
Abstract
Background
The trihelix gene family is a plant-specific transcription factor family that plays important roles in plant growth, development, and responses to abiotic stresses. However, to date, no systemic characterization of the trihelix genes has yet been conducted in wheat and its close relatives.
Results
We identified a total of 94 trihelix genes in wheat, as well as 22 trihelix genes in Triticum urartu, 29 in Aegilops tauschii, and 31 in Brachypodium distachyon. We analyzed the chromosomal locations and orthology relations of the identified trihelix genes, and no trihelix gene was found to be located on chromosome 7A, 7B, or 7D of wheat, thereby reflecting the uneven distributions of wheat trihelix genes. Phylogenetic analysis indicated that the 186 identified trihelix proteins in wheat, rice, B. distachyon, and Arabidopsis were clustered into five major clades. The trihelix genes belonging to the same clades usually shared similar motif compositions and exon/intron structural patterns. Five pairs of tandem duplication genes and three pairs of segmental duplication genes were identified in the wheat trihelix gene family, thereby validating the supposition that more intrachromosomal gene duplication events occur in the genome of wheat than in that of other grass species. The tissue-specific expression and differential expression profiling of the identified genes under cold and drought stresses were analyzed by using RNA-seq data. qRT-PCR was also used to confirm the expression profiles of ten selected wheat trihelix genes under multiple abiotic stresses, and we found that these genes mainly responded to salt and cold stresses.
Conclusions
In this study, we identified trihelix genes in wheat and its close relatives and found that gene duplication events are the main driving force for trihelix gene evolution in wheat. Our expression profiling analysis demonstrated that wheat trihelix genes responded to multiple abiotic stresses, especially salt and cold stresses. The results of our study built a basis for further investigation of the functions of wheat trihelix genes and provided candidate genes for stress-resistant wheat breeding programs.
Similar content being viewed by others


Background
Transcription factors (TFs) are extensively involved in the processes of plant growth and development through binding to specific cis-elements to modulate the expressions of target genes [1]. More than 60 TF families have been found in plants, many of which have been thoroughly studied to evaluate their functions in grasses [2]. As one of the first TFs discovered in plants, trihelix proteins widely participate in diverse development processes and abiotic stress responses [3]. Trihelix TFs are plant-specific, suggesting that they might be involved in plant-specific gene regulations [4]. However, the trihelix gene family has not been systematically identified in wheat or its closely related plant species. Trihelix TFs contain one or two trihelix DNA-binding domains which could specifically bind to the GT elements of promoters. The trihelix DNA-binding domain contains a typical trihelix structure (three α-helixes separated by two loops) and is similar to the Myb DNA-binding domains in sequence [5].
The first identified trihelix gene GT-1 was found in Pisum sativum, and orthologous genes were subsequently cloned in tobacco and Arabidopsis [6,7,8,9]. GT-1 protein directly interacts with pre-initiation complex and activates transcription. Early studies on Arabidopsis suggest that trihelix genes play multiple roles in diverse development processes. Arabidopsis ASIL1 targets GT-box-containing embryonic genes and represses the expression of embryonic seed maturation genes in vegetative tissues [10, 11]. The gain-of-function Arabidopsis mutant of the PTL (PETAL LOSS) gene causes male sterility and other pleiotropic phenotypes [12]. GTL1 has been reported to be involved in regulating ploidy-dependent cell growth in the Arabidopsis trichome [13].
In recent years, evidence has shown that trihelix proteins are extensively involved in the plant response to different abiotic stresses. Overexpression of GmGT-2A and GmGT-2B in Arabidopsis could improve its tolerance to abiotic stresses [14]. The OsGTγ-1 gene has also been proven to respond to salt stress in rice [15]. GTL1 affects plant water use efficiency and its tolerance to drought stress [16]. AtGT2L could interact with calmodulin and is involved in the abiotic stress response [17]. AtGT-4 modulates Arabidopsis salt tolerance by interacting with TEM2 [18]. TaGT2L1D regulates plant development and drought tolerance in wheat [19]. ShCIGT regulates the cold and drought tolerance of tomato by interacting with SnRK1 [20]. By binding to AGAG-Box, AtAST1 mediates Arabidopsis salt and osmotic stress tolerance [21].
