
Abstract
Background
The hard tick Hyalomma dromedarii is one of the most injurious ectoparasites affecting camels and apparently best adapted to deserts. As long-term blood feeders, ticks are threatened by host defense system compounds that can cause them to be rejected and, ultimately, to die. However, their saliva contains a cocktail of bioactive molecules that enables them to succeed in taking their blood meal. A recent sialotranscriptomic study uncovered the complexity of the salivary composition of the tick H. dromedarii and provided a database for a proteomic analysis. We carried out a proteomic-informed by transcriptomic (PIT) to identify proteins in salivary glands of both genders of this tick species.
Results
We reported the array of 1111 proteins identified in the salivary glands of H. dromedarii ticks. Only 24% of the proteins were shared by both genders, and concur with the previously described sialotranscriptome complexity. The comparative analysis of the salivary glands of both genders did not reveal any great differences in the number or class of proteins expressed their enzymatic composition or functional classification. Indeed, few proteins in the entire proteome matched those predicted from the transcriptome while others corresponded to other proteins of other tick species.
Conclusion
This investigation represents the first proteomic study of H. dromedarii salivary glands. Our results shed light on the differences between the composition of H. dromedarii male and female salivary glands, thus enabling us to better understand the gender-specific strategy to feed successfully.
Similar content being viewed by others

Background
Hard ticks (Ixodidae) are unique among hematophageous arthropods, mainly for their long-term feeding that can last up to two weeks [1]. As they feed on different animals, the ticks come under pressure from their host’s immune system, which led to their fast evolution [2]. During the feeding period, the host reacts to the injury inflicted by the tick bite by starting a wide range of mechanisms to prevent blood loss [3]. The feeding ticks are thus exposed to host defense system components including, not only immune ones, but also platelet aggregation, coagulation, and inflammation components [3]. All of these responses are designed to disrupt tick feeding and cause its rejection from the host’s skin [4]. To avoid host defenses, ticks secrete saliva at the bite site that contain many biologically active molecules that display anticoagulation, antiplatelet, vasodilatory, anti-inflammatory, and immunomodulatory activities [3, 5, 6]. Ticks are thus able to feed on their hosts for days and even weeks without being disturbed by their immune system [6]. Not only do tick salivary compounds facilitate tick feeding, but they may also promote the survival and dissemination of infectious agents in the host [6]. The enhancement of pathogen transmission by tick saliva, called saliva-assisted transmission, or SAT, has been documented for several tick-pathogen associations [7]. However, only a few of the salivary proteins implicated in pathogen transmission have been identified and characterized to date [6]. Overall, deciphering the composition of tick salivary glands could lead to the discovery of new potential targets for developing vaccines for tick control and/or blocking pathogen transmission and new pharmacological compounds with anti-hemostatic, anti-inflammatory and antibacterial activities [8,9,10].
Recent advances in new technologies, mainly Next Generation Sequencing, or NGS, through transcriptomic and proteomic approaches, has led to insights into the molecular mechanisms involved in tick hematophagy, pathogen transmission, and tick-host-pathogen interactions [11]. In addition, these technologies have revealed the complexity of tick salivary composition, which has hundreds of different proteins including many that are novel [12]. Apart from being diverse, these molecules are multipotent and were shown to be endowed with pharmacological features [13]. Accordingly, several transcript and protein profiles of tick salivary glands were carried out in different stages of development, for both genders and feeding behavior [2, 14, 15]. These studies were also conducted to compare the salivary gland secretion of both hard and soft ticks [16].
More interestingly, sialotranscriptomic analyses improved proteomic studies of unknown genome species that seek to identify pharmaceutically active proteins [17, 18]. Previously, proteomic studies relied on the information in sequence databases and were thus able to detect only those proteins that were encoded by known genes. As such, the identification of proteins by tandem mass spectrometry posed a big challenge for a non-model species of which there is no available genome [19]. Proteomic informed by transcriptomic (PIT) approach helped to solve this issue by generating protein databases based on the expressed mRNA sequencing [17]. The possibility of using sample-specific databases derived from RNA-seq data revolutionized large-scale proteomics [17]. This approach was used in many studies related to tick saliva, especially with the expansion of tick sialotranscriptomic analysis using next-generation sequencing methods [11]. Although PIT was applied to the study of the saliva from several tick species, most saliva proteins and their impact on the host tick interaction remain unknown [18].
The camel tick, Hyalomma dromedarii Koch, 1844 (Acari: Ixodidae) is considered to be the most closely associated with camels and is well adapted to the deserts where tick hosts live [20]. It is a common species in regions with Mediterranean steppe vegetation and in desert climates in Africa, the Near East, Middle East, Far East, India, Mongolia, and Tibet [21, 22]. During its blood meal, this tick species is involved in transmitting an array of pathogens including Theileria annulata [23], Rickettsia [24, 25] and Crimean-Congo haemorrhagic fever virus [26, 27]. It can also transmit Coxiella burnetii, the agent of Q fever, to camels [28]. Given this role in parasiting camels, H. dromedarii ticks were the object of several studies seeking to characterize the molecules isolated from their salivary gland extracts and saliva. These studies focused on isolating those some molecules whereas the set of other molecules remain unknown. For this reason, we recently analyzed the sialotranscriptome of H. dromedarii using NGS technology, highlighting the wide range of transcripts expressed in the salivary glands of this tick species [29]. The functional annotation of transcripts has provided information on predicted protein families. However, questions remain about proteins that are actually present in the salivary glands of this tick species. We therefore carried out the first comprehensive proteomic analysis of H. dromedarii salivary glands. We used proteomics informed by transcriptomics to identify H. dromedarii salivary gland proteins in both genders using LC-MS/MS. This approach relies on the translation of the predicted genes from the sialotranscriptome of H. dromedarii tick and generates databases of predicted proteins that were used by MS/MS-spectra search engines to identify peptides contained in these salivary glands.
Results and discussion
Hyalomma dromedarii salivary glands proteome
As a hematophagous ectoparasite mainly of camels, H. dromedarii tick has developed a complex cocktail of bioactive molecules that target and neutralize the molecule secreted by the host camel that allows for successful parasitism [28]. Only few reports have explored H. dromedarii salivary glands. Compared to other hematophagous parasites, relatively little information exists about the molecular composition of H. dromedarii salivary glands [27, 28, 30].
