
Abstract
Background
The Chinese giant salamander Andrias davidianus is an important amphibian species in China because of its increasing economic value, protection status and special evolutionary position from aquatic to terrestrial animal. Its large genome presents challenges to genetic research. Genetic linkage mapping is an important tool for genome assembly and determination of phenotype-related loci.
Results
In this study, we constructed a high-density genetic linkage map using ddRAD sequencing technology to obtain SNP genotyping data of members from an full-sib family which sex had been determined. A total of 10,896 markers were grouped and oriented into 30 linkage groups, representing 30 chromosomes of A. davidianus. The genetic length of LGs ranged from 17.61 cM (LG30) to 280.81 cM (LG1), with a mean inter-locus distance ranging from 0.11(LG3) to 0.48 cM (LG26). The total genetic map length was 2643.10 cM with an average inter-locus distance of 0.24 cM. Three sex-related loci and four sex-related markers were found on LG6 and LG23, respectively.
Conclusion
We constructed the first High-density genetic linkage map and identified three sex-related loci in the Chinese giant salamander. Current results are expected to be a useful tool for future genomic studies aiming at the marker-assisted breeding of the species.
Similar content being viewed by others

Background
The Chinese giant salamander Andrias davidianus, historically widely distributed in China, is the world’s largest extant amphibian. Due to its habitat destruction and hunting by man, it has been classified as endangered by the International Union for Conservation of Nature and Nature Resources since the 1980s [1]. To conserve the wild resources of giant salamander, artificial breeding technology was developed and succeeded [2, 3]. Because of its palatability and nutritional value, the Chinese salamander is commercially cultured and amphibians are now allowed to reach the market [4]. It is considered an important species to study because scientists see it as a living fossil present on Earth since 350 million years ago and linking aquatic to terrestrial organisms, with complex sex determination and differentiation characteristics. Until now, most studies on A. davidianus focused on the immune mechanisms [5,6,7,8], and only few papers were reported for the sex-differentiation issues [9,10,11].
Previous studies reported that the Chinese giant salamander has a huge genome (about 50 Gb) which was a big challenge to assemble [12], and to this direction the construction of a genetic linkage map will be a valuable tool. In the previous study, Simple sequence repeats (SSR) was the mainly basic tools to generate a linkage map in various species. With the development of biotechnology, single nucleotide polymorphisms (SNP) has become the most popular tools to construct the high-density linkage maps, because of their widely distribution in the genome and development of next genomic sequence technology. Recently, high-density genetic linkage maps have become increasingly important and widespread in cultured aquatic species. In fish, genetic linkage maps have been reported in Salmo salar [13, 14], Cyprinus carpio [15], Paralichthys olivaceus [16], Megalobrama amblycephala [17], Larimichthys polyactis [18, 19], Oreochromis niloticus [20], Ictalurus punctatus [21], and Lates calcarifer [22], all of which possess relatively small genomes. In amphibian, genetic linkage maps have been constructed in several species including Rana temporaria [23], Hyla arborea [24], and Lissotriton newts [25]. However, high-density genetics maps are not yet available in amphibians.
Quantitative trait locus (QTL) mapping is an effective means to relate phenotype to the genome. In previous study, QTL mapping has been used in various species for different traits location including loci related to ecomic phenotype [26] and resistance to disease [27, 28]. The composite interval mapping (CIM) is a common method for mapping QTLs based on linkage map [29] and it was used to located binary trait such as sex [30] and metamorphosis-related traits loci in fish [31]. To identify the sex chromosome of A. davidianus without a genome, the CIM method was effective approach. In the previous study, we explored the female-specific marker to identify the genetic sex of the salamander which was important to detect the sex reversal salamander treated by high temperature and sex hormone in the study of sex determination mechanism [32, 33]. we also characterized sex related gene in the sex differentiation [34, 35]. Studies on the genetic level are necessary to identify sex related regions and ascertain the sex determination mechanism. In the present study, we used a single cultured A. davidianus full-sib family to construct a high-density linkage map based on the ddRAD sequencing. Using the constructed linkage map. We identified three sex-related loci in different linkage groups. These results provide an effective genetic tool for A. davidianus genome assembly and marker-assisted breeding.
