
Abstract
Background
Barnacles are specialized marine organisms that differ from other crustaceans in possession of a calcareous shell, which is attached to submerged surfaces. Barnacles have a wide distribution, mostly in the intertidal zone and shallow waters, but a few species inhabit the deep-sea floor. It is of interest to investigate how such sessile crustaceans became adapted to extreme deep-sea environments. We sequenced the transcriptomes of a deep-sea barnacle, Glyptelasma gigas collected at a depth of 731 m from the northern area of the Zhongjiannan Basin, and a shallow-water coordinal relative, Octolasmis warwicki. The purpose of this study was to provide genetic resources for investigating adaptation mechanisms of deep-sea barnacles.
Results
Totals of 62,470 and 51,585 unigenes were assembled for G. gigas and O. warwicki, respectively, and functional annotation of these unigenes was made using public databases. Comparison of the protein-coding genes between the deep- and shallow-water barnacles, and with those of four other shallow-water crustaceans, revealed 26 gene families that had experienced significant expansion in G. gigas. Functional annotation showed that these expanded genes were predominately related to DNA repair, signal transduction and carbohydrate metabolism. Base substitution analysis on the 11,611 single-copy orthologs between G. gigas and O. warwicki indicated that 25 of them were distinctly positive selected in the deep-sea barnacle, including genes related to transcription, DNA repair, ligand binding, ion channels and energy metabolism, potentially indicating their importance for survival of G. gigas in the deep-sea environment.
Conclusions
The barnacle G. gigas has adopted strategies of expansion of specific gene families and of positive selection of key genes to counteract the negative effects of high hydrostatic pressure, hypoxia, low temperature and food limitation on the deep-sea floor. These expanded gene families and genes under positive selection would tend to enhance the capacities of G. gigas for signal transduction, genetic information processing and energy metabolism, and facilitate networks for perceiving and responding physiologically to the environmental conditions in deep-sea habitats. In short, our results provide genomic evidence relating to deep-sea adaptation of G. gigas, which provide a basis for further biological studies of sessile crustaceans in the deep sea.
Similar content being viewed by others


Background
Conditions on the deep-sea floor are poorly known but generally are considered too harsh for the survival of most organisms, e.g., high hydrostatic pressure, darkness, hypoxia, low temperature, and limited food availability [1,2,3,4,5]. However, a macrofauna consisting of a growing range of newly discovered animals adapted to deep-sea habitats has been reported, including crustaceans [6,7,8], polychaetes [9, 10], fishes [11, 12], and mollusks [13, 14]. Various mechanisms have adapted them for survival in deep-sea environments: e.g., squat lobsters and mussels have developed chemoautotrophic systems of symbiotic bacteria for inhabiting hydrothermal vents and cold seeps in the seafloor [15,16,17]; and snailfish have evolved special morphological and physiological characters to survive and thrive in the hadal zone [12]. Studies aimed at understanding survival strategies and adaptive evolution of organisms living in deep seas have also employed genomic or transcriptomic sequencing. For example, in the amphipod Hirondellea gigas, adaptation to the hadal environment is associated with gene family expansion and amino acid substitutions of specific proteins [6]; and the shrimp Rimicaris sp. upregulates genes associated with sulfur metabolism and detoxification to survive in deep-sea hydrothermal vent environments [8]. However, our understanding of deep-sea adaptation mechanisms remains incomplete, especially for sessile species. Although next-generation sequencing technology is now highly developed, and a few transcriptomic analyses of bio-adhesion mechanisms and development have been reported [18,19,20]. Merely genetic resources of adult barnacles were surveyed in Cirripedia except for Pollicipes pollicipes (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA394196) and Neolepas marisindica [21], none relate to adaptive mechanisms in barnacles.
Thoracica barnacles are a unique group of marine crustaceans, enclosed by a mantle and calcareous plates, whose adults are permanently attached to the substrate. Thoracica barnacles are well-known to the public as fouling organisms, adhering to many artificial structures, including vessels and submarine cables, causing structural damage, and increasing fuel consumption. Simultaneously, they are ecologically and economically important species and have been the focus of many studies in developmental biology, crustacean evolution, and ecotoxicology [22,23,24]. Most barnacles inhabit shallow or tidal marine waters [25, 26] but a few occur in deeper water, even in hadal zones and around hydrothermal vents or cold seeps [27,28,29,30,31]. Stalked barnacles in the family Poecilasmatidae are distributed from the shallow subtidal zone to depths > 3600 m [32]. Within this family, the genus Glyptelasma is a typical deep-sea inhabitant with G. gigas distributed in the Indo-West Pacific at depths ranging from 236 m to 1092 m. Conveniently, a coordinal shallow-water species, Octolasmis warwicki, is distributed in a neighboring region at depths < 100 m [32]. Recently, we successfully collected G. gigas and O. warwicki individuals at Zhongjiannan Basin and Weizhou Island respectively, and were able to investigate deep-sea adaptation mechanisms through transcriptome sequencing. Zhongjiannan Basin is a Cenozoic sedimentary basin located in the narrowest part of the South China Sea shelf with depths extending down to 4000 m [33, 34]. In the northern area of Zhongjiannan Basin, specific geological structures, including mud volcanoes and pockmarks, are common on the seafloor [35, 36]. Comparing genetic information between these two species could help explain how G. gigas has migrated and adapted to this complex environment.
In this study, we sequenced the transcriptomes of the deep-sea barnacle G. gigas and a shallow-water barnacle O. warwicki. Comparative transcriptomics analysis was performed on them to investigate the genetic changes associated with adaptation to the deep-sea habitat. The main objective of our project was to provide a genomic resource for deep-sea barnacles and to probe the genetic strategies and adaptation of G. gigas to the severe conditions of deep-sea environments.