In our current work, we obtained salivary glands from partially engorged males and females of H. dromedarii collected from camels from south Tunisia. We can therefore consider our proteomic results to provide a qualitative description of H. dromedarii salivary glands components since the glands were stimulated during the tick’s attachment on the host. As the genome of H. dromedarii is not yet sequenced, we chose the PIT approach as the best way to identify the proteins of H. dromedarii salivary glands based on the recently published sialotranscriptome [29]. We identified 854 previously known proteins from the Acari database and an additional 257 proteins predicted from the transcriptomics data. A few studies have previously reported the use of transcriptomics to inform proteomics in other tick species including the Dermacentor andersoni [18]. The first proteomic studies addressing tick saliva and salivary glands date to the first decade of the twenty-first century [16, 31]. Each of these studies had a different focus: comparing the saliva of hard and soft ticks [16], partially and fully engorged Rhipicephalus (Boophilus) microplus [14], and sexual differences in the sialomes of Rhipicephalus pulchellus and Ornithodoros moubata [15, 32]. In addition to their different objectives, these proteomic analyzes also used different approaches to identify proteins following LC-MS/MS analysis [2, 14, 15, 32, 33]. We were able to identify a total of 1111 different proteins, far from the 15,342 proteins predicted from the sialotranscriptome [29]. Several protein families were identified in common in both proteome and transcriptome, while few were exclusively found in one of them (Fig. 1). Besides, our results showed a correlation (r) of 0.33, which appears to be a weak correlation between proteins found in the sialoproteome compared to those found in the sialotranscriptome.

Correlation between protein families found in the transcriptome and proteome of salivary gland extracts from Hyalomma dromedarii tick
Several hypotheses can explain the discrepancy between the transcriptome and proteome obtained in our study. Firstly, even though the transcriptome and the proteome have been analyzed on adult H. dromedarii at almost the same feeding stage, it is possible that the proteome corresponds to proteins translated from transcripts that existed before the transcriptomic analyzes [17]. In addition, although mRNA profiling is an important tool in gene expression analysis, transcript abundance does not always correlate with protein expression levels, mainly because of translation regulation where cells only translate proteins when they are needed [34]. That means that not all the sets of mRNAs in a cell are necessarily translated into proteins. Secondly, constraints limiting transcriptomics and proteomics may also lead to discrepancies. Indeed, transcriptomics data do not provide information regarding post-translational modifications, subcellular location, or protein degradation, which is not associated with a decrease of transcripts [35]. Furthermore, in transcriptomics, the number of reads may not directly represent the level of expressed proteins [15]. This is sometimes due to the mass assembly of reads that produces fragmented transcripts resulting in more than one coding sequence (CDS) for a single protein [35]. In addition, some peptides extended over such fragmented regions will not be identified. Other proteins exclusively expressed in ticks may not be identified because their sequences are not included in the database [11]. These proteins have been reported in several transcriptomic studies that revealed a large percentage of transcripts without any sequence similarity to any known protein sequence and therefore no known function [3]. Lastly, regarding proteomics, lower sensitivity in detection limits restricts the number of proteins that can be detected. Thus, less abundant proteins may not be identified.
According to our results, only 24% of the identified proteins (n = 262) were shared by both genders. In addition, among total proteins (n = 1111), 40% (n = 443) were found exclusively in females and 36% (n = 406) were found only in males (Fig. 2). These proteomic results concur with differences between the genders observed in the sialotranscriptome of H. dromedarii [29]. Indeed, transcriptomic data provided a global view of the gene expression profile in tick salivary glands while the proteomics analysis provides information regarding mRNAs that are actually translated into proteins.

Venn diagram of SGE proteins identified in both genders of Hyalomma dromedarii ticks
We have identified almost the same amount of proteins in both genders but this does not rule out the difference observed in the transcriptomic study that is supported by the different protein identities shown in Additional file 1: Table S1 and Additional file 2: Table S2. Consequently, the so-called variation is assigned to the diversity of protein identities in the two genders. Such a difference was expected for several reasons (i) The anatomy and functions of Ixodidae salivary glands are known to be different between genders [36]. Females have three morphologically distinct acini types while males have additional specific acini [37]. Moreover, histological studies of tick salivary glands have described male-specific cells [38, 39] that were postulated to assist tick reproduction [40] (ii) Female ticks need days and even weeks to finish their extremely large blood meal on a single host where male ticks exhibit intermittent feeding during a shorter period of time and ingest small volumes of blood [41]. This means that the genders are exposed to different host defense constraints [41]. These differences between males and females was reflected in their proteomes, where the composition of their salivary glands is different [15, 42]. Several proteomic studies have therefore compared salivary gland compounds of both genders in hard and ticks [15, 32].
Description and classification of proteins in salivary gland extract of H. dromedarii
According to protein function, the identified proteins were classified into four main groups as described in previous sialome studies [43]: housekeeping class (H), possibly secreted proteins (S), transporters (T), and unknown class (U) (Fig. 3). Of the set of identified proteins, 68% (n = 760) belonged to the H class, 19% (n = 207) to the S class, 8% (n = 89) to the U class and 5% (n = 55) to the T class.