A total of 30 and 31 trihelix proteins have been identified in Arabidopsis and rice, respectively, and these proteins could be divided into five clades named GT-1, GT-2, GTγ, SH4, and SIP1 [4, 11]. Trihelix proteins were subsequently identified in many other plant species. Prior studies report 63 trihelix genes in Glycine max, 36 in Solanum lycopersicum, 20 in Chrysanthemum morifolium, 56 in Populus trichocarpa, 10 in Camellia sinensis, and 52 in Brassica rapa [22,23,24,25,26,27].
As an important cereal crop, wheat has a very high yield. According to the latest forecast from the Food and Agriculture Organization of the United Nations (FAO, http://www.fao.org/worldfoodsituation/csdb/en/), global cereal production in 2018 was 2.59 billion tonnes, of which 722.4 million tonnes was wheat yield. Crop productivity and food security are affected by many factors, including diverse environmental factors, such as drought, salt, and temperature stresses. As global climate change intensifies, the adverse effects of these abiotic stresses may be enhanced [28, 29]. Studies on wheat trihelix genes could contribute to stress-resistant crop breeding.
In the present study, the trihelix gene family was identified in hexaploid bread wheat (Triticum aestivum L.) and its relatives, including Triticum urartu, Aegilops tauschii, and Brachypodium distachyon. The chromosomal distributions, protein characteristics, gene structures, and conserved motif compositions of the identified trihelix genes were analyzed. We then identified orthology relations, analyzed gene duplication events, and constructed the phylogenetic trees of the identified trihelix genes. Using public RNA-seq data, we analyzed the spatial and temporal expressions and differential expression profiles of wheat trihelix genes under abiotic stresses. In addition, our qRT-PCR results validated the supposition that wheat trihelix genes participate in various abiotic stress responses. Our research provides valuable clues for future functional characterization of trihelix genes in wheat.
Results
Identification of trihelix genes
The Hidden Markov Model (HMM) profile of trihelix domain (PF13837) was used to search the trihelix domain in the wheat protein database (IWGSC RefSeq v1.0). The SMART and HMMER websites were used to confirm that all candidate genes identified contain the trihelix domain. A total of 94 non-redundant trihelix genes were identified in wheat (Additional file 1: Tables S1–S3). To confirm the reliability of the above identification, 31 published rice trihelix genes were used to search their homologous genes in wheat on NCBI by BLAST, and we found that all published wheat proteins containing the trihelix domain were included. The wheat trihelix genes were named from TaGT-1 to TaGT-94 based on their positions on the chromosomes. Additionally, TaGT2L1A, TaGT2L1B, and TaGT2L1D have been identified and named in previous studies [19], and these genes were found to be identical to TaGT-10, TaGT-41, and TaGT-73, respectively, in our 94 wheat trihelix genes.
T. urartu (diploid, AA) and Ae. tauschii (diploid, DD) are the progenitors of the hexaploid wheat (T. aestivum, AABBDD). The wild grass B. distachyon is the first sequenced member of the Pooideae subfamily. These grasses have a close genetic relationship with wheat. We identified 22, 29, and 31 trihelix genes in T. urartu, Ae. tauschii, and B. distachyon, respectively (Additional file 1: Tables S1–S3).
We predicted the isoelectric point (pI) and molecular weight (Mw) of trihelix genes by using the ExPASy pI/Mw tool. Wheat trihelix proteins were found to have large variations in length in the range of 197–851 amino acid residues. The trihelix proteins in wheat also varied greatly in pI (5.14–10.57) and Mw (21.6–94.4 kDa), and they showed similar variations in T. urartu, Ae. tauschii, and B. distachyon (Additional file 1: Table S1). Given that subcellular localization information could provide certain clues for the protein function study, subcellular localizations of trihelix proteins in wheat were predicted by using WOLF PSORT (Additional file 1: Table S1). The results of subcellular localization prediction of wheat trihelix proteins showed that most trihelix proteins are located in the nuclei, in accordance with their roles as TFs.