Results
Profile of transcriptome assembly and annotation
A total of 120,824,422 and 122,043,504 raw reads were generated from G. gigas and O. warwicki, respectively (Additional file 1: Table S1). After transcriptome assembly, there were 62,470 unigenes with an N50 length of 1708 bp for G. gigas. For O. warwicki, there were 51,585 unigenes with an N50 length of 2383 bp (Table 1). The quality of two assemblies was comparable or better than that of Pollicipes pollicipes (N50 length of 849 bp) and Neolepas marisindica (N50 length of 1596 bp). The two transcriptome assemblies were found to be highly complete including more than 95% of the core genes in the two species. Furthermore, 77.30 and 80.21% of the benchmarking universal single-copy orthologs (BUSCOs) were complete and single-copied in G. gigas and O. warwicki, respectively (Additional file 2: Table S2).
Among these unigenes, 25,627 (41.09%) and 21,143 (40.98%) were annotated in G. gigas and O. warwicki, respectively (Table 1). Generally, the distribution of unigenes in GO and KEGG classifications were similar between the two barnacle species (R2 = 0.9954), suggesting similar genetic structures. The unigenes were mainly assigned to the GO terms: single-organism process (GO:0044699); binding (GO:0005488); organelle (GO:0043226); metabolic process (GO:0008152) (Fig. 1a); and pathways related to signal transduction, translation, and the endocrine system (Fig. 1b). However, some differences were detected between the species. Relatively more unigenes of O. warwicki were distributed in the GO terms of regulation of biological process (GO:0050789) and regulation of metabolic process (GO:0019222), whereas in G. gigas, relatively more unigenes were distributed in nucleotide binding (GO:0000166) and transferase activity (GO:0016740) (Fisher’s exact test p < 0.05). These results were consistent with the findings of KEGG enrichment analysis, which indicated that G. gigas had relatively more unigenes with the functions of nucleotide metabolism and transcription (Fig. 1) (Fisher’s exact test p < 0.05).

GO (a) and KEGG (b) distributions of the unigenes of two barnacle species
Phylogenetic tree
A total of 24,204 gene families were identified in the comparative analysis of the six crustaceans. Among these gene families, all of the protein-coding genes of G. gigas and O. warwicki were distributed among 10,882 and 9904 gene families, respectively. A set of 566 single-copy gene families (370,657 amino acids) was selected for phylogenetic tree construction. The support values were mostly near 100% on each branch, suggesting high consensus (Fig. 2).

Maximum-likelihood phylogenetic tree based on the single-copy orthologs shared by the both barnacles as well as sequences of Branchiopoda Daphnia pulex, copepod Eurytemora affinis, and Malacostraca Parhyale hawaniensis and Litopenaeus vannamei. The estimated divergence times are displayed below the phylogenetic tree
Gene family expansion
Among the 24,204 gene families, 108 showed distinct expansion in both G. gigas and O. warwicki. Functional enrichment analysis indicated that these expanded gene families were particularly enriched in pathways of dorso-ventral axis formation, amino-acid biosynthesis and metabolism, and vascular smooth-muscle contraction (Additional file 3: Table S3). This pattern may be related to the unique morphological developmental and life pattern of barnacles, such as well-developed presoma, vestigial abdomen and sessile habit of the adult. Specifically, the major expanded gene family involved broad-complex core protein (Br-C) in the dorso-ventral axis formation pathway. Br-C is required for puffing and transcription of salivary gland late genes during metamorphosis, and is closely related to larval development in insects [37]. In G. gigas and O. warwicki, 42 and 36 Br-C genes, respectively, were detected. Phylogenetic analysis suggested that these Br-C genes had undergone lineage-specific expansion rather than species-specific expansion, with their distributions nested on the phylogenetic tree (Additional file 7: Fig. S1). Thus, it appears that these clusters of Br-C genes became expanded in the ancestor of G. gigas and O. warwicki and may have contributed to the adaptive evolution of barnacles.
In G. gigas, 26 species-specific expanded gene families were identified (Additional file 4: Table S4). These genes were mainly enriched in the pathways of focal adhesion (ko04510), ECM-receptor interaction (ko04512), PI3K-Akt signaling (ko04151), glycosaminoglycan biosynthesis-chondroitin sulfate (ko00532), hippo signaling (ko04391), and axon guidance (ko04360) (Table 2). In our results, tenascin was one of the major expanded gene families which are involved in focal adhesion, ECM-receptor interaction, and the PI3K-Akt signaling pathway. Phylogenetic analysis suggested that tenascin genes in G. gigas might have undergone expansion at least twice (Fig. 3). Tenascins are multimeric glycoproteins in the extracellular matrix (ECM) that play key functions in neuronal development, signaling, cell regulation, and axon growth and regeneration [38].

Phylogenetic tree of tenascin gene family. Bootstrap values (> 50%) are shown at branch nodes. ggi: Glyptelasma gigas, owa: Octolasmis warwicki, Dpul: Daphnia pulex, Eaff: Eurytemora affinis, LVAN: Litopenaeus vannamei, phaw: Parhyale hawaniensis
N-acetylgalactosamine-4-sulfate-6-O-sulfotransferase (CHST15), protocadherin fat 4/16/23 (FAT4/16/23), and plexin are three markedly expanded gene families involved in glycosaminoglycan biosynthesis-chondroitin sulfate, the hippo signaling pathway, and axon guidance, respectively. Furthermore, functional annotation revealed that the species-specific expanded gene families also contained X-ray repair cross-complementing protein 4 (XRCC4), very short patch repair (VSR) endonuclease, the nine-cysteines domain of family 3 (NCD3), and trehalose phosphatase (Table 3).
Positively selected genes
Genes under positive selection usually respond to natural selection. To identify positively selected genes, we collected 11,611 pairwise best-hit orthologs between G. gigas and O. warwicki, and performed adaptive evolutionary analyses on them. Our results identified 25 orthologs with ω values > 1.0 (Additional file 5: Table S5) and 118 with ω values > 0.5. These positively selected genes were mainly enriched in the pathways of focal adhesion (ko04510), cyanoamino acid metabolism (ko00460), and RNA transport (ko03013) (Additional file 6: Table S6). Specifically, transcription factor IIA and translation initiation factor eIF-2B were the two positively selected genes involved in RNA transport. Also identified as positively selected were genes encoding excision repair cross-complementation group 4 (ERCC4), calcitonin receptor-like protein, anoctamin-8, G protein-coupled receptor 125 (GPCR 125), discoidin domain receptor 2 (DDR2), WD domain, neurotransmitter-gated ion-channel transmembrane domain, and galactosyltransferase (Table 4).