Distribution of proteins identified in Hyalomma dromedarii SGE of both genders according to their functions
Housekeeping proteins (H class)
In H. dromedarii, 68% of the identified proteins for both genders were related mostly to the housekeeping class, which was the most abundant for both females (72%) and males (70%). This strongly corroborates the transcriptome data where transcripts coding for housekeeping proteins were the highly expressed ones [29]. As we are analyzing salivary gland extracts, we expected to find high amounts of housekeeping proteins, since they come from the leakage of intracellular components from degenerated or broken salivary gland cells [1]. Housekeeping proteins were defined as those required for the basal maintenance of tissues although it is also recognized that they might be secreted in the extracellular microenvironment especially since some of them were described in previous proteomic studies of saliva [32]. In this study, we organized housekeeping proteins into 20 groups by their functions and roles in the cell. All groups were the same in both genders except for transcription regulators and DNA replication regulators, which were only found in females. Proteins associated with nuclear regulation were most numerous in both genders. 15% in females and 23.2% in males of all proteins associated within the H class. These results suggest highly regulated gene transcription in the salivary glands of H. dromedarii. The second most abundant group for female ticks was the protein synthesis machinery group (14.9%), an expected result given the secretory nature of the organ. By contrast, cytoskeletal proteins (11.9%) represent the second most numerous group in males. Cytoskeletal proteins such as actin and tubulin were also found in females in large amounts (10.6%). These latter two proteins are largely conserved and were identified in almost all proteomes of other tick species [2, 32]. The remaining groups (n = 15), have nearly similar percentages as described in Fig. 4, and are involved, mainly, in the intracellular functions of tick salivary glands. As previously stated, sialomes have often noticed that some housekeeping intracellular proteins can be secreted by tick salivary glands where they play further extracellular function [44, 45]. Therefore, housekeeping proteins in H. dromedarii salivary glands may have further biological importance, particularly in the tick-host interface [14]. This could be explained by the presence of proteins in females (3.2%) and in males (4.2%) involved in the oxidant metabolism, as well as several enzymes linked to detoxification. For example, we identified superoxide dismutase, an essential enzyme involved in the mechanism of eliminating free radicals [46] and Glutathione S–transferase known for its catalysis of the conjugation of glutathione with several xenobiotic and endogenous substances [47]. These enzymes might be related to the decrease in the oxidative ability of phagocytes at the bite site, as reported in the saliva of the cattle tick Rhipicephalus (Boophilus) microplus [48]. Additional studies are thus needed to characterize their functions in the extracellular environment.

Classification of housekeeping proteins identified in Hyalomma dromedarii SGE of both genders, according to their functions and/or protein family
Possibly secreted proteins (S class)
Proteins of the S class were the second most numerous, with 19% from the set of identified proteins, and expressed equally (17%) in both genders. Secreted proteins were classified into 7 families as per a previous review [3]. Accordingly, most of the families described in Fig. 5 were identified in both genders, although a few were gender-exclusive. Our results showed a low number of the highly secretory protein families (2 Kunitz-type, 6 Serpins, and 2 Lipocalins) by contrast with the results observed in other hard ticks [2, 14, 15]. Moreover, no proteins from basic tail superfamily, mucins, Ixodegrin among others, were identified in the H. dromedarii proteome whereas these proteins are generally overexpressed in the proteomes of other ticks, including species from Hyalomma genus [2, 49]. It is important to emphasize that we cannot exclude the presence of these proteins in the H. dromedarii proteome given that we have previously identified these families in the transcriptome. It is probable that the heterogeneity observed for many of the principal secretory families in tick sialome [31, 43, 50, 51] is responsible for missing these proteins. Moreover, since we do not have the full repository of proteins from H. dromedarii, the lack of unidentified proteins can be explained by the absence of specific sequences in the databases needed for comparison.

Classification of possibly secreted proteins identified in Hyalomma dromedarii SGE of both genders, according to their functions and/or protein family
As reported in Fig. 5, total enzymes represent more than 73% in males and 46% in females of S class proteins. Several enzymatic families have been identified in the proteome of both genders, including proteases, lysosomal enzymes and several other enzymes. In contrast, ribonucleases were identified exclusively in males. Metalloproteases were also among the identified enzymes with 9% from the set of S class proteins in both of the genders. These enzymes require a metal ion, usually Zn2+, to catalyze the hydrolysis of a peptide bond [52]. These proteolytic enzymes have been found throughout almost the entire evolutionary scale from bacteria to mammals [53, 54]. In snakes, they are a crucial component of venom and many of them cause their prey to hemorrhage [55]. In tick saliva, they were found to be associated with several physiological processes such as inflammation, fibrinolysis, blood protein digestion, vitellogenesis, immunomodulation, and pathogen transmission [52]. Given their abundance in tick saliva, several studies have focused on characterizing the metalloproteases of various tick species [56,57,58]. Other studies have even used these enzymes as antigens for anti-tick vaccines [52]. Other unusually secreted enzymes such as aconitate hydratase, and enolases were also identified in both genders. The latter enzymes have also been identified in the saliva of Ornithodoros moubata and shown to act as a profibrinolytic plasminogen receptor, most likely helping the tick to maintain the fluidity of host blood during feeding [59]. In H. dromedarii, enolases might be specifically secreted into the saliva where they could act as anti-hemostatic, anti-inflammatory or immunomodulatory compounds [60].
Protease inhibitor proteins including serpins and Kunitz-type proteins were detected in the proteome of both genders. These two super families were abundantly expressed in the sialotranscriptome of H. dromedarii but they were the lowest in the S class in the proteome.
Members of the Kunitz-type family are particularly well characterized as inhibitors of a large number of serine endopeptidases [36]. Most Kunitz domain containing proteins are serine protease inhibitors, although some also block ion channels [37]. Only 2 proteins containing Kunitz domains were found in the current proteomic analysis. Interestingly, these inhibitors have been characterized from previous tick sialomes, as acting upon thrombin, factor Xa, factor XIIa, trypsin and elastase [38]. This raises the suggestion they contribute to H. dromedarii saliva anticoagulant activity [39, 40]. Thus, additional studies are therefore needed to unravel the pharmacological properties of these proteins.
We identified 3 serpins in males and 5 in females, 2 of which were common for both genders. Serpins are one of the most important ubiquitous serine proteases inhibitors that ticks rely on to control host hemostasis and immunity [61]. Thorough functional characterizations of several individual recombinant tick serpins such as Iris and IRS-2 have revealed their anticoagulant, anti-inflammatory and immunomodulatory properties [62, 63]. It is therefore possible that the identified serpins assist the feeding process in H. dromedarii ticks, especially in females that remain attached for long periods of time to become fully engorged. Previous studies have shown that serpins are involved not only in avoiding the host defense system but also in reproduction by contributing substantially to the seminal fluid content in some insect species [61]. It was therefore not surprising that one of the 6 serpins that we identified was only found in males. This serpin may play a role in tick reproduction as did serpins in the seminal fluid of Drosophila [64, 65]. In depth studies of their functions are needed.