Results
Library construction and sequencing
Ninety-seven RAD-seq libraries from two parents and 95 offspring were constructed and sequenced on an Illumina HiSeq X platform to generate raw reads. Based on the data trimming, 10.49 billion reads comprising ∼1516.07 Gb of sequencing data were individually partitioned into RAD tags according to their multiplex identifiers (GenBank accession No. SRP155453). Finally, female and male parent datasets which contained 210,000,000 and 190,000,000 clean reads, respectively, were correspondingly partitioned into 1,887,649 and 6,378,461 RAD tags. A total of 1458.71 Gb of data (mean 15.35 Gb per individual, 51 M reads pair per sample) were produced and divided into 302,909,245 RAD tags (ranging from 1,934,524 to 6,800,866 with a mean of 3,188,518.37) from the 95 offspring, for individual SNP discovery (Table S1).
SNP discovery and genotyping
RAD tags from each individual were clustered and compared, after stringent selection by the above-described method. A custom k-mer matching algorithm that excluded exact sequence matches (monomorphic loci) per 41 bp sequence was used to identify the SNP of the parents contained the RAD tags. A panel of 10,896 high-fidelity SNPs in both parents was identified, and alleles for each marker were assigned to their respective parent donor. The SNPs were analyzed across the 95 offspring with genotyping data and classified into three categories using an in-house script: maternal heterozygous (3343 SNPs), paternal heterozygous (6066 SNPs), and heterozygous in both (1487 SNPs) (Table S2).
High-density linkage map construction
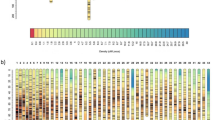
In the present study, Join-map and Lep-Map were used to construct the high-density genetics linkage map of A. davidianus. In both the maternal and paternal map, a total of 10,896 segregating SNPs were successfully classified into 30 linkage groups which was consistent with the karyotype 2n = 60 (Fig. 1). The maternal map contained 4830 SNPs with a total genetic distance of 2580.04 cM., The length of LGs ranged from 0 cM (LG27) to 298.26 cM (LG1) with an average genetic length of 86.00 cM (Table 1). Seven thousand five hundred fifty-three SNPs representing a total length of 2011.80 cM ranging from 0 cM (LG21, LG27, and LG30) to 244.33 cM (LG1) with an average genetic length of 67.06 cM were consisted the corresponding paternal map. The longest linkage group was LG1 which was 244.33 cM and 298.26 cM in maternal and paternal map, respectively. However, the shortest was 0 cM distributed in LG21, LG27, and LG30 in the paternal map and LG27 in the maternal map.

Integrated genetic linkage maps of Andreas davidianus comprising 10,896 markers assigned to 30 linkage groups (LG1–LG30). Genetic distances in Kosambi centiMorgans are listed on the left side of the linkage groups, and markers are listed on the right side of the linkage groups
Finally, the integrated map consisting of 30 LGs was constructed by 10,896 effective markers. The total length of the integrated map was 2643.10 cM. The average number of effective loci was 363 distributed in genetic distance of 88.10 cM with an average inter-locus distance of 0.24 cM. The genetic distance of LGs ranged from 17.61 cM (LG30) to 280.81 cM (LG1), and a mean inter-locus distance ranged from 0.11 (LG3) to 0.48 cM (LG26). The maximum density of SNP marker in linkage group was LG3 containing 878 effective loci and minimum density was LG30 containing 105 effective loci (Table S3).
Sex-related loci
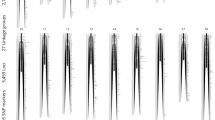
Sex-related loci of A. davidianus were detected using CIM on the maternal and paternal map by WinQTL cart 2.5. Total of 3 loci were found on the maternal map LG6 (qs-1, qs-2) and LG11(qs-3). The likelihood of odd (LOD) of the loci were 5.8, 4.3, and 3.1, which explained 29.37, 12.00, and 6.78% of the phenotypic variation, respectively. Genotyping results of SNP 97.nn_np_297384, 99.nn_np_294732, 99.nn_np_278109, and 90.hk_hk_96246 in the sex-related loci were correlated with phenotype by the single-marker analysis of the WinQTL cart 2.5 (Table 2, Fig. 2). Eight maternal SNPs were found in the sex-related loci qs-1 and qs-2. The correlation of the eight SNPs with phenotypic sex ranged from 57.81% (99.nn_np_278 109) to 68.18% (99.nn_np_282 501) (Table 3).