Differential gene expression between G. gigas and O. warwicki
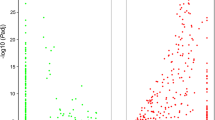
Generally, orthologous genes between two barnacle species should show similar expression patterns, but there may also be a subset of genes specifically highly expressed in G. gigas that were responsible for deep-sea adaptation. Thus, we calculated the relative expression levels (FPKM) of these orthologs to identify the genes that were highly expressed specifically in G. gigas and O. warwicki. As expected, the expression levels of these orthologs were generally similar between the two barnacle species. However, 480 genes in G. gigas and 791 genes in O. warwicki showed significantly higher expression levels relatively (Fig. 4). Functional enrichment analysis indicated that the highly expressed genes in G. gigas were strongly enriched in the GO terms of binding, especially in metal ion binding (GO:0043167) and protein binding (GO:0005515), and were also enriched in the pathways of galactose metabolism (ko00052), glycosphingolipid biosynthesis (ko00601), glycosaminoglycan biosynthesis (ko map00532), and ubiquitin-mediated proteolysis (ko04120) (p < 0.01) (Additional file 8: Fig. S2).

Differentially expressed genes in two barnacle species. The scarter plot in the left is the relative expression level of the orthologs between two barnacle species. The green dots indicated the genes showed highly expression in O. warwicki, while the red dots indicated the genes that highly expressed in Glyptelasma gigas. The right plot indicated the enriched GO terms of the highly expressed gene in Glyptelasma gigas
Discussion
Many potential mechanisms of adaptation to deep-sea environments have been identified. Lan et al. suggested that the expansion of cold-inducible proteins as well as zinc finger domains and positively selected genes related to β-alanine biosynthesis, energy metabolism and genetic information processing played important roles in adaptation to the hadal environment in the amphipod Hirondellea gigas [6]. Zhang et al. reported that the expression of the genes associated with sulfur metabolism and detoxification were upregulated in a deep-sea hydrothermal vent shrimp Rimicaris sp. [8]. In contrast to those free-swimming species in the deep sea, barnacles are confined to rather narrow zones, which would make adaptation to the deep-sea environment more difficult. In the phylogenetic (Fig. 2), G. gigas and O. warwicki formed a monophyletic clade that was highly divergent from other crustaceans. The time of divergence of the two barnacle species was estimated to be about 63 million years ago, which is consistent with the divergence of poecilasmatid barnacles [39, 40]. This divergence occurred after the Cretaceous–Palaeogene mass extinction, which was followed by an explosive radiation of organisms [41]. And this time is nearly contemporaneous with the early basement-forming stage of Zhongjiannan Basin [35, 42], suggesting that G. gigas was an invasive species in this basin. It has been proposed that deep-water barnacles originated from shallow waters [40, 43], and comparison with shallow-water barnacles might help explain how deep-sea barnacles have adapted to the harsh conditions on the deep-sea floor which characterized by high hydrostatic pressure, darkness, hypoxia, low temperature, and limited food availability.
Key KEGG pathways implicated in deep-sea adaptation
Our data indicate that focal-adhesion genes were specifically expanded and positively selected in G. gigas (Table 2, Additional file 6: Table S6), which suggests that changes in adhesion may have been involved in the adaption of G. gigas. For sessile organisms, adhesion is an important process in settlement and survival. In cell biology, focal adhesions are large macromolecular assemblies through which mechanical force and regulatory signals are transmitted between the ECM and an interacting cell. Focal adhesions lead cells to communicate and adhere with their extracellular matrix, and play essential roles in biological processes including cell motility, proliferation, differentiation, gene expression regulation and signal transmission [44]. Other major pathways with specifically expanded genes enriched in G. gigas included ECM-receptor interaction, the PI3K-Akt signaling pathway, glycosaminoglycan biosynthesis-chondroitin sulfate, the hippo signaling pathway, and axon guidance (Table 2). Functionally, ECM-receptor interaction, the PI3K-Akt and hippo signaling pathways are three key processes that participate in processing of environmental information [45,46,47]. Axon guidance represents a key stage in the formation of neuronal networks [48]. Glycosaminoglycan biosynthesis is engaged in glycan metabolic pathways whose products are also mediators of intercellular communication, cellular adhesion, and ECM maintenance [49]. In the meanwhile, cyanoamino acid metabolism and RNA transport were the other two pathways with positively selected genes enriched in G. gigas (Additional file 6: Table S6), and they two have roles in amino-acid metabolism and genetic information processing, respectively [50, 51]. Based on these observations, we speculate that G. gigas has developed an efficient and synergistic network for environmental perception (ECM-receptor interaction, PI3K-Akt signaling and hippo signaling pathways), Signal transmission (focal adhesion and axon guidance pathways) and physiological response (RNA transport, cyanoamino acid metabolism and glycosaminoglycan biosynthesis pathways). And this network finally results in functional and physiological adjustments that assist G. gigas in surviving in the severe and complex deep-sea environments. However, more research is needed to confirm this proposed network.
Key genes implicated in deep-sea adaptation
Specific genes that were expanded or positively selected in G. gigas could facilitate survival in the deep-sea environment. High hydrostatic pressure, hypoxia and low temperature, which characterize the deep-sea floor, could cause DNA damage [52,53,54,55] and result in mortality. XRCC4 is one of several break-repair and V(D)J recombination proteins, which could repair DNA double-strand breaks [56]; VSR is an essential component of the very short patch mismatch repair endonucleas, which specifically recognizes and exhibits strand-specific nicking at T-G deoxyribonucleic acid mismatches [57]; and DNA excision repair protein ERCC4 participates in nucleotide excision repair and DNA recombination [58]. In our study, XRCC4 and VSR genes were significantly expanded in G. gigas (Table 3), while ERCC4 gene was positively selected (Table 4). All of these genetic processing genes would ensure the structural integrity and normal function of DNA, which might be damaged in the deep-sea environment.