Like in the other tick salivary proteomes, proteins from Lipocalin family were found in H. dromedarii salivary glands. These proteins belong to a large family of proteins with low sequence conservation but characteristic structural features including an internal binding site [66]. Small hydrophobic molecules bind to this site and are generally transported to the extracellular environment [67]. Although they are among the most abundant transcripts in the transcriptome of H. dromedarii, lipocalins represent only 0.8% of the S class in both genders in the proteome. By contrast with our results, Lipocalins were the most abundant salivary proteins in the saliva proteome of O. moubata [32] and R. microplus [14]. Interestingly, the high content of Lipocalins in these tick species was suggested to be related to antihemostatic and immunomodulatory functions during feeding [68,69,70]. The amount of the identified Lipocalins does not appear to reflect the actual amount intended to be secreted into the saliva of H. dromedarii, making it likely that the functions of the Lipocalins we identified are similar to those proven in previous studies [70, 71].
Tick-specific proteins including Glycine-rich proteins were also expressed in the salivary glands of both genders. Together with cement proteins, they account for 4% of the S class in females and 3.5% in males. As glycine-rich do not produce suitable tryptic peptides [15], we believe that they might not all be identified in the proteome. Indeed, these proteins are known to play a crucial role in ticks. As a long-term blood feeder, H. dromedarii secrete a cement-like substance to strengthen their attachment to the hosts [37]. These proteins have been used as anti-tick vaccines isolated from other tick species [72,73,74]. Glycine-rich proteins may play roles other than in the tick-host relationship, especially in embryo development as was proven recently in Rhipicephalus microplus [72].
Tick-specific protein families were identified in the current study. Metastriate insulin growth factor-binding protein, a member of this family, was exclusively identified in female salivary glands of H. dromedarii. This proteins family was found in our transcriptomic analysis also [55], and was identified in several previous reviews of tick sialomes [56]. This family has two sets [57]. A shorter form includes only the IB domain, while the longer form has two additional domains, a Kazal domain and the SMART immunoglobulin C-2 type domain [57]. Interestingly, a human homolog proteins containing these three domains, named MC25, has several effects in tissue growth and differentiation [58, 59]. It has also been shown to inhibit vascular endothelial growth factor and keratinocyte growth [60, 61]. Therefore, proteins belonging to this family, identified in this proteome, could serve as binders of growth factors affecting angiogenesis, tissue repair, and immunity [57]. Deeper studies are needed to confirm these hypotheses.
The final difference is the immunity related proteins that account for 4.4% of possibly secreted proteins in males and 9.8% in female ticks. This difference may be explained by the different feeding behaviors of the two genders as previously described: H. dromedarii males do not remain attached to camels as long as females and therefore do not encounter the same host immune constraints. Both genders are thus expected to secrete a different arsenal of salivary molecules involved in encountering host defense. Interestingly, 6 of alpha 2 -macroglobulin (α2M) were identified in females compared to only one in males. These ubiquitous proteins have been identified in invertebrates and vertebrates. In vertebrates, α2M proteins have been found to regulate host cell apoptosis [75], inhibit several serum peptidases like thrombin [76], factor Xa [77] and kallikreins [78], mediate Tcell proliferation [79] and induce the proliferation and activation of macrophages [80]. Nevertheless, some studies on tick α2M have reported that they can intervene in inflammation and immunomodulation [79]. For this reason, we classified α2M proteins among the immunity related proteins in this study. It remains unclear, however, whether these α2M act as immunomodulators or as anticoagulants or as both: this role needs to be elucidated.
Conclusions
Using proteomics informed by transcriptomics, we have generated the most comprehensive set of proteins detected in H. dromedarii salivary glands to date. Several protein families previously found in H. dromedarii transcriptome were identified in proteome. The identification of such proteins was indicative of a broad and complex proteome and concurs with the complexity of the previously described Ixodid sialomes. Our results provide new information regarding H. dromedarii salivary gland composition that may serve to guide further studies seeking to characterize each single protein using molecular, biochemical and pharmacological approaches. This study may provide new information on the tick-host relationship and offer new perspectives for drug discovery.
Methods
Experimental design and PIT workflow
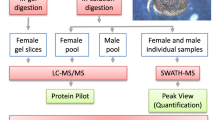
We used a PIT strategy to identify proteins from the salivary glands of both genders of H. dromedarii ticks, as summarized in Fig. 6.

Experimental design; Proteomics Informed by Transcriptomics workflow
Ticks collection and salivary glands dissection
H. dromedarii ticks were collected from camels in the Saharan bioclimatic zone of southern Tunisia (33°25′908″ N, 009°00′952″ E). The camels were thoroughly inspected especially in their inguinal region and legs. Partially engorged ticks were removed manually and placed in flasks. Each tick was identified using a taxonomic key [81]. Within the first hour of collection, the ticks were washed and fixed in paraffin by their legs, after which lateral cuts were made using a scalpel to remove the scutum. The salivary glands of all the collected ticks were immediately teased away from other organs using ultra-fine forceps. Salivary gland samples were organized into pools according to the tick’s gender, one pool for female and one pool for male ticks. After dissection, the salivary glands were gently washed in ice cold phosphate buffered saline (PBS), pH 7.2 and stored at − 80 °C.
Sample preparation and in gel digestion
The proteomic analysis was carried out in the SCSIE_university of Valencia Proteomics Unit, a member of ISCIII ProteoRed Proteomics Platform. Salivary glands were thawed, homogenized in PBS and centrifuged at 14000 rpm and 4 °C for 30 min. Salivary gland extracts (SGE) supernatants were pooled and the SGE protein concentration was determined according to the bicinchoninic acid method (BCA Protein assay, Pierce, Rockford, USA), as previously described [82]. Each solution (~ 50 μg) was diluted by Laemmli SB (1x) to 25 μL. The samples (samples in 1x Laemmli buffer; 5 min a 95 °C) were loaded in 1D PAGE TGX AnyKD (Biorad, Gernany) at 20 mA for 1 h 30 min. The gel was stained with QC colloidal coomasie stain. The gel lanes were sliced and each slice was subjected to in-gel tryptic digestion. Samples were digested with sequencing grade trypsin (Promega, Gernany) as described elsewhere [83]. The digestion was stopped with TFA (1% final concentration). A double extraction with ACN was done. The peptide mixtures were concentrated by speed vacuum to 50 μL. Only 3 μg of the sample was analyzed for both females and males by LC-MS/MS.