Sex-related loci of Andreas davidianus. The black horizontal line represents a linkage group-wise logarithm of odds significance threshold of 3.0
Discussion
Genetic linkage mapping is an important tool in genomic and genetic studies [36, 37]. Abundant molecular markers with an established order of arrangement provide an excellent resource for breeding research. Most amphibians possessed a large genome [38, 39], which exhibited a challenge in constructing a high-density genetic linkage map. Research exploring SNPs in A. davidianus has been reported, but identified markers were not used to build a linkage map [40]. Sessions et al. assembled thirty linkage groups in maternal, paternal, and integrated maps, showing consistency with the karyotype [41]. Andrias davidianus possesses a genome of ~ 50 G [12], hampering genomic and genetic study because of lack of large genome assembly technology and linkage map. The present study exhibited the first high-density genetic linkage map using SNPs of A. davidianus.
Thirty linkage groups were constructed with the average marker-interval of 0.24 cM, which was the highest density linkage map in amphibians. In common frog, 107 SSR markers was distributed in 15 linkage groups with total length 1698.8 cM [23]. In Lissotriton newts, total of 1146 markers was distributed in 12 linkage groups with an average marker interval of 1.29 cM [25]. The highest density linkage map of fish species was constructed for channel catfish, with the average marker interval of 0.22 cM [21]. Hence, the A. davidianus SNP linkage map displays high density even compared to fish species with relatively small genomes. In this study, we constructed 0 cM linkage groups in maternal and paternal map respectively. The main reason may be the specificity of A. davidianus genome. Some types of markers with different genotype distributed on different location in one chromosome. However, the other types of markers obtained the same genotype from same individual distributing on same location in linkage group (0 cM). These results indicated the LD differ in maternal and paternal linkage groups.
The high-density linkage map provided the means of fine mapping A. davidianus sex-related QTL. Three loci were identified on the maternal map LG6 (qs-1, qs-2) and LG11 (qs-3). The genetic distance of the QTL locus ranged from 0.1 cM to 3.5 cM suggested a narrow regions in the linkage group. This may provide valuable information for sex-related gene detection. While the same method was used to identify the loci on the maternal and paternal maps, all observed sex-related loci were on the maternal map. It is possible that some key sex-related markers were homozygous on the paternal map. Sex chromosomes have a greater number of repeat sequences, making the assembly of sex chromosomes and the construction of a sex linkage group more difficult, and sex is readily influenced by factors other than chromosome [42]. Some sex-related QTLs have been detected in other species, seven in the half-smooth tongue sole with five sex-linked markers [43], and 175 sex-related regions in turbot, most distributed among three LGs, most likely to be the sex chromosome [44]. In the present study, the major loci were detected only on linkage group LG6, providing evidence for the sex chromosome of A. davidianus. We analyzed sex related markers in this region and found correlation coefficient which ranged from 57.81 to 68.18% between phenotype and genotype in eight maternal SNP marker. Possibly due to low sequencing depth, some key SNPs in sex-related loci were not mapped on the linkage group. This region was considered as sex-related loci. So many SNPs in this region was related with sex too [17]. Verification could be carried out to confirm sex-linked genes and markers in A. davidianus through expanded single-linkage data in future. Further research requires expanded knowledge of the A. davidianus genome.
Conclusions
We constructed a high-density genetic linkage map using ddRAD sequencing technology to obtain SNP genotyping data from one full-sib family. A total of 10,896 markers were grouped and oriented into 30 linkage groups (LG), representing 30 chromosomes of A.davidianus. The genetic length of LGs ranged from 17.61 cM (LG30) to 280.81 cM (LG1), with a mean inter-locus distance ranging from 0.11(LG3) to 0.48 cM (LG26). The total genetic map length was 2643.10 cM with an average inter-locus distance of 0.24 cM. At last, we found three sex related loci on the maternal map LG6 (qs-1, qs-2) and LG11(qs-3). Genotyping results of four SNP markers (97.nn_np_297384, 99.nn_np_294732, 99.nn_np_278109, and 90.hk_hk_96246) in the sex-related loci were significantly correlated with phenotype which indicated the four markers were sex-related markers. We constructed a High-density genetic linkage map and mapped 3 sex-related loci, which provides the linkage map is a useful tool for genomic study and provides a genetic basis for marker-assisted breeding of Chinese giant salamander.
