
Abstract
Background
Cryptorchidism and scrotal/inguinal hernia are the most frequent congenital defects in pigs. Identification of genomic regions that control these congenital defects is of great interest to breeding programs, both from an animal welfare point of view as well as for economic reasons. The aim of this genome-wide association study (GWAS) was to identify single nucleotide polymorphisms (SNPs) that are strongly associated with these congenital defects. Genotypes were available for 2570 Large White (LW) and 2272 Landrace (LR) pigs. Breeding values were estimated based on 1 359 765 purebred and crossbred male offspring, using a binary trait animal model. Estimated breeding values were deregressed (DEBV) and taken as the response variable in the GWAS.
Results
Heritability estimates were equal to 0.26 ± 0.02 for cryptorchidism and to 0.31 ± 0.01 for scrotal/inguinal hernia. Seven and 31 distinct QTL regions were associated with cryptorchidism in the LW and LR datasets, respectively. The top SNP per region explained between 0.96% and 1.10% and between 0.48% and 2.77% of the total variance of cryptorchidism incidence in the LW and LR populations, respectively. Five distinct QTL regions associated with scrotal/inguinal hernia were detected in both LW and LR datasets. The top SNP per region explained between 1.22% and 1.60% and between 1.15% and 1.46% of the total variance of scrotal/inguinal hernia incidence in the LW and LR populations, respectively. For each trait, we identified one overlapping region between the LW and LR datasets, i.e. a region on SSC8 (Sus scrofa chromosome) between 65 and 73 Mb for cryptorchidism and a region on SSC13 between 34 and 37 Mb for scrotal/inguinal hernia.
Conclusions
The use of DEBV in combination with a binary trait model was a powerful approach to detect regions associated with difficult traits such as cryptorchidism and scrotal/inguinal hernia that have a low incidence and for which affected animals are generally not available for genotyping. Several novel QTL regions were detected for cryptorchidism and scrotal/inguinal hernia, and for several previously known QTL regions, the confidence interval was narrowed down.
Similar content being viewed by others

Background
Cryptorchidism and scrotal/inguinal hernias are the most frequent congenital defects in pigs with incidences that range from 0.27 to 0.90% and 0.39 to 1.09% for cryptorchidism and scrotal/inguinal hernia, respectively [1-3]. Cryptorchidism occurs when the testis fails to descend from its intra-abdominal location into the scrotum [4]. An inguinal hernia occurs when part of the small intestine passes through the internal inguinal ring and is present in the inguinal canal. A scrotal hernia occurs when part of the small intestine passes all the way through the inguinal canal and enters the scrotum [5,6]. Distinguishing between scrotal and inguinal hernias is not easy without clinical examination [7], therefore these two traits were considered as a single trait, hereafter called ‘hernia’.
Identification of genomic regions that control these congenital defects is of great interest to breeding programs, both from an animal welfare point of view and for economic reasons. Many studies have aimed at identifying regions in the genome that can, partially, explain the occurrence of these defects [5,8,9] but success was limited because of insufficient power in the analyses due to the small number of samples and/or the study design (e.g. parent-offspring trios, affected sib pairs).
Here, a novel approach was implemented to perform a genome-wide association study (GWAS) for congenital defects. To obtain “phenotypes” of the genotyped animals, we applied a pedigree-based method to calculate estimated breeding values (EBV). These EBV were subsequently deregressed (DEBV) and used as response variable in the GWAS analysis. The use of DEBV obtained with a binary trait model was considered to be a powerful approach for traits like cryptorchidism and hernia, which have a low incidence and for which affected animals are generally not genotyped [10]. Our aim was to identify single nucleotide polymorphisms (SNPs) associated with cryptorchidism and hernia using DEBV.
Methods
Ethics statement
The data used for this study was collected as part of routine data that are recorded in a commercial breeding program. Samples collected for DNA extraction were only used for routine diagnostic purposes of the breeding program. Data recording and sample collection were conducted strictly in line with the Dutch law on the protection of animals (Gezondheids- en welzijnswet voor dieren).