Liquid chromatography and tandem mass spectrometry (LC-MS/MS) analysis
Peptides resulting from tryptic digestion were loaded onto a trap column (NanoLC Column, 3 μ C18-CL, 350 μm × 0.5 mm; Eksigen) and desalted with 0.1% TFA at 3 μL/min during 5 min. The peptides were then loaded onto an analytical column (LC Column, 3 μ C18-CL, 75 μm × 12 cm, Nikkyo) equilibrated in 5% acetonitrile 0.1% FA (formic acid). Elution was carried out with a linear gradient from 5 to 35% B in A for 120 min. (A: 0.1% FA; B: ACN, 0.1% FA) at a flow rate of 300 nl/min. Peptides were analyzed in a mass spectrometer nanoESIqQTOF (5600 TripleTOF, ABSCIEX). The triple TOF was operated in information-dependent acquisition mode, in which a 0.25-s TOF MS scan from 350 to 1250 m/z was performed followed by 0.05-s product ion scans from 100 to 1500 m/z on the 50 most intense 2–5 charged ions.
Database search
Protein pilot v4.5. Search engine (ABSciex)
The data obtained for each sample were analyzed and combined for a database search. Protein Pilot default parameters were used to generate a peak list directly from 5600 TripleTofwiff files. The Paragon algorithm of Protein Pilot was used to search the NCBI protein database with the following parameters: trypsin specificity, Iodoacetamidecys-alkylation, and the search effort set to rapid. To avoid using the same spectral evidence in more than one protein, the identified proteins were grouped based on MS/MS spectra by the Protein Pilot Progroup algorithm. Proteins sharing MS/MS spectra were therefore grouped regardless of the peptide sequence assigned. The protein within each group that can explain more spectral data with confidence was shown as the group’s primary protein. Only the proteins of the group for which there was individual evidence (unique peptides with enough confidence) were also listed, usually toward the end of the protein list. Proteins showing a Protein Pilot unused score above 1.3 were identified with greater than 95% confidence and considered significant.
Search engine used for protein identification
The raw files generated by Protein Pilot were used for protein searches using Peaks Studio Software (Bioinformatics Solutions Inc., Canada). Database searches (including a post-translational modification (PTM) search and a sequence variant search) were performed on the Acari database, constructed with proteins retrieved from the “Acari” term. Additionally, tandem MS and MS/MS spectra were searched against a recently published protein fasta database derived from H. dromedarii sialotranscriptome [29]. Searches were done with tryptic specificity allowing one missed cleavage and a tolerance on the mass measurement of 0.1 Da in MS mode and 0.1 Da for MS/MS ions. Carbamidomethylation of Cys was used as a fixed modification and oxidation of Met and deamidation of Asn and Gln as variable modifications. Proteins showing a score higher than homology or significance threshold were identified with greater than 95% confidence.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Abbreviations
- ACN:
-
AcetonitrileA
- BCA:
-
bicinchoninic acid
- CDS:
-
Coding domain sequence
- FA:
-
Formic acid
- LC:
-
Liquid chromatography
- MS:
-
Mass spectometry
- NGS:
-
Next Generation Sequencing
- PAGE:
-
Polyacrylamide gel electrophoresis
- PIT:
-
Proteomic informed by transcriptomic
- PTM:
-
Post-translational modification
- RNA-seq:
-
Ribonucleic acid-sequencing
- SAT:
-
saliva-assisted transmission or
- SGE:
-
Salivary gland extracts
- TFA:
-
Trifluoroacetic acid
- α2M:
-
alpha 2 –macroglobulin
References
Allen JR. Immunology of interactions between ticks and laboratory animals. Exp Appl Acarol. 1989;7:5–13.
Francischetti IMB, Anderson JM, Manoukis N, Pham VM, Ribeiro JMC. An insight into the sialotranscriptome and proteome of the coarse bontlegged tick, Hyalomma marginatum rufipes. J Proteome. 2011;74:2892–908.
Kazimírová M, Štibrániová I. Tick salivary compounds: their role in modulation of host defences and pathogen transmission. Front Cell Infect Microbiol. 2013;3:43.
Francischetti IMB, Sa-Nunes A, Mans BJ, Santos IM, Ribeiro JMC. The role of saliva in tick feeding. Front Biosci. 2009;14:2051–88.
Chmelar J, Calvo E, Pedra JHF, Francischetti IMB, Kotsyfakis M. Tick salivary secretion as a source of antihemostatics. J Proteome. 2012;75:3842–54.
Šimo L, Kazimirova M, Richardson J, Bonnet SI. The essential role of tick salivary glands and saliva in tick feeding and pathogen transmission. Front Cell Infect Microbiol. 2017;7:281.
Nuttall PA, Labuda M. Saliva-assisted transmission of tick-borne pathogens. In: Bowman AS, Nuttall PA, editors. Ticks. Cambridge: Cambridge University Press; 2008. p. 205–19. http://ebooks.cambridge.org/ref/id/CBO9780511551802A017. Accessed 20 Jan 2017.
Assumpção TC, Mizurini DM, Ma D, Monteiro RQ, Ahlstedt S, Reyes M, et al. Ixonnexin from tick saliva promotes fibrinolysis by interacting with plasminogen and tissue-type plasminogen activator, and prevents arterial thrombosis. Sci Rep. 2018;8:4806.
Assumpção TC, Ma D, Mizurini DM, Kini RM, Ribeiro JMC, Kotsyfakis M, et al. In vitro mode of action and anti-thrombotic activity of Boophilin, a multifunctional Kunitz protease inhibitor from the midgut of a tick vector of Babesiosis, Rhipicephalus microplus. PLOS Neglected Tropical Diseases. 2016;10:e0004298.
Díaz-Martín V, Manzano-Román R, Oleaga A, Pérez-Sánchez R. New salivary anti-haemostatics containing protective epitopes from Ornithodoros moubata ticks: assessment of their individual and combined vaccine efficacy. Vet Parasitol. 2015;212:336–49.
Chmelař J, Kotál J, Karim S, Kopacek P, Francischetti IMB, Pedra JHF, et al. Sialomes and Mialomes: a systems-biology view of tick tissues and tick–host interactions. Trends Parasitol. 2016;32:242–54.
Chmelař J, Kotál J, Kopecký J, JHF P, Kotsyfakis M. All For One and One For All on the Tick-Host Battlefield. Trends in Parasitology. 2016.
Steen NA, Barker SC, Alewood PF. Proteins in the saliva of the Ixodida (ticks): pharmacological features and biological significance. Toxicon. 2006;47:1–20.