Methods
Mapping population
Total of 97 apparently healthy A. davidianus including two parents (8 year old) and 95 salamander larvae (body weight 34.24 ± 12.13 g; body length 17.85 ± 1. 76 cm) from one F3 full-sib family were collected from Zhejiang Yongqiang Chinese Giant Salamander Limited Company (Jinhua, Zhejiang province, China). The salamanders were soaked in 4 L MS222 with the concentration of 0.5 g/L for 10mins to anaesthetize. After all the salamanders were unconscious, the vertebra was broken from the neck, then the skin of the neck was removed, and muscle tissue was collected. All operations were carried out as per Yangyze River Fisheries Research Institute Care Committee (no. 2013001). The muscle tissue was collected from each salamander using the tissue DNA extracting kit (Omega, USA) to extract genomic DNA following manufacturer’s instruction. The concentration and quality of DNA were detected by Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) and 1% agarose gel electrophoresis. Genetic sex of each offspring was identified using a sex-specific marker explored by our lab [32].
RAD library construction, sequencing, and genotyping
The RAD library construction, sample indexing, and pooling followed the natural population [43, 45]. The high quality genomic DNA (250 ng) from each of the 97 individuals were used to construct libraries. The genomic DNA were digested by enzyme EcoRI and NIaIII (New England Biolabs) at 37 °C for 1 h and then at 65 °C for 30 min to inactivate the enzyme. Illumina HiSeq X on a total throughput of 24 lanes (~60Gb ddRAD data for each lane) were used to perform the Pair-end (150 bp) sequencing. Raw sequence reads were trimmed to 110 nucleotides from the 3′ end to ensured a quality value >Q30 in more than 98% of the nucleotides (equals 0.1% sequencing error). To produce unique candidate alleles for each RAD locus, Stacks [46] had been used to cluster the trimmed reads by sequence similarity. A maximum base-pair mismatch of one was allowed in this step for the genetic mapping population. Stacks were used under default parameters to collapse the RAD-tags into clusters for SNP calling. High-Quality and Bi-allelic SNPs were identified under following criteria which were 1) > =6 and < =100 reads for each loci, otherwise this loci will be considered as missing data, 2) present in at least 60% (< 39 genotyping-deletion rate) of the individuals.
Linkage map construction
RAD-based SNPs were first tested against the expected segregation ratio at the beginning of the linkage analysis. Markers were filtered out by a chi-square test for separation ratio at the intersection sites with P < 0.001. The linkage map was constructed using joinmap and Lep-Map. Joinmap was used to divide markers into groups using a grouping tree function in CP model with LOD = 8.0 [47] and Lep-MAP was used to determine marker order [48]. Kosambi mapping function was used to estimate of each linkage group distances.
Sex-related loci mapping
CIM method of WinQTL Cart 2.5 software was used to map the sex-related loci on the male and female linkage maps respectively [49]. Model six with three parameters as 10-cM window size, five control markers, and a step size of 1-cM was used to analyze the forward and backward stepwise regression. The location of each QTL was determined based on its LOD peak location and surrounding region with the threshold value of 3.0. Single-marker association tests with WinQTL Cart 2.5 was used to analysis the association between phenotype and genotype for each marker [49]. All maternal SNPs in the located sex-related region were analyzed to detect correlation with the phenotype to find sex-related SNPs.
Availability of data and materials
All data generated or analyzed during this study are included in this article and its supplementary information files. GenBank accession No. SRP155453.
Abbreviations
- LG:
-
Linkage group
- QTL:
-
Quantitative trait locus
- ddRAD:
-
Double digest restriction-site associated DNA
- SNP:
-
Single-nucleotide polymorphism
- LOD:
-
Likelihood of odd
- CIM:
-
Composite interval mapping
References
Zhou ZY, Geng Y, Liu XX, Ren SY, Zhou Y, Wang KY, et al. Characterization of a ranavirus isolated from the Chinese giant salamander ( Andrias davidianus, Blanchard, 1871) in China. Aquaculture. 2013;384:66–73.
Li HZ., Liu LX,, Liu XL.,Guo JS. Compared study on artificial insemination methods of Andrias davidianus. Guangdong Agric Sci,2013; 11: 128–129. [In Chinese].
Xiao H., Liu J., Yang Y., Lin X. Artificial propagation of tank-cultured Chinese giant salamander Andrias davidianus. Acta Hydrobiol Sin 2006; 30: 530–534. [In Chinese].