Phenotypes
The phenotypes consisted of 1 360 453 records of purebred and crossbred male offspring of genotyped animals from two sow lines: Large White (LW) and Landrace (LR). Phenotypic data were collected from 57 different farms. Presence of cryptorchidism or hernia was recorded as either 1 (affected) or 0 (not-affected). In total, 2947 pigs were recorded with cryptorchidism and were offspring of 832 sires and 2287 dams with one to 34 affected offspring per sire and one to six affected offspring per dam. In total, 5251 pigs were recorded with hernia and were offspring of 995 sires and 3859 dams with one to 115 affected offspring per sire and one to eight affected offspring per dam. Of the 184 239 litters, 2599 were affected by cryptorchidism and 4446 were affected by hernia with one to six affected pigs per litter for both defects. None of the pigs were affected by both cryptorchidism and hernia.
Genotypes
For the GWAS, animals that were genetically related with the phenotyped animals were genotyped. Thus, 2570 LW pigs (608 males and 1962 females) and 2272 LR pigs (835 males and 1437 females) were genotyped using the Porcine SNP60 Beadchip of Illumina (San Diego, CA, USA) [11]. Of the 2570 genotyped LW pigs, 2264 had records from their own offspring. Genotyped sires had on average 1093 individual offspring records that ranged from one to 6128 per sire, while genotyped dams have on average 38 individual offspring records that ranged from one to 150 per dam. Of the 2272 genotyped LR pigs, 1825 had records from their own offspring. Genotyped sires had on average 903 individual offspring records that ranged from four to 5438 per sire, while genotyped dams had on average 32 individual offspring records that ranged from one to 119 per dam.
Genotype data were filtered in a two-step quality control process. First, SNPs were removed if their GenCall score was less than 0.15, if they were located on SSCY (SSC for Sus scrofa chromosome), or if their position on the genome build10.2 was unknown [12]; animals were removed if they had a missing genotype frequency of 0.30 or higher. Then, SNPs with a call rate less than 0.95, a minor allele frequency less than 0.01 and/or a strong deviation from Hardy Weinberg equilibrium (χ 2 > 600) were removed. For males, SNPs that were outside the pseudoautosomal region on SSCX were also removed if the frequency of heterozygous calls was greater than 0.05. Animals with a missing genotype frequency higher than 0.05 were removed.
Genotyped parent-offspring pairs were checked for Mendelian inconsistencies. Offspring and parents with more than 1% Mendelian inconsistencies with each of their parents or offspring were excluded.
After quality control, genotyping data of the LW and LR populations consisted of 2102 and 1926 animals, and 38 632 and 39 508 SNPs, respectively. The final call rate was 99.6% for both populations.
Breeding value estimation
For all phenotyped animals, pedigree information was collected up to 20 generations back. The pedigree data included 1 434 713 animals. A binary single trait animal model was used to estimate heritabilities and EBV by restricted maximum likelihood methodology implemented in the software ASReml [13]. The following model was applied:
where Y ijkmn was the phenotype for cryptorchidism or hernia observed in animal n, μ was the overall mean, HYS i was the fixed effect of herd-year-season of birth i (1194 classes), TNB j was the fixed effect of number of littermates j (30 classes), P k was the fixed effect of the parity k of the mother (seven classes) and LL l was the fixed effect of litter type l (four classes i.e. LWxLW, LWxLR, LRxLR, LRxLW). Random effects included the common litter effect (\( {c}_m^2 \)) assumed to be normally distributed ~ N(0, I \( {\sigma}_{\mathrm{c}}^2 \)), where I is an identity matrix and \( {\sigma}_{\mathrm{c}}^2 \) is the unknown variance between litters. The additive genetic effect (an) assumed to be normally distributed ~ N(0, A \( {\sigma}_{\mathrm{a}}^2 \)), where A was a known matrix of additive genetic relationship among animals and \( {\sigma}_{\mathrm{a}}^2 \) was the unknown genetic variance between animals. The residual error (e ijklmn ) was defined on the logistic scale by setting the residual variance to 1.