Low temperature and high hydrostatic pressure also lead to the depression of ligand binding and ion channel function in organisms [59,60,61,62], which would decrease the efficiency of signal transduction. To counteract the negative effects of low temperature and high hydrostatic pressure on signal transduction, genes encoding ligands and receptors were expanded or positively selected in G. gigas. For example, the expanded NCD3 genes encode the nine-cysteines domain of family 3 (Table 3), which is a G protein-coupled receptor (GPCR). The calcitonin receptor-like protein gene and GPCR 125 gene were also positively selected (Table 4). GPCRs constitute a large protein family with essential nodes in signal transduction between the interior and exterior of cells. They bind various ligands, including hormones, neurotransmitters, ions, and other stimuli [63,64,65]. Genes related to ion channel proteins were also positively selected, e.g., the anoctamin-8 and neurotransmitter-gated ion-channel transmembrane domain (Table 4). The former is a key tether protein that helps Ca2+ across membrane transport and assembles all core Ca2+-signaling proteins at the endoplasmic reticulum and plasma membrane junctions [66]; the latter is a key domain of ion channels that allows ions, such as Na+, K+, Ca2+, and/or Cl− to pass through the membrane when binding a neurotransmitter [67]. Jointly, the expanded and positively selected of genes concerned with ligand binding and ion channel proteins would help G. gigas maintain signal transmission in the deep-sea environment.
Compared with shallow waters, food availability is limited in the deep-sea, which may have encouraged evolution of more efficient energy metabolism [6, 7]. Among the orthologous genes of the two barnacle species, those related to carbohydrate metabolism showed relatively higher expression in G. gigas than in O. warwicki, including the genes from the pathways of galactose metabolism, glycosphingolipid biosynthesis, and glycosaminoglycan biosynthesis (Fig. 4, Additional file 8: Fig. S2). Accordingly, our results suggested that three key genes that participate in energy metabolic processes, including glycometabolism and lipometabolism, were expanded or positively selected. Trehalose is present in high concentration in insect hemolymph and is consumed during flight [68]. And this sugar has been shown be involved in low-temperature resistance in Escherichia coli [69]. Trehalose-phosphatase (Table 3) is used to hydrolyze trehalose-6-phosphate which could be directly converted to glucose and participate in glycolysis. We speculate that trehalose represents a form of energy storage in G. gigas, as in insects [70], but more evidence is needed to confirm this conjecture. Galactosyltransferase (Table 4) is a key protein acting in the biosynthesis of disaccharides, oligosaccharides and polysaccharides [71]. DDR2 (Table 4) is a receptor tyrosine kinase activated by collagens, it has diverse functions in cell proliferation, adhesion, migration, extracellular matrix remodeling and reproduction. Remarkably, evidence suggests that it can promote lipid metabolism, although the mechanism is unclear [72]. Corporately, the highly expressed genes, expanded gene families and genes under positive selection involved in energy metabolism may help G. gigas use energy efficiently in the harsh conditions of the deep-sea floor.
Conclusions
The present study is the first to report the transcriptome of a deep-sea barnacle that is compared with that of a shallow-water coordinal species. Our data indicate that G. gigas and O. warwicki diverged about 63 million years ago, and that G. gigas was an invasive species of the Zhongjiannan Basin. By specific gene-family expansion and positive selection of key genes, the deep-sea barnacle G. gigas probably evolved an efficient network concerned with environmental perception and physiological response, and acquired adaptive abilities in neural signal transduction, genetic information processing, and energy metabolism. All of these genetic strategies would facilitate confrontation of stress factors and survival in the severe environment of the deep-sea floor. Nevertheless, the present results are preliminary, and the evolutionary mechanisms and precise functional roles of the amplified genes and positively selected genes in genetic adaptation to the deep-sea environment require further confirmation and investigation. This work provides a genomic resource and clues to the genetic adaptation of a deep-sea barnacle that will be helpful for future studies on deep-sea invertebrates.
Methods
Sample collection, RNA extraction and sequencing
Specimens of G. gigas were collected from the northern area of Zhongjiannan Basin (15°19.17′N, 110°37.84′E, depth ~ 731 m) in May 2018, during a scientific cruise of the manned submersible Shenhaiyongshi. Large numbers of G. gigas attached to a limb of a gorgonian coral were acquired (Fig. 5a, b, c). On board, the specimens were immediately frozen in liquid nitrogen and stored at − 80 °C. Specimens of O. warwicki were collected from the nearshore waters of Weizhou Island, South China Sea (20°53.95′N, 109°0.61′E, depth ~ 3 m), using a fishing net from a fishing-boat in July 2018. Several specimens of O. warwicki attached to the carapace of a crab (Fig. 5a, d) were obtained. After collection, they were immediately immersed in RNAlater solution (Takara, Tokyo, Japan) and stored at − 80 °C.

Location of the sampling site and in situ photos of barnacles. a Location of the sampling site. b, c Glyptelasma gigas attached to the limb of a gorgonian coral. d Octolasmis warwicki attached on the carapace of a crab. The base map (a) is created by ArcGIS 10 (ESRI, Redlands, CA). Photographs b and c were taken by the sixth author XZL, and photograph d was taken by the third author XML, the map and pictures belong to the authors
Three adult individuals randomly selected from each species were pooled to provide sufficient RNA for transcriptome sequencing, a total of six specimens were used for the present research. TRIzol kit (Invitrogen, Carlsbad, CA) was used to extract total RNA following the manufacturer’s instructions. RNA quality was examined by 1% agarose gel electrophoresis, RNA purity was checked using the NanoPhotometer® spectrophotometer (IMPLEN, Westlake Village, CA), RNA concentration was measured using Qubit® RNA Assay Kit in Qubit® 2.0 Fluorimeter (Life Technologies, Carlsbad, CA), and RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, Palo Alto, CA). From each sample, 1.5 μg of RNA was prepared for sequencing. Sequencing libraries were generated using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA) following the manufacturer’s instructions and sequenced on an Illumina® HiSeq platform (San Diego, CA). Finally, paired-end reads with length 150 bp were generated. The raw transcriptomic data for G. gigas and O. warwicki are deposited in NCBI SRA database with accession numbers SRR10523768 and SRR10527303, respectively.