Zhang K., Wang X., Wu W., Wang ZH., Song H., et al., Advances in conservation biology of Chinese giant salamander. Biodivers Sci2002; 10: 291–297. [In Chinese].
Chen Z, Gui J, Gao X, Pei C, Hong YJ, Zhang QY. Genome architecture changes and major gene variations of Andrias davidianus ranavirus (ADRV). Vet Res. 2013;44(1):101. https://doi.org/10.1186/1297-9716-44-101.
Zhang Q, Gui JF. Virus genomes and virus-host interactions in aquaculture animals. Sci China Life Sci. 2015;58(2):156–69. https://doi.org/10.1007/s11427-015-4802-y.
Qi ZT, Zhang QH, Wang ZS, Ma TY, Zhou J, Holland JW, et al. Transcriptome analysis of the endangered Chinese giant salamander (Andrias davidianus): immune modulation in response to Aeromonas hydrophila infection. Vet Immunol Immunopathol. 2016;169:85–95. https://doi.org/10.1016/j.vetimm.2015.11.004.
Yang H, Lan QQ, Liu RR, Cui D, Liu HX, Xiong DM, et al. Characterization of galectin-1 from Chinese giant salamanders Andrias davidianus and its involvements during immune response. Dev Comp Immunol. 2017;70:59–68. https://doi.org/10.1016/j.dci.2017.01.004.
Hu QM, Tian HF, Li W, Meng Y, Wang QH, Xiao HB. Identification of critical sex-biased genes in Andrias davidianus by de novo transcriptome. Mol Gen Genomics. 2019;294(2):287–99. https://doi.org/10.1007/s00438-018-1508-4.
Hu QM, Meng Y, Wang D, Tian HF, Xiao HB. Characterization and function of the T-box 1 gene in Chinese giant salamander Andrias davidianus. Genomics. 2019;111(6):1351–9. https://doi.org/10.1016/j.ygeno.2018.09.007.
Chen R, Du J, Ma L, Wang LQ, Xie SS, Yang CM, et al. Comparative microRNAome analysis of the testis and ovary of the Chinese giant salamander. Reproduction. 2017;154(3):169–79. https://doi.org/10.1530/REP-17-0109.
Geng XF, Li WS, Shang HT, Gou Q, Zhang FC, Zang XY, et al. A reference gene set construction using RNA-seq of multiple tissues of Chinese giant salamander, Andrias davidianus. GigaScience. 2017;6(3):1–7. https://doi.org/10.1093/gigascience/gix006.
Lien S, Gidskehaug L, Moen T, Hayes BJ, Berg PR, Davidson WS, et al. A dense SNP-based linkage map for Atlantic salmon (Salmo salar) reveals extended chromosome homeologies and striking differences in sex-specific recombination patterns. BMC Genomics. 2011;12(1):615. https://doi.org/10.1186/1471-2164-12-615.
Tsai HY, Robledo D, Lowe NR, Bekaert M, Taggart JB, Bron JE, et al. Construction and Annotation of a High Density SNP Linkage Map of the Atlantic Salmon (Salmo salar) Genome. G3 (Bethesda). 2016;6:2173–9.
Peng W, Xu J, Zhang Y, Feng J, Dong C, Jiang L, et al. An ultra-high density linkage map and QTL mapping for sex and growth-related traits of common carp (Cyprinus carpio). Sci Rep. 2016;6(1):26693. https://doi.org/10.1038/srep26693.
Shao C, Niu Y, Rastas P, Liu Y, Xie Z, Li H, et al. Genome-wide SNP identification for the construction of a high-resolution genetic map of Japanese flounder (Paralichthys olivaceus): applications to QTL mapping of Vibrio anguillarum disease resistance and comparative genomic analysis. DNA Res. 2015;22(2):161–70. https://doi.org/10.1093/dnares/dsv001.
Wan SM, Liu H, Zhao BW, Nie CH, Wang WM, Gao ZX. Construction of a high-density linkage map and fine mapping of QTLs for growth and gonad related traits in blunt snout bream. Sci Rep. 2017;7(1):46509. https://doi.org/10.1038/srep46509.
Ao J, Li J, You X, Mu Y, Ding Y, Mao K, et al. Construction of the high-density genetic linkage map and chromosome map of large yellow croaker (Larimichthys crocea). Int J Mol Sci. 2015;16(11):26237–48. https://doi.org/10.3390/ijms161125951.