The EBV obtained with model 1 were deregressed according to Garrick et al. [14], whereby parental average effects were removed as part of the deregression process to obtain a more accurate estimate of the expected phenotype. In addition, following the approach of Garrick et al. [14], a weighting factor (w) was estimated based on the reliability of the calculated DEBV by considering a value of 0.5 for the scalar c.
Association analysis
A single SNP GWAS was performed with the software ASReml [13] by applying the following model:
where DEBV ij is the DEBV for genotyped animal j, μ is the overall DEBV mean of the genotyped animals, SNP i is the genotype of the SNP i coded as 0, 1 or 2 copies of one of the alleles, a j is the additive genetic effect and e ij the residual error. The weighting factor w was used in the GWAS to account for differences in the amount of available information on offspring to estimate DEBV [14].
Only animals with a w value higher than 0 and a minimum reliability of DEBV of 0.06 for cryptorchidism and 0.08 for hernia were included in the GWAS. DEBV of 1528 LW and 1229 LR pigs for cryptorchidism and DEBV of 1361 LW and 1405 LR pigs for hernia were included in the GWAS.
To account for multiple testing, a false discovery rate (FDR) implemented in the R package ‘qvalue’ [15] was applied. A FDR ≤ 0.20 was set to define significant associations.
QTL regions
QTL regions were defined based on the location of the significant SNPs. All significant SNPs located within a region of 10 Mb were considered to belong to the same QTL region. For multiple QTL regions to be considered present on the same chromosome, the distance between consecutive significant SNPs had to be larger than 10 Mb. The genetic variance explained by each QTL region (\( {\sigma}_{SNP}^2 \)) was calculated as follows:
where p is the observed allele frequency of the top SNP in each QTL region, q = 1-p and α is the estimated allele substitution effect obtained from model 2. The result was expressed as the percentage of the total variance explained by the \( \mathrm{S}\mathrm{N}\mathrm{P}\left(\frac{\upsigma_{\mathrm{SNP}}^2}{\upsigma_{\mathrm{P}}^2} \times 100\right) \).
Candidate genes
Putative candidate genes within the QTL regions and in the neighbouring upstream and downstream 2-Mb regions were identified based on the Sscrofa10.2 genome assembly, using the NCBI Map Viewer (http://www.ncbi.nlm.nih.gov/projects/mapview/).
Results and discussion
For GWAS to be successful, it is essential to obtain a relevant dataset, which in the case of congenital defects is especially challenging because of their low incidence, and because affected animals are generally culled when diagnosed and thus not included in sampling and genotyping. For this study, we combined phenotypes from many thousands of animals with genotypes of their ancestors. To connect genotype to phenotype data, phenotypic observations of individuals were used to estimate the EBV of their genotyped ancestors.
Breeding value estimation and variance components
Incidences of cryptorchidism and hernia were on average 0.22% and 0.39%, respectively. The incidence of cryptorchidism was lower in LW than in LR pigs (0.14 vs. 0.33%), while the incidence of hernia was slightly higher in LW than in LR pigs (0.42% vs. 0.34%) (Table 1). Concerning litter type, the highest incidence of cryptorchidism and hernia was observed in purebred litters of both lines compared to their corresponding crossbred litters (Table 1).
Heritabilities for cryptorchidism and for hernia were estimated at 0.26 ± 0.02 and 0.31 ± 0.01, respectively, using an animal model (Table 2). In the literature, heritability estimates reported for congenital defects range from 0.03 to 0.27 [1-3,9,16] depending on the population sampled and the statistical model (usually obtained with a sire-model [1-3,9] except for [16]). Estimation of the heritability of binary traits is not straightforward and contradictory results have been reported, although the factors responsible for these differences remain unclear [17]. Moreover, Moreno et al. [18] showed that, in models for binary traits, inferences about the variance component of random effects are biased when there is little information on fixed effects, which leads to poor estimates of variance components. The mean of the posterior distribution of the variance component when using the animal model, is biased upwards and leads to overestimation of the heritability. With our large dataset, we obtained more precise estimates of fixed effects (herd-year-season, number of littermates, litter line, and parity), thus reducing the potential for biased estimates of variance components.
