochemical parameters in Large White pigs. Anim Genet. 2020; 51(4):601–6.
Wimmers K, Murani E, Schellander K, Ponsuksili S. QTL for traits related to humoral immune response estimated from data of a porcine F2 resource population. Int J Immunogenet. 2009; 36(3):141–51.
Uddin MJ, Grosse-Brinkhaus C, Cinar MU, Jonas E, Tesfaye D, Tholen E, Juengst H, Looft C, Ponsuksili S, Wimmers K, et al.Mapping of quantitative trait loci for mycoplasma and tetanus antibodies and interferon-gamma in a porcine F2 Duroc x Pietrain resource population. Mamm Genome. 2010; 21(7):409–18.
Uddin MJ, Cinar MU, Große-Brinkhaus C, Tesfaye D, Tholen E, Juengst H, Looft C, Wimmers K, Phatsara C, Schellander K. Mapping quantitative trait loci for innate immune response in the pig. Int J Immunogenet. 2011; 38(2):121–31.
Lu X, Gong YF, Liu JF, Wang ZP, Hu F, Qiu XT, Luo YR, Zhang Q. Mapping quantitative trait loci for cytokines in the pig. Anim Genet. 2011; 42(1):1–5.
Lu X, Liu J, Fu W, Zhou J, Luo Y, Ding X, Liu Y, Zhang Q. Genome-wide association study for cytokines and immunoglobulin G in swine. PLoS One. 2013; 8(10):e74846.
Wu ZC, Liu Y, Zhao QH, Zhu SP, Huo YJ, Zhu GQ, Wu SL, Bao WB. Association between polymorphisms in exons 4 and 10 of the BPI gene and immune indices in Sutai pigs. Genet Mol Res. 2015; 14(2):6048–58.
Ramos AM, Crooijmans RPMA, Affara NA, Amaral AJ, Archibald AL, Beever JE, Bendixen C, Churcher C, Clark R, Dehais P, et al.Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS One. 2009; 4(8):e6524.
Seutter U. Einfluss von Rasse, Haltung, Fütterung, Management, Alter und Reproduktionsstadium auf hämatologische und klinisch-chemische Parameter beim Schwein. PhD Thesis, Ludwig-Maximilians-Universität München. 1995.
Widowski TM, Curtis SE, Graves CN. The neutrophil: lymphocyte ratio in pigs fed cortisol. Can J Anim Sci. 1989; 69(2):501–4.
Lu X, Fu W-X, Luo Y-R, Ding X-D, Zhou J-P, Liu Y, Liu J-F, Zhang Q. Genome-wide association study for T lymphocyte subpopulations in swine. BMC Genomics. 2012; 13:488.
Sutherland MA, Rodriguez-Zas SL, Ellis M, Salak-Johnson JL. Breed and age affect baseline immune traits, cortisol, and performance in growing pigs. J Anim Sci. 2005; 83(9):2087–95.
Fabregat A, Sidiropoulos K, Viteri G, Forner O, Marin-Garcia P, Arnau V, D’Eustachio P, Stein L, Hermjakob H. Reactome pathway analysis: a high-performance in-memory approach. BMC Bioinformatics. 2017; 18:142.
Fabregat A, Sidiropoulos K, Viteri G, Marin-Garcia P, Ping P, Stein L, D’Eustachio P, Hermjakob H. Reactome diagram viewer: data structures and strategies to boost performance. Bioinformatics. 2018; 34(7):1208–14.
Kumar KP, Nicholls AJ, Wong CHY. Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018; 371(3):551–65.
Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford). 2010; 49(9):1618–31.
Conrad S, Younsi A, Bauer C, Geburek F, Skutella T. Mesenchymal stem cell-derived extracellular vesicles as mediators of anti-inflammatory effects In: Pham P, editor. Stem Cell Transplantation for Autoimmune Diseases and Inflammation, Stem Cells in Clinical Applications. Basel: Springer International Publishing: 2019. p. 89–123.
Cheverud JM. The genetic architecture of pleiotropic relations and differential epistasis In: Wagner GP, editor. The Character Concept in Evolutionary Biology. San Diego: Academic Press: 2001. p. 411–34.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al.Gene ontology: tool for the unification of biology. Nat Genet. 2000; 25:25–9.
The Gene Ontology Consortium. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019; 47(D1):330–8.
Sun G, Cao Y, Qian C, Wan Z, Zhu J, Guo J, Shi L. Romo1 is involved in the immune response of glioblastoma by regulating the function of macrophages. Aging (Albany NY). 2020; 11(2):1114–27.
Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004; 15:177–82.
Dawson HD, Sang Y, Lunney JK. Porcine cytokines, chemokines and growth factors: 2019 update. Res Vet Sci. 2020; 131:266–300.
Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S, the AmiGO Hub, the Web Presence Working Group. AmiGO: online access to ontology and annotation data. Bioinformatics. 2009; 25(2):288–9.
Jiao S, Maltecca C, Gray KA, Cassady JP. Feed intake, average daily gain, feed efficiency, and real-time ultrasound traits in Duroc pigs: II, Genomewide association. J Anim Sci. 2014; 92(7):2846–60.
Kim E-S, Ros-Freixedes R, Pena RN, Baas TJ, Estany J, Rothschild MF. Identification of signatures of selection for intramuscular fat and backfat thickness in two Duroc populations. J Anim Sci. 2015; 93(7):3292–302.
Davoli R, Luise D, Mingazzini V, Zambonelli P, Braglia S, Serra A, Russo V. Genome-wide study on intramuscular fat in Italian Large White pig breed using the PorcineSNP60 BeadChip. J Anim Breed Genet. 2016; 133:277–82.