Chromosomal distribution of trihelix genes
The positions of trihelix genes were obtained from the genome annotation files. A total of 30, 32, and 31 wheat trihelix genes were located on subgenomes A, B, and D (Fig. 1, Additional file 1: Table S1), respectively. TaGT-94 was located on chromosome TaUn because of the incomplete wheat genome sequence. Given that TaGT-26 and TaGT-54 are homologous with TaGT-94, we speculated that the actual location of TaGT-94 might be located on the distal of chromosome 4DL or 5DL. Chromosome 2D had eight genes, the largest number of wheat trihelix genes found in a single chromosome. No trihelix gene was located on chromosome 7A, 7B, or 7D. The numbers of trihelix genes distributed in the remaining chromosomes showed little difference, and relatively high densities were detected at the distal of each chromosome.

Locations and duplication events of trihelix genes on wheat chromosomes. Red boxes indicate tandem duplications, and red lines indicate segmental duplications. The image was drawn via MapInspect
The trihelix genes in T. urartu, Ae. tauschii, and B. distachyon were named according to their positions and tended to distribute at the distal of each chromosome (Additional file 2: Figures S1–S3, Additional file 1: Table S1). No trihelix gene was located on chromosome 7A of T. urartu or 7D of Ae. tauschii. Trihelix genes existed on all 5 chromosomes of B. distachyon and 11 genes located on chromosome Bd3, which showed the largest number of trihelix genes.
Identification of orthologs between wheat and its relatives
Given that orthology relations reflect species phylogenies and can be used to transfer annotations from a known gene to another newly-sequenced genome, ortholog identification has become very important [30, 31]. Here, we used OrthoGNC software to predict pairwise orthologous genes among wheat, T. urartu, Ae. tauschii, B. distachyon, and rice (Table 1).
Wheat is an allohexaploid species with a complex genetic background derived from two naturally interspecific hybridization events of three diploid donor species [32, 33]. Therefore, each wheat gene usually has three homologous copies. Using BLASTP, orthologous relationships among all wheat trihelix genes were identified and are described in Table 1. Likely due to gene loss or the incomplete genome sequence, no gene was identified on subgenome A as a homologous gene with TaGT-31 and TaGT-63, and no gene was identified on subgenome A or B as homologous with TaGT-74. Of particular interest here is that TaGT-42 and TaGT-75 had a homologous gene TraesCS2A02G436000.1 located on subgenome A, which reveals fragment deletion of 170 amino acids at the N-terminal containing a trihelix domain; by contrast, nearly no amino acid difference was found at the C-terminal. Based on this finding, TraesCS2A02G436000.1 was not identified as a trihelix gene in our study.
Phylogenetic analysis and genome synteny analysis of trihelix genes
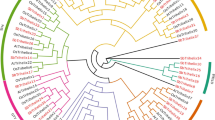
Using MEGA7, we constructed an unrooted phylogenetic tree following the full amino acid sequences of 186 identified trihelix proteins in Arabidopsis, rice, B. distachyon, and wheat (Fig. 2). Trihelix proteins were clustered into five major clades, consistent with the results in B. rapa and P. trichocarpa [22, 24]. The five major clades were named GT-1, GT-2, SIP1, SH4, and GTγ based on studies on rice and Arabidopsis [4], and these clades respectively contained 10, 15, 38, 15, and 16 wheat trihelix proteins (Additional file 1: Table S1). The distribution trends were similar to those in Arabidopsis and rice: the SIP1 clade contained the maximum number of members and the GT-1 clade contained the minimum number of members.

Phylogenetic tree of trihelix proteins in wheat, B. distachyon, rice, and Arabidopsis. The unrooted tree was drawn by MEGA 7.0 with the sequences of 186 identified trihelix proteins. The bootstrap value was set to 1000 replicates
Both tandem and segmental duplications are essential to gene family evolution for adapting to various environmental conditions [34, 35]. In this study, five pairs of genes among 94 trihelix genes of wheat were identified as tandem duplications, and three pairs of genes were considered segmental duplications (Fig. 1). TuGT-12/TuGT-13 was the only pair of tandem duplication genes identified in T. urartu (AA) (Additional file 2: Figure S1), and these genes were orthologous with TaGT-13/TaGT-14 in wheat subgenome A. AetGT-13/AetGT-14 were the only pair of tandem duplication genes identified in Ae. tauschii (DD) (Additional file 2: Figure S2), and these genes were orthologous with TaGT-77/TaGT-78 in wheat subgenome D. TaGT-13/TaGT-14 were homologous with TaGT-77/TaGT-78. These results suggest that the tandem duplication genes TaGT-13/TaGT-14 formed before the interspecific hybridization events of three diploid donor species. During the speciation and evolution of bread wheat, a new duplication event occurred, and the tandem duplication genes TaGT-12/TaGT-13 were generated. No segmental duplication gene was identified in T. urartu. We identified AetGT-5/AetGT16 as a pair of segmental duplication genes in Ae. tauschii. No trihelix gene in B. distachyon was identified as a tandem duplication, and 10 pairs of genes were found to be segmental duplications (Additional file 2: Figure S3).