Tirloni L, Reck J, Terra RMS, Martins JR, Mulenga A, Sherman NE, et al. Proteomic analysis of cattle tick Rhipicephalus (Boophilus) microplus saliva: a comparison between partially and fully engorged females. PLoS One. 2014;9:e94831.
Tan AWL, Francischetti IMB, Slovak M, Manjunatha KR, JMC R. Sexual differences in the sialomes of the zebra tick, Rhipicephalus pulchellus. J Proteomics. 2015.
Mans BJ, Andersen JF, Francischetti IMB, Valenzuela JG, Schwan TG, Pham VM, et al. Comparative sialomics between hard and soft ticks: implications for the evolution of blood-feeding behavior. Insect Biochem Mol Biol. 2008;38:42–58.
Evans VC, Barker G, Heesom KJ, Fan J, Bessant C, Matthews DA. De novo derivation of proteomes from transcriptomes for transcript and protein identification. Nat Meth. 2012;9:1207–11.
Mudenda L, Pierlé SA, Turse JE, Scoles GA, Purvine SO, Nicora CD, et al. Proteomics informed by transcriptomics identifies novel secreted proteins in Dermacentor andersoni saliva. Int J Parasitol. 2014;44:1029–37.
Collins LJ, Biggs PJ, Voelckel C, Joly S. An approach to transcriptome analysis of non-model organisms using short-read sequences. Genome Inform. 2008;21:3–14.
Apanaskevich DA, Schuster AL, Horak IG. The genus Hyalomma: VII. Redescription of all parasitic stages of H.(Euhyalomma) dromedarii and H.(E.) schulzei (Acari: Ixodidae). J Med Entomol. 2008;45:817–31.
Estrada-Peña A. Ticks of domestic animals in the Mediterranean region: a guide to identification of species. University of Zaragoza; 2004.
Alanazi AD, Al-Mohammed HI, Alyousif MS, Said AE, Salim B, Abdel-Shafy S, et al. Species diversity and seasonal distribution of hard ticks (Acari: Ixodidae) infesting mammalian hosts in various districts of Riyadh Province. Saudi Arabia J Med Entomol. 2019.
de Kok JB, d’Oliveira C, Jongejan F. Detection of the protozoan parasite Theileria annulata in Hyalomma ticks by the polymerase chain reaction. Exp Appl Acarol. 1993;17:839–46.
Abdullah HHAM, El-Molla A, Salib FA, Allam NAT, Ghazy AA, Abdel-Shafy S. Morphological and molecular identification of the brown dog tick Rhipicephalus sanguineus and the camel tick Hyalomma dromedarii (Acari: Ixodidae) vectors of rickettsioses in Egypt. Veterinary World. 2016;9:1087–101.
Karim S, Budachetri K, Mukherjee N, Williams J, Kausar A, Hassan MJ, et al. A study of ticks and tick-borne livestock pathogens in Pakistan. PLoS Negl Trop Dis. 2017;11:e0005681.
Bensaoud C, Abdelkafi-Koubaa Z, Ben Mabrouk H, Morjen M, Hmila I, Rhim A, et al. Hyalomma dromedarii (Acari: Ixodidae) salivary gland extract inhibits angiogenesis and exhibits in vitro antitumor effects. J Med Entomol. 2017. https://doi.org/10.1093/jme/tjx153.
Ibrahim MA, Masoud HMM. Thrombin inhibitor from the salivary gland of the camel tick Hyalomma dromedarii. Exp Appl Acarol. 2018;74:85–97.
Abdullah HHAM, El-Shanawany EE, Abdel-Shafy S, Abou-Zeina HAA, Abdel-Rahman EH. Molecular and immunological characterization of Hyalomma dromedarii and Hyalomma excavatum (Acari: Ixodidae) vectors of Q fever in camels. Vet World. 2018;11:1109–19.
Bensaoud C, Nishiyama MY, Ben Hamda C, Lichtenstein F, Castro de Oliveira U, Faria F, et al. De novo assembly and annotation of Hyalomma dromedarii tick (Acari: Ixodidae) sialotranscriptome with regard to gender differences in gene expression. Parasit Vectors. 2018;11:314.
Marzouk AS, Darwish ZE. Changes in the salivary glands of female Hyalomma (Hyalomma) dromedarii during and after feeding. J Egypt Soc Parasitol. 1994;24:39–57.
Francischetti IMB, Meng Z, Mans BJ, Gudderra N, Hall M, Veenstra TD, et al. An insight into the salivary transcriptome and proteome of the soft tick and vector of epizootic bovine abortion. Ornithodoros coriaceus J Proteomics. 2008;71:493–512.
Díaz-Martín V, Manzano-Román R, Valero L, Oleaga A, Encinas-Grandes A, Pérez-Sánchez R. An insight into the proteome of the saliva of the argasid tick Ornithodoros moubata reveals important differences in saliva protein composition between the sexes. J Proteome. 2013;80:216–35.
Oliveira CJ, Anatriello E, de Miranda-Santos IK, Francischetti IM, Sá-Nunes A, Ferreira BR, et al. Proteome of Rhipicephalus sanguineus tick saliva induced by the secretagogues pilocarpine and dopamine. Ticks Tick Borne Dis. 2013;4:469–77.
Siwiak M, Zielenkiewicz P. Co-regulation of translation in protein complexes. Biol Direct. 2015;10:18.
Nagalakshmi U, Waern K, Snyder M. RNA-Seq: a method for comprehensive transcriptome analysis. Curr Protoc Mol Biol. 2010;Chapter 4:Unit 4.11.1–13.
Anderson JF, Magnarelli LA. Biology of ticks. Infect Dis Clin N Am. 2008;22:195–215 v.
Bowman AS, Sauer JR. Tick salivary glands: function, physiology and future. Parasitology. 2005;129:S67.
Chinery WA. Studies on the various glands of the tick Haemaphysalis spinigera Neumann 1897. Acta Trop. 1965;22:235–66.
Furquim KCS, Bechara GH, Camargo Mathias MI. Morpho-histochemical characterization of salivary gland cells of males of the tick Rhipicephalus sanguineus (Acari: Ixodidae) at different feeding stages: description of new cell types. Exp Appl Acarol. 2010;50:59–70.
Feldman-Muhsam B, Borut S, Saliternik-Givant S. Salivary secretion of the male tick during copulation. J Insect Physiol. 1970;16:1945–9.