Sevillano CA, Lopes MS, Harlizius B, Hanenberg EHAT, Knol EF, Bastiaansen JWM. Genome-wide association study using deregressed breeding values for cryptorchidism and scrotal/inguinal hernia in two pig lines. Genet Sel Evol. 2015; 47:18.
Wang K, Liu D, Hernandez-Sanchez J, Chen J, Liu C, Wu Z, Fang M, Li N. Genome wide association analysis reveals new production trait genes in a male duroc population. PLoS One. 2015; 10(9):e0139207.
Fontanesi L, Schiavo G, Gallo M, Baiocco C, Galimberti G, Bovo S, Russo V, Buttazzoni L. Genome-wide association study for ham weight loss at first salting in Italian Large White pigs: towards the genetic dissection of a key trait for dry-cured ham production. Anim Genet. 2017; 48:103–7.
Onteru SK, Fan B, Du Z-Q, Garrick DJ, Stalder KJ, Rothschild MF. A whole-genome association study for pig reproductive traits. Anim Genet. 2012; 43:18–26.
Chen Z, Ye S, Teng J, Diao S, Yuan X, Chen Z, Zhang H, Li J, Zhang Z. Genome-wide association studies for the number of animals born alive and dead in duroc pigs. Theriogenology. 2019; 139:36–42.
Zhang Z, Chen Z, Ye S, He Y, Huang S, Yuan X, Chen Z, Zhang H, Li J. Genome-wide association study for reproductive traits in a Duroc pig population. Animals (Basel). 2019; 9:732.
Hornung RW, Reed LD. Estimation of average concentration in the presence of nondetectable values. Appl Occup Environ Hyg. 1990; 5(1):46–51.
Uh H-W, Hartgers FC, Yazdanbakhsh M, Houwing-Duistermaat JJ. Evaluation of regression methods when immunological measurements are constrained by detection limits. BMC Immunol. 2008; 9:59.
Aulchenko YS, Ripke S, Isaacs A, Van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007; 23(10):1294–6.
Gilmour AR, Gogel BJ, Cullis BR, Welham SJ, Thompson R. ASReml User Guide Release 4.1 Structural Specification. Hemel Hempstead: VSN international ltd.; 2015.
Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999; 55:997–1004.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995; 57(1):289–300.
Storey JD. A direct approach to false discovery rates. J R Stat Soc Series B Stat Methodol. 2002; 64(3):479–98.
Kendall M, Stuart A. The Advanced Theory of Statistics. Vol, 1: Distribution Theory, 4th ed. London: Griffin; 1977.
Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, et al.Ensembl 2020. Nucleic Acids Res. 2020; 48(D1):682–8.
Acknowledgements
The authors thank the Bundeshybridzuchtprogramm (BHZP GmbH) for providing data sets and the staff members of the BHZP GmbH for animal selection, sample collection and providing the phenotypes. We also wish to thank Nadine Leyer, Helga Brodeßer and Birgit Koch-Fabritzius for laboratory work.
Funding
The study was performed within the ’pigFit’ project which was supported by funds of the German Government’s Special Purpose Fund held at Landwirtschaftliche Rentenbank (FKZ28-RZ-3-72.038). The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
CMD performed the statistical analysis, interpreted the results and wrote the first draft of the manuscript. CGB, ET, KS, HH and AKA set up the experimental design. HH and AKA selected the animals and organized the blood sampling. MPC set up the serum sample management and performed cytokine analyses. KR and EMH contributed to the data management and statistical analyses. CGB, ET and MPC oversaw the analysis and contributed to the interpretation of the results and writing the manuscript. ET, CGB and KS supervised the study. All authors contributed to the discussion of the results and development of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All applicable international, national, and institutional guidelines for the care and use of animals were followed. Animals were reared on the BHZP nucleus herds in compliance with national regulations pertaining to livestock production and according to the procedures approved by the German Animal Protection Law (TierSchG). These animals are not to be considered as experimental animals as defined in EU directive 2010/63 and the Animal Protection Law (TierSchG). Blood sampling was performed by veterinarians as part of the routinely health and hygiene monitoring of the BHZP nucleus herds according to the Pig Keeping Hygiene Requirements Ordinance (SchHaltHygV) § 7 (1) Sentence 2. There was no need for ethical approval of this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
Complete GWAS results in Landrace. Significant SNPs in Landrace associated with immune relevant traits identified by GWAS. MCHC: Mean corpuscular haemoglobin concentration, IL: Interleukin, SSC: Sus Scrofa Chromosome, MAF: Minor allele frequency, *: genome-wide significance, adj. p-value: adjusted p-value after Bonferroni correction, B5: Additionally significant after Bonferroni correction with adjusted p-value <0.05, B10: Additionally significant after Bonferroni correction with adjusted p-value <0.1.
Additional file 2
Complete GWAS results in Large White. Significant SNPs in Large White associated with immune relevant traits identified by GWAS. MCHC: Mean corpuscular haemoglobin concentration, IFN: Interferon, IL: Interleukin, TNF: Tumor necrosis factor, SSC: Sus Scrofa Chromosome, MAF: Minor allele frequency, *: genome-wide significance, adj. p-value: adjusted p-value after Bonferroni correction, B5: Additionally significant after Bonferroni correction with adjusted p-value <0.05, B10: Additionally significant after Bonferroni correction with adjusted p-value <0.1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dauben, C.M., Pröll-Cornelissen, M.J., Heuß, E.M. et al. Genome-wide associations for immune traits in two maternal pig lines. BMC Genomics 22, 717 (2021). https://doi.org/10.1186/s12864-021-07997-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07997-1