Transcriptome assembly and annotation
Clean reads were obtained by removal of reads with adaptors and those of low quality from the raw reads (low-quality bases with Qphred ≤5 were > 50% in a read). De novo transcriptome assembly was conducted with Trinity (v2.5.1) using default parameters [73]. The longest transcript of each transcription group was regarded as a unigene for the following analyses. The assembly unigenes of two species were deposited on NCBI TSA database with the accession numbers of GIJX00000000 and GIJW00000000. BUSCO (v1.22) was used to check the quality of the assembly against the database of arthropoda_odb9 [74]. All unigenes were annotated through blasting against public databases, including NCBI non-redundant protein (Nr, E-value 1E-5), Swiss-Prot (E-value 1E-5), and euKaryotic Ortholog Group (KOG, E-value 1E-3) using DIAMOND (v0.8.22) [75], and the NCBI nucleotide database (Nt, E-value 1E-5) using BLAST (v2.2.28+) [76]. Kyoto Encyclopedia of Genes and Genomes (KEGG) classification was performed using the KEGG Automatic Annotation Server (KAAS) with an E-value of 1E-10 [77]. Protein family (Pfam) alignments were carried out using the HMMER (v3.0, http://hmmer.org/) with an E-value of 1E-2, and the Gene Ontology (GO) classification was conducted based on the results of Nr and Pfam using Blast2GO (v2.5) with an E-value of 1E-6 [78]. BLASTx searches were performed for unigenes against Nr database, and followed by conjoining fragmental alignments using SOLAR [79]. Thus, a partial or full open reading frame (ORF) of each unigene was obtained and translated into amino acid sequences.
Gene family clustering and phylogenetic analysis
Genomic resources for adult barnacles are limited and, therefore, to perform comparative transcriptomic and phylogenetic analyses, the full protein coding genes of four crustaceans, Daphnia pulex (PRJNA12756), Eurytemora affinis (PRJNA423276); Parhyale hawaiensis (PRJNA306836), and Litopenaeus vannamei (http://www.shrimpbase.net/lva.download.html) were obtained from NCBI and other databases [80,81,82,83]. Pair-wise BLASTp alignment was performed to align all-to-all with an E-value cutoff of 1E-07, and all genes were clustered into gene families using OrthoMCL v2.0.3 [84]. Then, single-copy genes of these species were collected for phylogenetic analysis using maximum-likelihood (ML) methods. Sequence alignment was performed using MUSCLE 3.6 [85]. ML analysis was performed on PhyML with the substitution model WAG + gamma + Inv [86]. One thousand bootstrap replicates were conducted to produce the branch support values. The divergence time was estimated by Bayesian relaxed molecular clock approaches implemented in TIMETREE in MEGA v7.0 [87], with the time calibrations according to the findings of Zhang et al. and Yuan et al. [83, 88]. The expanded and contracted gene families on each branch of the phylogenetic tree were calculated by CAFE [89].
Identification of positively selected genes
Adaptive evolution was assessed by comparing the nonsynonymous/synonymous substitution ratios (ω = dN/dS). Orthologs of G. gigas and O. warwicki were collected by pair-wise best-hit BLAST. Sequence alignment was performed using MUSCLE, and all gaps were removed from the alignment. The ω value of each ortholog was calculated using the program yn00 of PAML v4.48a [90]. Genes with ω > 1.0 were considered fast evolving genes, and ω > 0.5 was considered potential positively selected genes.
Differential gene expression
Relative levels of gene expression were calculated by mapping clean reads to the assembled unigenes using RSEM [91]. The read counts were calculated using uniquely mapped reads and normalized to the expected number of fragments per kilobase of unigene sequence per million (FKPM). Differential expression analysis was performed on the orthologs of G. gigas and O. warwicki. Genes with fold change values > 4 were considered to be differentially expressed.
GO and KEGG enrichment analysis
GO and KEGG enrichment analysis was performed on the expanded gene families, positively selected and differentially expressed genes using Omicshare CloudTools (http://www.omicshare.com/tools/?l=en-us). Enriched GO terms and KEGG Orthology (KO) terms were calculated relative to the background of all unigenes.
Availability of data and materials
All data presented in this work are provided either in the article or additional files, and the raw transcriptomic data and assembly unigenes for Glyptelasma gigas and Octolasmis warwicki are deposited in NCBI database (https://www.ncbi.nlm.nih.gov/sra) with accession numbers SRR10523768 and SRR10527303, GIJX00000000 and GIJW00000000, respectively.
Abbreviations
- BUSCOs:
-
Benchmarking universal single-copy orthologs
- CHST15:
-
N-acetylgalactosamine-4-sulfate-6-O-sulfotransferase
- DDR2:
-
Discoidin domain receptor 2
- ECM:
-
Extracellular matrix
- ERCC4:
-
Excision repair cross-complementation group 4
- FAT:
-
Protocadherin fat
- FKPM:
-
Fragments per kilobase of unigene sequence per million
- GO:
-
Gene Ontology
- GPCR:
-
G protein-coupled receptor
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- ko:
-
KEGG Orthology
- ML:
-
Maximum-likelihood
- NCD3:
-
The nine-cysteines domain of family 3
- Nr:
-
Non-redundant protein
- ORF:
-
Open reading frame
- Pfam:
-
Protein family
- VSR:
-
Very short patch repair
- XRCC4:
-
X-ray repair cross-complementing protein 4
References
Somero GN. Adaptations to high hydrostatic pressure. Annu Rev Physiol. 1992;54:557–77.
Macdonald AG. Hydrostatic pressure as an environmental factor in life processes. Comp Biochem Physiol A Physiol. 1997;116(4):291–7.
Helly JJ, Levin LA. Global distribution of naturally occurring marine hypoxia on continental margins. Deep-Sea Res I Oceanogr Res Pap. 2004;51(9):1159–68.
Lü C, Hao T, Lin J, Qiu X. The role of rifting in the development of the continental margins of the southwest subbasin, South China Sea: insights from an OBS experiment. Mar Geophys Res. 2017;38(1–2):105–23.
Ruhl HA, Smith KL. Shifts in deep-sea community structure linked to climate and food supply. Science. 2004;305:513–5.