Kong S, Ke Q, Chen L, Zhou Z, Pu F, Zhao J, et al. Constructing a high-density genetic linkage map for large Yellow Croaker (Larimichthys crocea) and mapping resistance trait against Ciliate Parasite Cryptocaryon irritans. Mar Biotechnol (NY). 2019;21:262–75.
Joshi R, Árnyasi M, Lien S, Gjøen HM, Alvarez AT, Kent M. Development and validation of 58K SNP-array and high-density linkage map in Nile tilapia (O. niloticus). Front Genet. 2018;9:472.
Li Y, Liu S, Qin Z, Waldbieser G, Wang R, Sun L, et al. Construction of a high-density, high-resolution genetic map and its integration with BAC-based physical map in channel catfish. DNA Res. 2014;22:39–52.
Liu P., Wang L., Wong SM., , Yue GH. Fine mapping QTL for resistance to VNN disease using a high-density linkage map in Asian seabass. Sci Rep2016; 24: 32122.
Cano JM, Li MH, Laurila A, Vilkki J, Merilä J. First-generation linkage map for the common frog Rana temporaria reveals sex-linkage group. Heredity. 2011;107(6):530–6. https://doi.org/10.1038/hdy.2011.39.
Dufresnes C, Brelsford A, Perrin N. First-generation linkage map for the European tree frog (Hyla arborea) with utility in congeneric species. BMC Res Notes. 2014;26:850.
Niedzicka M, Dudek K, Fijarczyk A, Zieliński P, Babik W. Linkage map of lissotriton newts provides insight into the genetic basis of reproductive isolation. G3 (Bethesda). 2017;5:2115–24.
Besnier F, Glover KA, Lien S, Kent M, Hansen MM, Shen X, et al. Identification of quantitative genetic components of fitness variation in farmed, hybrid and native salmon in the wild. Heredity. 2015;115(1):47–55. https://doi.org/10.1038/hdy.2015.15.
Gonen S, Baranski M, Thorland I, Norris A, Grove H, Arnesen P, et al. Mapping and validation of a major QTL affecting resistance to pancreas disease (salmonid alphavirus) in Atlantic salmon (Salmo salar). Heredity. 2015;115(5):405–14. https://doi.org/10.1038/hdy.2015.37.
Geng X, Sha J, Liu S, Bao L, Zhang J, Wang R, et al. A genome-wide association study in catfish reveals the presence of functional hubs of related genes within QTLs for columnaris disease resistance. BMC Genomics. 2015;17:196.
Zeng Z. Precision mapping of quantitative trait loci. Genetics. 1994;136(4):1457–68. https://doi.org/10.1093/genetics/136.4.1457.
Song W, Li Y, Zhao Y, Liu Y, Niu Y, Pang R, et al. Construction of a high-density microsatellite genetic linkage map and mapping of sexual and growth-related traits in half-smooth tongue sole (Cynoglossus semilaevis). PLoS One. 2012;7(12):e52097. https://doi.org/10.1371/journal.pone.0052097.
Liu Y, Wei M, Guo H, Shao C, Meng L, Xu W, et al. Locus mapping, molecular cloning, and expression analysis of rps6kb2, a novel metamorphosis-related gene in Chinese Tongue Sole (Cynoglossus semilaevis). Mar Biotechnol (NY). 2017;19:497–516.
Hu Q, Chang C, Wang Q, Tian H, Qiao Z, Wang L, et al. Genome-wide RAD sequencing to identify a sex-specific marker in Chinese giant salamander Andrias davidianus. BMC Genomics. 2019a;23:415.
Hu Q, Tian H, Xiao H. Effects of temperature and sex steroids on sex ratio, growth, and growth-related gene expression in the Chinese giant salamander Andrias davidianus. Aquat Biol. 2019b;28:79–90. https://doi.org/10.3354/ab00710.
Hu Q, Meng Y, Tian H, Zhang Y, Xiao H. Sexually dimorphic expression of Foxl2 and Ftz-F1 in Chinese Giant salamander Andrias Davidianus. J Exp Zool B Mol Dev Evol. 2016;326(6):363–74. https://doi.org/10.1002/jez.b.22693.
Hu Q, Xiao H, Wang Q, Tian H, Meng Y. Identification and expression of forkhead box genes in the Chinese giant salamander Andrias davidianus. Reprod Fertil Dev. 2017;30:634–42.