To analyze the synteny relationships of trihelix genes between T. aestivum, B. distachyon, and rice, we used the Multiple Collinearity Scan toolkit (MCScanX). Approximately 80.6% (25 of 31) the rice trihelix genes exhibited synteny with trihelix genes in wheat (Additional file 2: Figure S4, Additional file 1: Table S4). Furthermore, 74.2% (23 of 31) of the trihelix genes in B. distachyon were found to have synteny with wheat trihelix genes (Additional file 2: Figure S5, Additional file 1: Table S4).
Motif composition and gene structure analysis of the trihelix genes
The conserved motifs of trihelix genes in Arabidopsis, rice, B. distachyon, and wheat were analyzed by using MEME Suite tool. Motif logos were also obtained (Additional file 2: Figure S6). A total of 13 conserved motifs were identified. Gene Structure Display Server 2.0 was used to analyze and visualize the exon and intron structures of trihelix genes in wheat and B. distachyon.
Trihelix genes belonging to the same clade usually have similar motif compositions and exon/intron structures (Fig. 3), thereby indicating that they may have similar functions. Except for GTγ clade genes, all trihelix proteins contained motif 1 and motif 2. GTγ clade genes and some SH4 clade genes featured motif 8 at their C-terminal, and all SIP1 clade genes contained motif 10 at their C-terminal. GT-2 clade genes had two trihelix domains, with motif 3 and motif 12 in the middle of the protein sequence. The majority of trihelix genes (90% in wheat, 87% in B. distachyon) had very few introns (0–2). All GTγ clade genes had no intron (one exon). Some members belonging to GT-1 and GT-2 clades had more than 10 introns. TaGT-28, TaGT-61, and TaGT-91 are homologs and the longest trihelix genes in wheat, containing 16 introns. In B. distachyon, BdGT-20 and BdGT-17 are the longest trihelix genes, containing 19 and 16 introns, respectively.

Phylogenetic tree, motif compositions, and gene structures of trihelix genes. a Unrooted neighbor-joining phylogenetic tree. The tree was built using trihelix protein sequences of Arabidopsis, rice, B. distachyon, and wheat. b Schematic representation of conserved motifs. Colored boxes indicate different conserved motifs. c Exon/intron organization. Exons are shown as yellow boxes, and introns are shown as gray lines
Tissue-specific expression analysis of trihelix genes in wheat
To analyze the tissue-specific expression profiles of 94 wheat trihelix genes, public RNA-seq data of wheat cv. Chinese Spring was obtained from the expVIP website. The data covered gene expression profiles of different tissues including root, stem, leaf, spike, and grain throughout the entire life cycle of wheat. Hierarchical cluster analysis was conducted based on the log2 of transcript per million (TPM) values of 94 wheat trihelix genes (Fig. 4, Additional file 1: Table S5).

Tissue-specific expression profiles of wheat trihelix genes. The heatmap was drawn using R. The color scale indicates the log2 values of transcript per million. The colors red and blue represent higher and lower expression levels, respectively
The tissue expression profiles of trihelix genes in wheat were divided into three groups based on their expression characteristics. Group I contained 27 genes, and their average expression levels in TPM ranged from 3.47 to 10.14 (average value = 5.82). The expression levels of these genes showed remarkable differences at different stages in different tissues. Group II included 60 genes that had relatively low expression (average value = 1.23) in most tissues, and this group could be divided into three subgroups. Subgroup IIA comprised 17 genes with tissue-specific expressions. Subgroup IIB consisted of 41 genes with nearly negligible expression in all the tissues. Subgroup IIC only included 2 genes, which were specifically highly expressed in grain. Group III was composed of 7 genes, and these genes were highly expressed in almost all the tissues (average value = 13.63).