Lan Y, Sun J, Tian R, Bartlett DH, Li R, Wong YH, et al. Molecular adaptation in the world’s deepest-living animal: insights from transcriptome sequencing of the hadal amphipod Hirondellea gigas. Mol Ecol. 2017;26:3732–43.
Kobayashi H, Hatada Y, Tsubouchi T, Nagahama T, Takami H. The hadal amphipod Hirondellea gigas possessing a unique cellulose for digesting wooden debris buried in the deepest seafloor. PLoS One. 2012;7:e42727.
Zhang J, Sun QL, Luan ZD, Lian C, Sun L. Comparative transcriptome analysis of Rimicaris sp reveals novel molecular features associated with survival in deep-sea hydrothermal vent. Sci Rep. 2017;7:2000.
Zhang Y, Sun J, Chen C, Watanabe HK, Feng D, Zhang Y, et al. Adaptation and evolution of deep-sea scale worms (Annelida: Polynoidae): insights from transcriptome comparison with a shallow-water species. Sci Rep. 2017;7:46205.
Holder T, Basquin C, Ebert J, Randel N, Jollivet D, Conti E, et al. Deep transcriptome-sequencing and proteome analysis of the hydrothermal vent annelid Alvinella pompejana identifies the CvP-bias as a robust measure of eukaryotic thermostability. Biol Direct. 2013;8:2.
Lan Y, Sun J, Xu T, Chen C, Tian R, Qiu JW, et al. De novo transcriptome assembly and positive selection analysis of an individual deep-sea fish. BMC Genomics. 2018;19:394.
Wang K, Shen Y, Yang Y, Gan X, Liu G, Hu K, et al. Morphology and genome of a snailfish from the Mariana trench provide insights into deep-sea adaptation. Nat Ecol Evol. 2019;3:823–33.
Bettencourt R, Pinheiro M, Egas C, Gomes P, Afonso M, Shank T, et al. High-throughput sequencing and analysis of the gill tissue transcriptome from the deep-sea hydrothermal vent mussel Bathymodiolus azoricus. BMC Genomics. 2010;11:559.
Zheng P, Wang M, Li C, Sun X, Wang X, Sun Y, et al. Insights into deep-sea adaptations and host-symbiont interactions: a comparative transcriptome study on Bathymodiolus mussels and their coastal relatives. Mol Ecol. 2017;26:5133–48.
Fisher CR. Chemoautotrophic and methanotrophic symbioses in marine invertebrates. Rev Aquat Sci. 1990;2:399–613.
Baeza JA. The biology of squat lobsters. In: Poore GCB, Ahyong ST, Taylor J, editors. Squat lobsters as symbionts and in chemo-autotrophic environments. Melbourne: CSIRO Publishing; 2011. p. 249–70.
Duperron S, Bergin C, Zielinski F, Blazejak A, Pernthaler A, McKiness ZP, et al. A dual symbiosis shared by two mussel species, Bathymodiolus azoricus and Bathymodiolus puteoserpentis (Bivalvia: Mytilidae), from hydrothermal vents along the northern mid-Atlantic ridge. Environ Microbiol. 2006;8(8):1441–7.
Chandramouli KH, Al-Aqeel S, Ryu T, Zhang H, Seridi L, Ghosheh Y, et al. Transcriptome and proteome dynamics in larvae of the barnacle Balanus Amphitrite from the Red Sea. BMC Genomics. 2015;16:1063.
Machado AM, Sarropoulou E, Castro LFC, Vasconcelos V, Cunha I. An important resource for understanding bio-adhesion mechanisms: cement gland transcriptomes of two goose barnacles, Pollicipes pollicipes and Lepas anatifera (Cirripedia, Thoracica). Mar Genomics. 2018;45:16–20.
Yan XC, Chen ZF, Sun J, Matsumura K, Wu RS, Qian PY. Transcriptomic analysis of neuropeptides and peptide hormones in the barnacle Balanus amphitrite: evidence of roles in larval settlement. PLoS One. 2012;7(10):e46513.
Ryu T, Woo S, Lee N. The first reference transcriptome assembly of the stalked barnacle, Neolepas marisindica, from the Onnuri vent field on the central Indian ridge. Mar Genomics. 2019;48:100679.
Chan BKK, Prabowo RE, Lee KS. Crustacean fauna of Taiwan: barnacles, volume I-Cirripedia: Thoracica excluding the Pyrgomatidae and Acastinae. 1st ed. Keelung: National Taiwan Ocean University; 2009.
Da Silva ET, Ridd M, Klumpp D. The barnacle Balanus amphitrite as a biomonitor for Cd: radiolabelled experiments. Mar Environ Res. 2009;67(4–5):177–82.
Rivera A, Gelcich S, García-Florez L, Alcázar JL, Acuña JL. Co-management in Europe: insights from the gooseneck barnacle fishery in Asturias, Spain. Mar Policy. 2014;50:300–8.
Newman WA, Ross A. Antarctic Cirripedia. Ant Res Ser. 1971;14:1–257.
Jones DS. Australian barnacles (Cirripedia: Thoracica), distributions and biogeographical affinities. Integr Comp Biol. 2012;52(3):366–87.
Southward AJ, Jones DS. A revision of stalked barnacles (Cirripedia: Thoracica: Scalpellomorpha: Eolepadidae: Neolepadinae) associated with hydrothermalism, including a description of a new genus and species from a volcanic seamount off Papua New Guinea. Senckenberg Marit. 2003;32(1–2):77–93.
Yamaguchi T, Newman WA, Hashimoto J. A cold seep barnacle (Cirripedia: Neolepdinae) from Japan and the age of the vent/seep fauna. J Mar Biol Assoc U K. 2004;84:111–20.
Chan BK, Prabowo RE, Lee KS. North West Pacific deep-sea barnacles (Cirripedia, Thoracica) collected by the TAIWAN expeditions, with descriptions of two new species. Zootaxa. 2010;2405:1–47.
Shalaeva K, Boxshall G. An illustrated catalogue of the scalpellid barnacles (Crustacea: Cirripedia: Scalpellidae) collected during the HMS challenger expedition and deposited in the Natural History Museum, London. Zootaxa. 2014;3804:1–63.