Anderson JF. The natural history of ticks. Med Clin North Am. 2002;86:205–18.
de Castro MH, de Klerk D, Pienaar R, Latif AA, Rees DJG, Mans BJ. De novo assembly and annotation of the salivary gland transcriptome of Rhipicephalus appendiculatus male and female ticks during blood feeding. Ticks Tick Borne Dis. 2016;7:536–48.
Francischetti IMB, Sa-Nunes A, Mans BJ, Santos IM, Ribeiro JMC. The role of saliva in tick feeding. Front Biosci (Landmark Ed). 2009;14:2051–88.
Aguilera L, Ferreira E, Giménez R, Fernández FJ, Taulés M, Aguilar J, et al. Secretion of the housekeeping protein glyceraldehyde-3-phosphate dehydrogenase by the LEE-encoded type III secretion system in enteropathogenic Escherichia coli. Int J Biochem Cell Biol. 2012;44:955–62.
Bendtsen JD, Jensen LJ, Blom N, Von Heijne G, Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel. 2004;17:349–56.
Ibrahim MA, Mohamed MM, Ghazy A-HM, Masoud HMM. Superoxide dismutases from larvae of the camel tick Hyalomma dromedarii. Comp Biochem Physiol B: Biochem Mol Biol. 2013;164:221–8.
Rosa de Lima MF, Sanchez Ferreira CA, Joaquim de Freitas DR, Valenzuela JG, Masuda A. Cloning and partial characterization of a Boophilus microplus (Acari: Ixodidae) glutathione S-transferase. Insect Biochem Mol Biol. 2002;32:747–54.
da Silva VI, Torino Lermen T, Michelon A, Sanchez Ferreira CA, Joaquim de Freitas DR, Termignoni C, et al. Effect of acaricides on the activity of a Boophilus microplus glutathione S-transferase. Vet Parasitol. 2004;119:237–45.
Ribeiro JMC, Slovák M, Francischetti IMB. An insight into the sialome of Hyalomma excavatum. Ticks Tick Borne Dis. 2016;8:201–7.
Karim S, Singh P, Ribeiro JMC. A deep insight into the Sialotranscriptome of the Gulf coast tick. Amblyomma maculatum PLoS ONE. 2011;6:e28525.
Francischetti IMB, Mans BJ, Meng Z, Gudderra N, Veenstra TD, Pham VM, et al. An insight into the sialome of the soft tick. Ornithodorus parkeri Insect Biochem Mol Biol. 2008;38:1–21.
Ali A, Khan S, Ali I, Karim S, da Silva VI, Termignoni C. Probing the functional role of tick metalloproteases. Physiol Entomol. 2015;40:177–88.
Macours N, Hens K. Zinc-metalloproteases in insects: ACE and ECE. Insect Biochem Mol Biol. 2004;34:501–10.
Giebeler N, Zigrino P. A Disintegrin and Metalloprotease (ADAM): Historical Overview of Their Functions. Toxins (Basel). 2016;8. https://doi.org/10.3390/toxins8040122.
Tasoulis T, Isbister GK. A Review and Database of Snake Venom Proteomes. Toxins (Basel). 2017;9.
Ali A, Tirloni L, Isezaki M, Seixas A, Konnai S, Ohashi K, et al. Reprolysin metalloproteases from Ixodes persulcatus, Rhipicephalus sanguineus and Rhipicephalus microplus ticks. Experimental & Applied Acarology. 2014;63:559–78.
Ali A, Fernando Parizi L, Garcia Guizzo M, Tirloni L, Seixas A, da Silva Vaz Jr I, et al. Immunoprotective potential of a Rhipicephalus (Boophilus) microplus metalloprotease. Vet Parasitol. 2015;207:107–14.
Francischetti IMB, Mather TN, Ribeiro JMC. Cloning of a salivary gland metalloprotease and characterization of gelatinase and fibrin (ogen) lytic activities in the saliva of the Lyme disease tick vector Ixodes scapularis. Biochem Biophys Res Commun. 2003;305:869–75.
Díaz-Martín V, Manzano-Román R, Oleaga A, Encinas-Grandes A, Pérez-Sánchez R. Cloning and characterization of a plasminogen-binding enolase from the saliva of the argasid tick Ornithodoros moubata. Vet Parasitol. 2013;191:301–14.
Xu X-L, Cheng T-Y, Yang H. Enolase, a plasminogen receptor isolated from salivary gland transcriptome of the ixodid tick Haemaphysalis flava. Parasitol Res. 2016;115:1955–64.
Meekins DA, Kanost MR, Michel K. Serpins in arthropod biology. Semin Cell Dev Biol. 2016;62:105–19.
Páleníková J, Lieskovská J, Langhansová H, Kotsyfakis M, Chmelař J, Kopecký J. Ixodes ricinus salivary serpin IRS-2 affects Th17 differentiation via inhibition of the interleukin-6/STAT-3 signaling pathway. Infect Immun. 2015;83:1949–56.
Toyomane K, Konnai S, Niwa A, Githaka N, Isezaki M, Yamada S, et al. Identification and the preliminary in vitro characterization of IRIS homologue from salivary glands of Ixodes persulcatus Schulze. Ticks Tick Borne Dis. 2016;7:119–25.
Walker MJ, Rylett CM, Keen JN, Audsley N, Sajid M, Shirras AD, et al. Proteomic identification of Drosophila melanogaster male accessory gland proteins, including a pro-cathepsin and a soluble gamma-glutamyl transpeptidase. Proteome Sci. 2006;4:9.
Karr TL, Southern H, Rosenow MA, Gossmann TI, Snook RR. The old and the new: discovery proteomics identifies putative novel seminal fluid proteins in Drosophila. Mol Cell Proteomics. 2019;18(Suppl 1):S23–33.
Flower DR. The lipocalin protein family: structure and function. Biochem J. 1996;318(Pt 1):1–14.
Ganfornina MD, Gutiérrez G, Bastiani M, Sánchez D. A phylogenetic analysis of the lipocalin protein family. Mol Biol Evol. 2000;17:114–26.
Paesen GC, Adams PL, Harlos K, Nuttall PA, Stuart DI. Tick histamine-binding proteins: isolation, cloning, and three-dimensional structure. Mol Cell. 1999;3:661–71.