The common litter environment (c2) was found to explain an important part of the variance for cryptorchidism and hernia. The proportion of variance due to c2 was estimated at 0.64 for cryptorchidism and 0.53 for hernia. Thaller et al. [3] reported a significant c2 effect that improved remarkably the fit of a binary model to data on congenital defects in pigs. They also suggested that intrauterine and possibly postnatal environments substantially influence the occurrence of these disorders. If the sow is exposed to disease during gestation, and subsequently to medical treatment, or if it is in contact with toxic products such as mycotoxins in the feed, the development of the piglets can be affected, and an increase in the incidence of congenital defects in those litters can be observed.
QTL regions and candidate genes
Cryptorchidism
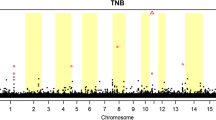
The GWAS for cryptorchidism identified 22 and 83 significant SNPs (FDR ≤ 0.20) in the LW and LR pig datasets, respectively (Figures 1 and 2). The quantile-quantile plots (QQ plot) for these GWAS are shown in Additional file 1: Figure S1. For the LW dataset, the 22 significant SNPs were distributed over seven QTL regions located on SSC2, 8, 10, 13 and 14 (Table 3). The most significant SNP of each QTL region explained between 0.96% and 1.10% of the total variance of cryptorchidism incidence in the LW population (Table 3). For the LR dataset, the 83 significant SNPs were distributed over 31 QTL regions located on all autosomes with the exception of SSC2 and SSC12 (Table 4). The most significant SNP of each QTL region explained between 0.48% and 2.77% of the total variance of cryptorchidism incidence in the LR population (Table 4).

Association between cryptorchidism and 38 632 genome-wide SNPs in a Large White pig population. Each dot represents one SNP; on the y-axis are -log10(P-values) and on the x-axis are the physical positions of the SNPs by chromosome; blue dots represents SNPs that surpassed the FDR ≤ 0.20 threshold.

Association between cryptorchidism and 39 508 genome-wide SNPs in a Landrace pig population. Each dot represents one SNP; on the y-axis are -log10(P-values), and on the x-axis are the physical positions of the SNPs by chromosome; blue dots represents SNPs that surpassed the FDR ≤ 0.20 threshold.
One QTL region located on SSC8 between 61 and 73 Mb was found to overlap between the LW and LR datasets. This region contains the gonadotropin-releasing hormone receptor (GNRHR) and alpha-fetoprotein (AFP) genes. In mouse, a mutation in GNRHR was shown to initiate undescended testes in male carriers [19]. The AFP protein is known to interact with oestrogens and may modulate foetal responses to oestrogens. Thus, high foetal AFP levels may have a direct role in the occurrence of cryptorchidism; but also, indirectly, by inducing placental dysfunction [20]. Because this region on SSC8 was found to be associated with cryptorchidism in both LW and LR populations and because it contained strong candidate genes that have been shown to be involved in testes descent, it is likely that it has a role in the incidence of cryptorchidism in pigs.
On SSC13, a QTL region was identified for cryptorchidism in the LW dataset and a peak was also observed in this same region in the LR dataset, but it did not surpass the FDR threshold. This region contains a candidate gene i.e. the GATA binding protein 2 (GATA2) that is located at 79 Mb and has been shown to play a critical role in urogenital development [21]. In addition, expression of the GATA2 protein is regulated by FOG2 (friend of GATA2), a transcriptional cofactor that is able to differentially regulate the expression of GATA-target genes and has been shown to be involved in normal testis development in a mouse model [22].