Gan ZB, Li XZ. Report on four deep-water barnacles (Cirripedia, Thoracica) from the north west Pacific, with remarks on Trianguloscalpellum regium (Wyville-Thomson, 1873). Zootaxa. 2019;4565(2):201–12.
Jones DS, Hosie AM. A checklist of the barnacles (Cirripedia: Thoracica) of Singapore and neighbouring waters. Raffles Bull Zool. 2016;34:241–311.
Qiu Y, Yao B, Li T, Bao C, Gong X, Zhong H. Geologic and tectonic features and hydrocarbon potential of the Zhongjiannan Basin in South China Sea. Geologic Res S China Sea. 1997;9:37–53.
Gao HF, Wang YT, Guo LH. Petroleum geological conditions and prospects in the Zhongjiannan Basin in the western South China Sea. Geol China. 2007;34(4):592–8.
Chen J, Song H, Guan Y, Yang S, Pinheiro LM, Bai Y, et al. Morphologies, classification and genesis of pockmarks, mud volcanoes and associated fluid escape features in the northern Zhongjiannan Basin, South China Sea. Deep-Sea Res II Top Stud Oceanogr. 2015;122:106–17.
Wan Z, Yao Y, Chen K, Zhong S, Xia B, Sun Y. Characterization of mud volcanoes in the northern Zhongjiannan Basin, western South China Sea. Geol J. 2019;54(1):177–89.
Uhlirova M, Foy BD, Beaty BJ, Olson KE, Riddiford LM, Jindra M. Use of Sindbis virus-mediated RNA interference to demonstrate a conserved role of broad-complex in insect metamorphosis. Proc Natl Acad Sci U S A. 2003;100:15607–12.
Hsia H, Schwarzbauer J. Meet the tenascins: multifunctional and mysterious. J Biol Chem. 2005;280(29):26641–4.
Linse K, Jackson JA, Fitzcharls E, Sands CJ, Buckeridge JS. Phylogenetic position of Antarctic scalpelliformes (crustacea: cirripedia: thoracica). Deep-Sea Res I Oceanogr Res Pap. 2013;73:99–116.
Herrera S, Watanabe H, Shank TM. Evolutionary and biogeographical patterns of barnacles from deep-sea hydrothermal vents. Mol Ecol. 2015;24:673–89.
Halliday TJD, Upchurch P, Goswami A. Eutherians experienced elevated evolutionary rates in the immediate aftermath of the cretaceous–Palaeogene mass extinction. Proc R Soc B Biol Sci. 2016;283:20153026.
Lin C, Chu F, Gao J, Tan Y. On tectonic movement in the South China Sea during the Cenozoic. Acta Oceanol Sin. 2007;29(4):87–96.
Bingham BL, Young CM. Larval phototaxis in barnacles and snails associated with bathyal sea urchins. Deep-Sea Res I Oceanogr Res Pap. 1993;40(1):1–12.
Lo SH. Focal adhesions: what's new inside. Dev Biol. 2006;294(2):280–91.
Bosman FT, Stamenkovic I. Functional structure and composition of the extracellular matrix. J Pathol. 2003;200:423–8.
Kim D, Chung J. Akt: versatile mediator of cell survival and beyond. J Biochem Mol Biol. 2002;35:106–15.
Oh H, Irvine KD. Yorkie: the final destination of Hippo signaling. Trends Cell Biol. 2010;20:410–7.
Negishi M, Oinuma I, Katoh H. Plexins: axon guidance and signal transduction. Cell Mol Life Sci. 2005;62:1363.
Sugahara K, Kitagawa H. Recent advances in the study of the biosynthesis and functions of sulfated glycosaminoglycans. Curr Opin Struct Biol. 2000;10:518–27.
Andersen MD, Busk PK, Svendsen I. Cytochromes P-450 from cassava (Manihot esculentaCrantz) catalyzing the first steps in the biosynthesis of the Cyanogenic Glucosides Linamarin and Lotaustralin. Cloning, functional expression in Pichia pastoris and substrate specificity of the isolated recombinant enzymes. J Biol Chem. 2000;275:1966–75.
Asano K, Clayton J, Shalev A, Hinnebusch AG. A multifactor complex of eukaryotic initiation factors, eIF1, eIF2, eIF3, eIF5, and initiator tRNAMet is an important translation initiation intermediate in vivo. Genes Dev. 2000;14:2534–46.
Aertsen A, Houdt RV, Vanoirbeek K, Michiels CW. An SOS response induced by high pressure in Escherichia coli. J Bacteriol. 2004;186:6133–41.
Aertsen A, Michiels CW. Mrr instigates the SOS response after high pressure stress in Escherichia coli. Mol Microbiol. 2005;58:1381–91.
Qiu J, Wang WN, Wang LJ, Liu YF, Wang AL. Oxidative stress, DNA damage and osmolality in the Pacific white shrimp, Litopenaeus vannamei exposed to acute low temperature stress. Comp Biochem Physiol C Toxicol Pharmacol. 2011;154:36–41.
Li Y, Wei L, Cao J, Qiu L, Jiang X, Li P, et al. Oxidative stress, DNA damage and antioxidant enzyme activities in the pacific white shrimp (Litopenaeus vannamei) when exposed to hypoxia and reoxygenation. Chemosphere. 2016;144:234–40.
Oksenych V, Kumar V, Liu X, Guo C, Schwer N, Zha S, et al. Functional redundancy between the XLF and DNA-PKcs DNA repair factors in V (D) J recombination and nonhomologous DNA end joining. Proc Natl Acad Sci U S A. 2013;110:2234–9.
Hennecke F, Kolmar H, Bründl K, Fritz HJ. The vsr gene product of E. coli K-12 is a strand-and sequence-specific DNA mismatch endonuclease. Nature. 1991;353:776–8.
Gregg SQ, Robinson AR, Niedernhofer LJ. Physiological consequences of defects in ERCC1–XPF DNA repair endonuclease. DNA Repair. 2011;10:781–91.
Shinozaki K, Yamaguchi-Shinozaki K. Molecular responses to dehydration and low temperature: differences and cross-talk between two stress signaling pathways. Curr Opin Plant Biol. 2000;3:217–23.