Rodriguez-Valle M, Moolhuijzen P, Piper EK, Weiss O, Vance M, Bellgard M, et al. Rhipicephalus microplus lipocalins (LRMs): genomic identification and analysis of the bovine immune response using in silico predicted B and T cell epitopes. Int J Parasitol. 2013;43:739–52.
Beaufays J, Adam B, Decrem Y, Prévôt P-P, Santini S, Brasseur R, et al. Ixodes ricinus tick Lipocalins: identification, cloning, Phylogenetic Analysis and Biochemical Characterization. PLoS ONE. 2008;3:e3941.
Beaufays J, Adam B, Menten-Dedoyart C, Fievez L, Grosjean A, Decrem Y, et al. Ir-LBP, an Ixodes ricinus tick salivary LTB4-binding Lipocalin, Interferes with Host Neutrophil Function. PLoS ONE. 2008;3:e3987.
Leal BF, Alzugaray MF, Seixas A, Da Silva Vaz I, Ferreira CAS. Characterization of a glycine-rich protein from Rhipicephalus microplus: tissue expression, gene silencing and immune recognition. Parasitology. 2018;145:927–38.
Bishop R, Lambson B, Wells C, Pandit P, Osaso J, Nkonge C, et al. A cement protein of the tick Rhipicephalus appendiculatus, located in the secretory e cell granules of the type III salivary gland acini, induces strong antibody responses in cattle. Int J Parasitol. 2002;32:833–42.
Maruyama SR, Anatriello E, Anderson JM, Ribeiro JM, Brandão LG, Valenzuela JG, et al. The expression of genes coding for distinct types of glycine-rich proteins varies according to the biology of three metastriate ticks, Rhipicephalus (Boophilus) microplus, Rhipicephalus sanguineus and Amblyomma cajennense. BMC genomics. 2010;11:363.
De Souza EM, Meuser-Batista M, Batista DG, Duarte BB, Araújo-Jorge TC, Soeiro MNC. Trypanosoma cruzi: Alpha-2-macroglobulin regulates host cell apoptosis induced by the parasite infection in vitro. Exp Parasitol. 2008;118:331–7.
Cvirn G, Gallistl S, Koestenberger M, Kutschera J, Leschnik B, Muntean W. Alpha 2-macroglobulin enhances prothrombin activation and thrombin potential by inhibiting the anticoagulant protein C/protein S system in cord and adult plasma. Thromb Res. 2002;105:433–9.
Meijers JC, Tijburg PN, Bouma BN. Inhibition of human blood coagulation factor Xa by alpha 2-macroglobulin. Biochemistry. 1987;26:5932–7.
Harpel PC. Human plasma alpha 2-macroglobulin. An inhibitor of plasma kallikrein. J Exp Med. 1970;132:329–52.
Banks RE, Evans SW, Van Leuven F, Alexander D, McMahon MJ, Whicher JT. Measurement of the “fast” or complexed form of alpha 2 macroglobulin in biological fluids using a sandwich enzyme immunoassay. J Immunol Methods. 1990;126:13–20.
Bonacci GR, Cáceres LC, Sánchez MC, Chiabrando GA. Activated alpha(2)-macroglobulin induces cell proliferation and mitogen-activated protein kinase activation by LRP-1 in the J774 macrophage-derived cell line. Arch Biochem Biophys. 2007;460:100–6.
Bouattour A. Cle dichotomique et identification des tiques (Acari: Ixodidae) parasites du betail au Maghreb. Arch Inst Pasteur Tunis. 2002;79:43–50.
Brown RE, Jarvis KL, Hyland KJ. Protein measurement using bicinchoninic acid: elimination of interfering substances. Anal Biochem. 1989;180:136–9.
Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, et al. Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc Natl Acad Sci U S A. 1996;93:14440–5.
Acknowledgments
We would like to thank Dr. Deborah Glassman for her English corrections and constructive comments on the manuscript.
Funding
This work was conducted with financial support from the Collaborative internal project PCI-05/2012-IPT and the Tunisian Ministry for Higher Education, Scientific Research, and Technology. The authors (Juliana Mozer Sciani, Fernanda Faria, and Ana Marisa Chudzinski-Tavassi) received financial support from CAPES (Auxpe-Toxinologia 1207/2011) and FAPESP (2013/07467–1). The funders had no role in design, data collection and analysis decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
CB contributed to the collection and identification of ticks, performing the experiments drafting the manuscript. HA contributed in drafting the manuscript. JMS contributed in the data analysis and drafting the paper. FF contributed to the design of work, supervision of the work and correction of the manuscript. AB contributed to correction of the manuscript. AMC contributed to the supervision of the work. YM contributed to the design and supervision of the work and correction of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Commission of Ethics and Animal Welfare of the Institut Pasteur of Tunis have approved the study with the given number IPT/LR03/Project PCI/25. All technical procedures were in accordance with the National and the European legislation regarding animal welfare and have met the International Guiding Principles for Biomedical Research Involving Animals by the Council for the International Organizations of Medical Sciences.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Classification of proteins identified in the salivary glands of Hyalomma dromedarii females. (XLSX 98 kb)
Additional file 2:
Table S2. Classification of proteins identified in the salivary glands of Hyalomma dromedarii males. Description of additional files. Accession: protein ID in the database. %coverage: percentage Coverage_ The percentage of all the amino acids in the protein sequence that were covered by identified peptides detected in the sample. -10logP: the P-value is converted to -10log10 (P-value). In PEAKS, this value is denoted by -10lgP as lg is the ISO reserved notation for log10. By this conversion, a more significant match will have a higher -10lgP value. Additionally, a P-value of 1% is equivalent to -10lgP of 20. #Peptides: number of peptides identified following LC-MS/MS analysis for a single protein. #Unique: Unique peptides_peptides with the same amino acid sequence but different charges or with different modifications are grouped together and counted only once. Avg.Mass: the average mass of a single protein obtained by summing the average atomic masses of the constituent elements. Description: description of the identified proteins in the database. Class: housekeeping class (H); possibly secreted proteins (S); transporters (T); and unknown class (U). (XLSX 94 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bensaoud, C., Aounallah, H., Sciani, J.M. et al. Proteomic informed by transcriptomic for salivary glands components of the camel tick Hyalomma dromedarii. BMC Genomics 20, 675 (2019). https://doi.org/10.1186/s12864-019-6042-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-6042-1