In the LW dataset, a second region on SSC8 around 43 Mb was identified that contains the platelet-derived growth factor receptor-alpha like (PDGFRA) and tyrosine-protein kinase (v-KIT) genes. Phosphorylation of PDGFRA and v-KIT have a role in the development and function of male gonads and are associated with cryptorchidism in humans [23,24] and with gonadal hypoplasia in cattle [25]. No evidence was found for additional candidate genes in the QTL regions identified for both datasets.
Previously, Stinckens et al. [9] reported an association study for cryptorchidism using the Porcine SNP60 Beadchip in different populations of LW and LR pigs. There is no overlap between our results and those of Stinckens et al. [10], who found associations on SSC12 and SSCX (A. Stinckens, personal communication). This may be explained by the different structure of their dataset that included affected parent-offspring trios and by the different populations used, although of the same breed.
Hernia
The GWAS for hernia identified 10 and 22 significant SNPs in the LW and LR datasets, respectively (Figures 3 and 4). The QQ plots for these GWAS are shown in Additional file 1: Figure S1. For the LW dataset, the 10 significant SNPs were distributed over five QTL regions on SSC3, 5, 7, 8 and 13 (Table 5). The most significant SNP of each QTL region explained between 1.22% and 1.60% of the total variance of hernia incidence in the LW population (Table 5). For the LR dataset, the 22 significant SNPs were distributed over five QTL regions on SSC1, 2, 4, 10, and 13 (Table 6). The most significant SNP of each QTL region explained between 1.15% and 1.46% of the total variance of hernia incidence in the LR population (Table 6).

Association between hernia and 38 632 genome-wide SNPs in a Large White pig population. Each dot represents one SNP; on the y-axis are -log10(P-values), and on the x-axis are the physical positions of the SNPs by chromosome; blue dots represents SNPs that surpassed the FDR ≤ 0.20 threshold.

Association between hernia and 39 508 genome-wide SNPs in a Landrace pig population. Each dot represents one SNP; on the y-axis are -log10(P-values), and on the x-axis are the physical positions of the SNPs by chromosome; blue dots represents SNPs that surpassed the FDR ≤ 0.20 threshold.
One QTL region located on SSC13 between 34 and 37 Mb was found to overlap between the LW and LR datasets. This region was previously reported by Grindflek et al. [5] and Ding et al. [8]. It contains the ras homolog family member A (RHOA) gene, which regulates the contraction and shortening of smooth muscle tissues [26]. Smooth muscle cells play an important role in the obliteration of processus vaginalis, which is the embryological protrusion of the peritoneum that precedes testicular descent into the scrotum [27,28]. Incomplete obliteration of the processus vaginalis leads to cryptorchidism and hernia [28,29]. Altered expression of RHOA was identified in a rat strain with inherited cryptorchidism [30]. Since the region on SSC13 was shown to be associated with hernia in both LW and LR datasets studied here and in other previous studies [5,8], and because it contains a candidate gene that is connected with the process of obliteration of the processus vaginalis, it is likely to play a role in the incidence of hernia in pigs.
The region on SSC7 detected in the LW dataset overlaps with a region previously identified by Grindflek et al. [5]. In their paper, Grindflek et al. [5] suggested that cytochrome P450 family 19A1 (CYP19A1) may be a candidate gene for this QTL, based on its location on the homoeologous chromosome in humans. However, according to the Ensembl Sscrofa10.2 database (http://www.ensembl.org/Sus_scrofa/Info/Index), the CYP19A1 gene is located on SSC1 and not SSC7.
To our knowledge, it is the first time that the other three QTL regions identified on SSC3, 5, and 8 are found to be associated with hernia, although some studies [5,8] have reported that different regions on these chromosomes harbour QTL for hernia. The QTL region on SSC8 contains two candidate genes i.e., pro-epidermal growth factor precursor (EGF) and lymphoid enhancer-binding factor 1 (LEF1). Mutations in EGF have been related to connective-tissue problems, such as inguinal hernia [31]. Regarding the putative role of the LEF1 gene in hernia, it has been shown that the LEF1 protein constitutively associates with β-catenin which mediates the action of the anti-mullerian hormone (AMH) [32], itself involved in the gubernaculum swelling reaction that occurs during the first phase of testicular descent [33,34]. No other candidate genes were identified.