The wheat trihelix genes belonging to the same clade did not have the same expression profiles. However, three homologous copies often had similar tissue expression profiles. For example, TaGT-26, TaGT-54, and TaGT-94 are homologous genes belonging to group I, and they were all highly expressed in seedling leaves and lowly expressed in roots and grain. Homologous genes TaGT-21, TaGT-49, and TaGT-82, which belong to group II, were barely expressed in nearly all tissues. Homologous genes TaGT-20, TaGT-50, and TaGT-83, which belong to group III, had relatively high transcript levels in the majority of tissues. Among the trihelix genes in wheat, TaGT-83 had the highest average transcript level.
Expression profiling analysis of trihelix genes in wheat
Evidence has shown that the transcription levels of some members belonging to the GT-2 clade decline in white light [36, 37]. To investigate the expression patterns of members in GT-2 clade in photoperiod sensing, qRT-PCR analyses were conducted. We analyzed the changes of the expression levels of TaGT-1, TaGT-10, TaGT-26, TaGT-27, and TaGT-30 in two-week-old wheat seedling leaves under a 12 h light/12 h dark cycle (Fig. 5). The results showed that these five trihelix genes in GT-2 clade shared relatively similar expression profiles in response to photoperiod. The transcript levels of these genes began to decrease rapidly from 0 h and then gradually increased. These results are similar to those of previous studies. Except for TaGT-27, the transcript levels of the four other genes reached their first peaks at 9–12 h, then reached a second peak in the dark at 15–18 h, and then reached a third peak at approximately 24 h. The expression profile of TaGT-27 was simpler because it only had two peaks (at 15 and 24 h). Differences in expression profiles between TaGT-27 and the remaining four GT-2 clade genes may be related to their different motif compositions. TaGT-27 has a motif composition similar to that of Arabidopsis GT-2 clade genes (At5g28300 and At5g47660) and features a trihelix domain at the C-terminal, similar to all other members belonging to the GT-2 clade; however, this gene lacks the trihelix domain at the N-terminal, which trihelix genes usually have.

Expression patterns of wheat trihelix genes in response to photoperiod. Two-week-old seedling leaves were sampled at 0, 3, 6, 9, 12, 15, 18, 21, and 24 h under a 12-h light/12-h dark cycle. Bars reflect the means ± SD of three replicates
Recent studies indicate that trihelix genes play crucial roles in response to phytohormones and abiotic stresses. Using RNA-seq data based on IWGSC 1.1 genome annotations obtained from the expVIP website, differentially expressed wheat trihelix genes were analyzed under cold and drought stresses, and MA plots were drawn (Additional file 2: Figures S7–S9, Additional file 1: Table S6). Then, ten trihelix genes were selected to validate their responses against abiotic stresses further. We analyzed the transcript levels of these genes in the roots and leaves of two-week-old wheat seedlings under different abiotic stresses, including drought (PEG), salt (NaCl), cold (4 °C), and H2O2 stress treatments. Since the ABA signaling pathway is key in plant response to drought and salt stresses [29, 38], we also analyzed the expression profiles of these genes under exogenous ABA treatment (Figs. 6 and 7).

Expression profiles of wheat trihelix genes in seedling leaves under different treatments. Two-week-old seedling leaves were collected at 0, 1, 3, 6, 12, and 24 h under stress conditions of 20% (v/v) PEG6000, 200 mM NaCl, cold (4 °C), 10 mM H2O2, and 100 mM ABA. Three independent experiments were conducted. Error bars indicate SD. Statistically significant differences from the mock group are indicated as *P < 0.05; **P < 0.01

Expression profiles of wheat trihelix genes in seedling roots under different treatments. Two-week-old seedling roots were collected at 0, 1, 3, 6, 12, and 24 h under stress conditions of 20% (v/v) PEG6000, 200 mM NaCl, cold (4 °C), 10 mM H2O2, and 100 mM ABA. Three independent experiments were conducted. Error bars indicate SD. Statistically significant differences from the mock group are indicated as *P < 0.05; **P < 0.01
Based on the analysis results of RNA-seq data of leaves under cold stress, TaGT-10, TaGT-26, and TaGT-27 were significantly up-regulated, and TaGT-8 and TaGT-20 were significantly down-regulated. The expression level of ShCIGT (homologous with TaGT-42) increased in tomato under cold and drought stresses [20]. AtGT2L is homologous with TaGT-27 and up-regulated under cold, salt, and ABA treatments in Arabidopsis [17]. The expression profiles of the above wheat trihelix genes were confirmed in our qRT-PCR experiment. TaGT-42 in roots was significantly up-regulated under cold stress. Interestingly, its expression level in roots showed no significant difference during the initial period of salt stress but was remarkably up-regulated by nearly 7-fold 12 h after salt treatment. The expression profiles of TaGT-27 conformed to its homologous gene AtGT2L under cold, salt, and ABA treatments in roots. Furthermore, TaGT-26 and TaGT-27 showed very similar expression profiles under different treatments.