Wang Y, Sun SL, Liu B, Wang H, Deng J, Liao YC, et al. A sequence-based genetic linkage map as a reference for Brassica rapa pseudochromosome assembly. BMC Genomics. 2011;12(1):239. https://doi.org/10.1186/1471-2164-12-239.
Zheng X, Kuang Y, Zhang X, Lu C, Cao D, Li C, et al. A genetic linkage map and comparative genome analysis of common carp (Cyprinus carpio L.) using microsatellites and SNPs. Mol Gen Genomics. 2011;286(3-4):261–77. https://doi.org/10.1007/s00438-011-0644-x.
Roth G, Nishikawa KC, Wake DB. Genome size, secondary simplification, and the evolution of the brain in salamanders. Brain Behav Evol. 1997;50(1):50–9. https://doi.org/10.1159/000113321.
Smith JJ, Timoshevskaya N, Timoshevskiy VA, Keinath MC, Hardy D, Voss SR. A chromosome-scale assembly of the axolotl genome. Genome Res. 2019;29(2):317–24. https://doi.org/10.1101/gr.241901.118.
Yan F, Lü J, Zhang B, Yuan Z, Zhao H, Huang S, et al. The Chinese giant salamander exemplifies the hidden extinction of cryptic species. Curr Biol. 2019;21:R590–2.
Sessions SK, Leon PE, Kezer J. Cytogenetics of the chinese giant salamander, Andrias davidianus (Blanchard): the evolutionary significance of cryptobranchoid karyotypes. Chromosoma. 1982;86(3):341–57. https://doi.org/10.1007/BF00292262.
Li XY, Gui JF. Diverse and variable sex determination mechanisms in vertebrates. Sci China Life Sci. 2018;61(12):1503–14. https://doi.org/10.1007/s11427-018-9415-7.
Recknagel H, Elmer KR, Meyer A. A Hybrid genetic linkage map of two ecologically and morphologically divergent midas cichlid fishes (Amphilophus spp.) obtained by massively parallel DNA sequencing (ddRADSeq). G3 (Bethesda). 2008;3:65–74.
Wang W., Hu Y., Ma Y., Xu L, Guan J, Kong J. High-Density Genetic Linkage Mapping in Turbot (Scophthalmus maximus L.) Based on SNP Markers and Major Sex- and Growth-Related Regions Detection. PloS One. 2015; 10: e0120410.a.
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One. 2012;7:e37135.
Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks: Building and Genotyping Loci De Novo from Short-Read Sequences. G3 (Bethesda). 2011;1(3):171–82.
Van Ooijen JW. Multipoint maximum likelihood mapping in a full-sib family of an out breeding species. Genet Res. 2011;93(5):343–9. https://doi.org/10.1017/S0016672311000279.
Rastas P, Paulin L, Hanski I, Lehtonen R, Auvinen P. Lep-MAP: fast and accurate linkage MAP construction for large SNP datasets. Bioinformatics. 2013;15:3128–34.
Wang S, Basten CJ, Zeng ZB. Windows QTL cartographer 2.5. Department of Statistics. Raleigh: North Carolina State University; 2007.
Acknowledgements
We acknowledge the assistance for data analysis by LinEn laboratory.
Funding
This work was supported by National Nature Science Foundation of China (31502155); Key Laboratory of Freshwater Aquatic Biotechnology and Breeding, Ministry of Agriculture, Heilongjiang Fisheries Research Institute, Chinese Academy of Fishery Sciences (KF-2017-06). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
QMH conceived and designed the experiments; QMH, YL, XLL, XSJ, HBX analyzed the data; HFT, JJZ, QMH collected the samples and performed the experiments; QMH and YL wrote the paper. All authors reviewed the manuscript and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The owners granted informed consent by written. Handling and husbandry of salamander and other relevant protocols used in this study were approved by Yangtze River Fisheries Research Institute Care Committee protocols 2013001. The methods used in this study were performed in strict accordance with the Laboratory Animal Management Principles of China.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. Original data of each sample for RAD sequencing
Additional file 2: Table S2
. Genotyping information and location of each SNP on the 30 LGs
Additional file 3: Table S3
Distribution of each SNP on the 30 LGs
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hu, Q., Liu, Y., Liao, X. et al. A high-density genetic map construction and sex-related loci identification in Chinese Giant salamander. BMC Genomics 22, 230 (2021). https://doi.org/10.1186/s12864-021-07550-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07550-0