In the LR dataset, the QTL region on SSC4 overlaps with a previously reported QTL by Ding et al. [8]. To our knowledge, it is the first time that the three other regions identified on SSC1, 2 and 10 are found to be associated with hernia, although previous studies [5,7,8] have reported different regions on these chromosomes as harbouring QTL for hernia but no candidate genes were detected.
Genetic differences between the populations analysed and differences in marker density and/or power of resolution between our study and those of Grindflek et al. [5] and Ding et al. [8] may explain the differences in the regions detected. The only previous analysis on hernia in pigs which used the Porcine SNP60 Beadchip is that of Stinckens et al. [9] but on other LW and LR populations. They reported a QTL for hernia on SSC5 without specifying its location and some associations on SSC6 and SSCX, which were not confirmed in our study (A. Stinckens, personal communication). As mentioned before, differences in the structure of their dataset and in the populations analysed may explain the absence of detection of overlapping regions.
In general, the proportion of total variance described by strongly associated SNPs for cryptorchidism and hernia is low. Moreover, the number of regions that were found to be significantly associated with cryptorchidism and hernia is large, which indicates that cryptorchidism is a disease affected by many genes with small effects as suggested by Stinckens et al. [9] and Thaller et al. [3]. Our results suggest a similar architecture for hernia. The polygenic character of these two traits makes it is difficult to identify the causative variations that are involved.
Conclusions
The use of DEBV in combination with a binary trait model was found to be a powerful approach for the GWAS of difficult traits such as cryptorchidism and hernia that have a low incidence and for which affected animals are generally not available for genotyping.
We detected several novel QTL regions and the confidence interval of some previously known QTL regions was narrowed down. Overlapping QTL regions between the two pig datasets was limited to one QTL for each trait.
We identified several significant SNPs that can contribute to an index of SNPs for selection against cryptorchidism and hernia in pigs. However, given the small proportion of variance explained by these SNPs, it will be necessary to use genomic estimated breeding values to expedite the reduction of cryptorchidism and hernia incidence in pig populations.
References
Larzul C, Delaunay I, Schwob S, Mercat MJ. Paramètres génétiques des principales anomalies congénitales porcines. In: 40ème Journées de la Recherche Porcine: 5–6 February 2008. Paris: Ed. IFIP, INRA; 2008. pp. 141–142. http://www.journees-recherche-porcine.com/texte/index.htm.
Mattsson P. Prevalence of congenital defects in Swedish Hampshire, Landrace and Yorkshire pig breeds and opinions on their prevalence in Swedish commercial herds. M.S. thesis. Uppsala: Department of Animal Breeding and Genetics, Swedish University of Agricultural Sciences; 2011. http://urn.kb.se/resolve?urn=urn:nbn:se:slu:epsilon-6-474.
Thaller G, Dempfle L, Hoeschele I. Investigation of the inheritance of birth defects in swine by complex segregation analysis. J Anim Breed Genet. 1996;113:77–92.
Zhao X, Du ZQ, Rothschild MF. An association study of 20 candidate genes with cryptorchidism in Siberian Husky dogs. J Anim Breed Genet. 2010;127:327–31.
Grindflek E, Moe M, Taubert H, Simianer H, Lien S, Moen T. Genome-wide linkage analysis of inguinal hernia in pigs using affected sib pairs. BMC Genet. 2006;7:25.
Zhao X, Du ZQ, Vukasinovic N, Rodriguez F, Clutter AC, Rothschild MF. Association of HOXA10, ZFPM2, and MMP2 genes with scrotal hernias evaluated via biological candidate gene analyses in pigs. Am J Vet Res. 2009;70:1006–12.