Holden CP, Storey KB. Signal transduction, second messenger, and protein kinase responses during freezing exposures in wood frogs. Am J Phys Regul Integr Comp Phys. 1996;271:1205–11.
Zonia L, Munnik T. Life under pressure: hydrostatic pressure in cell growth and function. Trends Plant Sci. 2007;12(3):90–7.
Siebenaller JF. The effects of hydrostatic pressure on signal transduction in brain membranes of deep-sea fishes of the genus Coryphaenoides. Fish Physiol Biochem. 2000;23:99–106.
Liu X, He Q, Studholme DJ, Wu Q, Liang S, Yu L. NCD3G: a novel nine-cysteine domain in family 3 GPCRs. Trends Biochem Sci. 2004;29:458–61.
Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–63.
Aiyar N, Rand K, Elshourbagy NA, Zeng Z, Adamou JE, Bergsma DJ, et al. A cDNA encoding the calcitonin gene-related peptide type 1 receptor. J Biol Chem. 1996;271(19):11325–9.
Jha A, Chung WY, Vachel L, Maleth J, Lake S, Zhang G, et al. Anoctamin 8 tethers endoplasmic reticulum and plasma membrane for assembly of Ca2+ signaling complexes at the ER/PM compartment. EMBO J. 2019;38:e101452.
Le Novère N, Changeux JP. The ligand gated ion channel database. Nucleic Acids Res. 1999;27(1):340–2.
Becker A, Schloder P, Steele JE, Wegener G. The regulation of trehalose metabolism in insects. Experientia. 1996;52(5):433–9.
Kandror O, De Leon A, Goldberg AL. Trehalose synthesis is induced upon exposure of Escherichia coli to cold and is essential for viability at low temperatures. Proc Natl Acad Sci. 2002;99(15):9727–32.
Candy DJ, Kilby BA. The biosynthesis of trehalose in the locust fat body. Biochem J. 1961;78:531–6.
Campbell JA, Davies GJ, Bulone V, Henrissat B. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem J. 1997;326:929–42.
Kawai I, Matsumura H, Fujii W, Naito K, Kusakabe K, Kiso Y, et al. Discoidin domain receptor 2 (DDR2) regulates body size and fat metabolism in mice. Transgenic Res. 2014;23(1):165–75.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:5936.60.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:182–5.
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–35.
Rota-Stabelli O, Kayal E, Gleeson D, Daub J, Boore JL, Telford MJ, et al. Ecdysozoan mitogenomics: evidence for a common origin of the legged invertebrates, the Panarthropoda. Genome Biol Evol. 2010;2:425e440.
Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, et al. The ecoresponsive genome of Daphnia pulex. Science. 2011;331(6017):555–61.
Regier JC, Shultz JW, Zwick A, Hussey A, Ball B, Wetzer R, et al. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature. 2010;463:1079–83.
Kao D, Lai AG, Stamataki E, Rosic S, Konstantinides N, Jarvis E, et al. The genome of the crustacean Parhyale hawaiensis, a model for animal development, regeneration, immunity and lignocellulose digestion. Elife. 2016;5:e20062.
Zhang X, Yuan J, Sun Y, Li S, Gao Y, Yu Y, et al. Penaeid shrimp genome provides insights into benthic adaptation and frequent molting. Nat Commun. 2019;10(1):356.
Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Yuan J, Zhang X, Liu C, Sun X, Sivaramasamy E, Li F, Xiang J. Comparative genomics analysis of decapod shrimps in the Pancrustacea clade. Biochem Syst Ecol. 2016;64:111–21.
De Bie T, Cristianini N, Demuth JP, Hahn MW. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22(10):1269–71.
Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–6.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323.
Acknowledgments
We are grateful to all the scientists and crew of the vessel Xiangyanghong 9 and the submersible Shenhaiyongshi, for their help in the collection of the Glyptelasma gigas specimens.
Funding
This work was financially supported by the National Key R & D Program of China (No. 2018YFC0310800) and the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB06010101). None of the funding bodies participated in the research design, sample collection, experimental operation, and data analysis, or writing the manuscript.
Author information
Authors and Affiliations
Contributions
XZL, ZBG and FUL conceived and designed the project. XZL collected Glyptelasma gigas samples, and XML collected Octolasmis warwicki samples. ZBG, XML and DD performed the experiments, ZBG and JBY analyzed data and drafted the manuscript. XZL, FUL, XML and DD revised the manuscript. All authors approved the final version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The cruise and field study were approved by the Chinese Academy of Sciences and the National Natural Science Foundation of China. This work was approved by the Committee on the Ethics of Animal Experiments of the Institute of Oceanology, Chinese Academy of Sciences, and all the experiments involving animals were conducted with approval from the Institutional Animal Care and Use Committee (IACUC) of the Chinese Academy of Sciences.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Table S1. Summary of the transcriptome sequencing data.
Additional file 2:
Table S2. BUSCO evaluation of the transcriptome assembly.
Additional file 3:
Table S3. KEGG pathway annotation of expanded gene families both in Glyptelasma gigas and Octolasmis warwicki.
Additional file 4:
Table S4. Complete list of significantly expanded gene families in Glyptelasma gigas.
Additional file 5:
Table S5. Complete list of positively selected genes in Glyptelasma gigas.
Additional file 6:
Table S6. KEGG pathway annotation of the positively selected genes of Glyptelasma gigas.
Additional file 7:
Figure S1. Phylogenetic tree of Br-C gene family. Bootstrap values (> 50%) are shown at branch nodes. ggi: Glyptelasma gigas, owa: Octolasmis warwicki, Dpul: Daphnia pulex, Eaff: Eurytemora affinis, LVAN: Litopenaeus vannamei, phaw: Parhyale hawaniensis.
Additional file 8:
Figure S2. GO (A) and KEGG (B) distribution of the highly expressed genes in Glyptelasma gigas.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gan, Z., Yuan, J., Liu, X. et al. Comparative transcriptomic analysis of deep- and shallow-water barnacle species (Cirripedia, Poecilasmatidae) provides insights into deep-sea adaptation of sessile crustaceans. BMC Genomics 21, 240 (2020). https://doi.org/10.1186/s12864-020-6642-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-6642-9