Du ZQ, Zhao X, Vukasinovic N, Rodriguez F, Clutter AC, Rothschild MF. Association and haplotype analyses of positional candidate genes in five genomic regions linked to scrotal hernia in commercial pig lines. PLoS One. 2009;4:e4837.
Ding NS, Mao HR, Guo YM, Ren J, Xiao SJ, Wu GZ, et al. A genome-wide scan reveals candidate susceptibility loci for pig hernias in an intercross between White Duroc and Erhualian. J Anim Sci. 2009;87:2469–74.
Stinckens A, Janssens S, Spincemaille G, Buys N: A whole genome association study using the 60 K porcine SNP beadchip reveals candidate susceptibility loci for scrotal hernia in pigs. In Book of Abstracts of the 62th Annual Meeting of the EAAP: 29 August-2 September 2011; Stavanger. Wageningen Academic Publishers; 2011, 17:412.
Ostersen T, Christensen OF, Henryon M, Nielsen B, Su G, Madsen P. Deregressed EBV as the response variable yield more reliable genomic predictions than traditional EBV in pure-bred pigs. Genet Sel Evol. 2011;43:38.
Ramos AM, Crooijmans RP, Affara NA, Amaral AJ, Archibald AL, Beever JE, et al. Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS One. 2009;4:e6524.
Groenen MAM, Archibald AL, Uenishi H, Tuggle CK, Takeuchi Y, Rothschild MF, et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature. 2012;491:393–8.
Gilmour AR, Gogel BJ, Cullis BR, Thompson R. 2009 ASReml User Guide Release 3.0. VSN International Ltd, Hemel Hempstead, HP1 1ES, UK. www.vsni.co.uk.
Garrick DJ, Taylor JF, Fernando RL. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet Sel Evol. 2009;41:55.
Dabney A, Storey J, Warnes G. qvalue: Q-value estimation for false discovery rate control. R package version 1.24.0. 2010.
Ranberg IMA, Tajet H. Health traits in the breeding goal for Norsvin Landrace and Norsvin Duroc. In: Book of Abstracts of the 58th Annual Meeting of the EAAP: 26–29 August 2007. Dublin: Wageningen Academic Publishers; 2007. p. 78.
Villemereuil P, Gimenez O, Doligez B. Comparing parent-offspring regression with frequentist and Bayesian animal models to estimate heritability in wild populations: a simulation study for Gaussian and binary traits. Methods Ecol Evol. 2012;4:260–75.
Moreno C, Sorensen D, García-Cortés LA, Varona L, Altarriba J. On biased inferences about variance components in the binary threshold model. Genet Sel Evol. 1997;29:145–60.
Pask AJ, Kanasaki H, Kaiser UB, Conn PM, Janovick JA, Stockton DW, et al. A novel mouse model of hypogonadotrophic hypogonadism: N-ethyl-N-nitrosourea-induced gonadotropin-releasing hormone receptor gene mutation. Mol Endocrinol. 2005;19:972–81.
Boyd HA, Myrup C, Wohlfahrt J, Westergaard T, Nørgaard-Pedersen B, Melbye M. Maternal serum alpha-fetoprotein level during pregnancy and isolated cryptorchidism in male offspring. Am J Epidemiol. 2006;164:478–86.
Zhou Y, Lim KC, Onodera K, Takahashi S, Ohta J, Minegishi N, et al. Rescue of the embryonic lethal hematopoietic defect reveals a critical role for GATA-2 in urogenital development. EMBO J. 1998;17:6689–700.
Finelli P, Pincelli AI, Russo S, Bonati MT, Recalcati MP, Masciadri M, et al. Disruption of friend of GATA 2 gene (FOG-2) by a de novo t(8;10) chromosomal translocation is associated with heart defects and gonadal dysgenesis. Clin Genet. 2007;71:195–204.
Basciani S, Brama M, Mariani S, De Luca G, Arizzi M, Vesci L, et al. Imatinib mesylate inhibits leydig cell tumor growth: evidence for In vitro and In vivo activity. Cancer Res. 2005;65:1897–903.
Rapley EA, Hockley S, Warren W, Johnson L, Huddart R, Crockford G, et al. Somatic mutations of KIT in familial testicular germ cell tumours. Br J Cancer. 2004;90:2397–401.
Venhoranta H, Pausch H, Wysocki M, Szczerbal I, Hänninen R, Taponen J, et al. Ectopic KIT copy number variation underlies impaired migration of primordial germ cells associated with gonadal hypoplasia in cattle Bos taurus. PLoS One. 2013;8:e75659.
Zhang W, Huang Y, Gunst SJ. The small GTPase RhoA regulates the contraction of smooth muscle tissues by catalyzing the assembly of cytoskeletal signaling complexes at membrane adhesion sites. J Biol Chem. 2012;287:33996–4008.
Hosgor M, Karaca I, Ozer E, Erdag G, Ulukus C, Fescekoglu O, et al. The role of smooth muscle cell differentiation in the mechanism of obliteration of processus vaginalis. J Pediatr Surg. 2004;39:1018–23.
van Veen RN, van Wessem KJP, Halm JA, Simons MP, Plaisier PW, Jeekel J, et al. Patent processus vaginalis in the adult as a risk factor for the occurrence of indirect inguinal hernia. Surg Endosc. 2007;21:202–5.
Tanyel FC. Obliteration of processus vaginalis: aberrations in the regulatory mechanism result in an inguinal hernia, hydrocele or undescended testis. Turk J Pediatr. 2004;46:18–27.
Barthold JS, McCahan SM, Singh AV, Knudsen TB, Si X, Campion L, et al. Altered expression of muscle and cytoskeleton related genes in a rat strain with inherited cryptorchidism. J Androl. 2008;29:352–66.
Schrijver I, Liu W, Brenn T, Furthmayr H, Francke U. Cysteine substitutions in epidermal growth factor-like domains of fibrillin-1: distinct effects on biochemical and clinical phenotypes. Am J Hum Genet. 1999;65:1007–20.
Allard S, Adin P, Gouedard L, di Clemente N, Josso N, Orgebin-Crist MC, et al. Molecular mechanisms of hormone-mediated Mullerian duct regression: involvement of beta-catenin. Development. 2000;127:3349–60.
Kubota Y, Temelcos C, Bathgate RAD, Smith KJ, Scott D, Zhao C, et al. The role of insulin 3, testosterone, Müllerian inhibiting substance and relaxin in rat gubernacular growth. Mol Hum Reprod. 2002;8:900–5.
Hutson JM, Davidson PM, Reece LA, Baker ML, Zhou BY. Failure of gubernacular development in the persistent müllerian duct syndrome allows herniation of the testes. Pediatr Surg Int. 1994;9:544–6.
Acknowledgements
We are grateful to Topigs Norsvin for providing the data needed to perform this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CAS conducted the statistical analysis, prepared figures and tables, and wrote the first draft of the manuscript. MSL participated in the design of the study, design of the selection of phenotypes, genotypes and pedigree information, was involved in discussions on statistical issues. BH conceived the study, participated in its design and coordination. EHATH participated in the design of the selection of phenotypes and was involved in discussions of the results. EFK conceived the study and participated in its coordination. JWMB participated in the design of the study, was involved in the discussion and evaluation of statistical issues. All authors read and approved the final manuscript.
Additional file
Additional file 1: Figure S1.
Quantile-quantile plots for cryptorchidism for the Large White (a) and Landrace datasets (b) and for hernia for the Large White (c) and Landrace datasets (d). Observed distribution of -log10(P-values) on the y-axis compared to the expected distribution of -log10(P-values) on the x-axis across the 38 632 and 39 508 SNPs for Large White and Landrace, respectively.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Sevillano, C.A., Lopes, M.S., Harlizius, B. et al. Genome-wide association study using deregressed breeding values for cryptorchidism and scrotal/inguinal hernia in two pig lines. Genet Sel Evol 47, 18 (2015). https://doi.org/10.1186/s12711-015-0096-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12711-015-0096-6