The results of RNA-seq data analysis of drought stress in leaves reveal that TaGT-10 was significantly up-regulated, whereas TaGT-6, TaGT-9, TaGT-26, and TaGT-27 were significantly down-regulated. The expression profiles of these five trihelix genes were validated by the results of qRT-PCR. Arabidopsis GT-4, which is homologous with TaGT-9, was reported up-regulated under salt stress [18]. Our qRT-PCR results showed that TaGT-9 was down-regulated in roots to a certain extent at 0–6 h under salt stress but remarkably up-regulated after 6 h. The transcript level of TaGT-9 was found to have remarkably increased in seedling leaves under cold stress.
Trihelix genes of GTγ clade in rice respond to diverse abiotic stresses, especially to salt stress [15]. qRT-PCR experiments were conducted to analyze the expression profiles of 3 GTγ clade genes (TaGT-5, TaGT-20, TaGT-23) under abiotic stresses and exogenous ABA treatments. TaGT-5 and TaGT-23 were down-regulated both in leaves and roots under salt stress, whereas TaGT-20 was significantly up-regulated in roots. Furthermore, the results showed that all 3 genes responded to cold stress in seedling leaves.
Discussion
As sessile organisms, wheat undergoes a variety of abiotic stresses, including salinity, drought, and extreme temperatures [29]. Wheat shares the same ancestor with B. distachyon and rice [39], and it is derived from two naturally interspecific hybridization events of three diploid donor species, two of which are the ancestors of T. urartu and Ae. tauschii [40, 41]. In our study, 94, 22, 29, and 31 trihelix genes were identified in T. aestivum, T. urartu, Ae. tauschii, and B. distachyon, respectively. The chromosomal distributions showed that no trihelix gene was located on wheat chromosome 7A, 7B, or 7D (Fig. 1); these results are consistent with the results of T. urartu and Ae. tauschii (no trihelix gene located on chromosome 7A of T. urartu or 7D of Ae. tauschii).
Due to the incompleteness of genome sequencing results, some trihelix gene family members probably are not identified in T. urartu or Ae. tauschii. The average number of trihelix genes on each subgenome of wheat is 31, which is similar to the previous studies in Arabidopsis (30) and rice (31). Ninety-four trihelix genes were clustered in 5 clades. The members belonging to the same clades usually share similar motif compositions and exon/intron structures, and they may have similar functions (Fig. 3). Our results showed the conservation in wheat trihelix gene evolution. Interspecific comparisons among the genomes of grass plants revealed more intrachromosomal gene duplication events in the wheat genome than in the genomes of other grass species [33]. In our study, five pairs of tandem duplication genes were identified in the wheat trihelix gene family, and only one gene corresponding to that presented in the equivalent genomic regions of rice and B. distachyon. Tandem duplication events occurred twice among TaGT-12, TaGT-13, and TaGT-14, thereby formed a cluster of three tandem duplication genes. Thus, gene duplication events are the main driving force for the trihelix gene evolution during the speciation and evolution of bread wheat.
The tissue-specific expression of genes usually reflects their corresponding functions. Our results indicated that the tandem duplication genes TaGT-12, TaGT-13, and TaGT-14 show nearly no expression in all tested wheat tissues (Fig. 4, Additional file 1: Table S5). The homologous genes TaGT-44/TaGT-77 and TaGT-45/TaGT-78 had similar expression profiles but were specifically expressed in grains, thereby showing that their functions may be associated with grain development. Further expression profile analysis of wheat trihelix homologous genes revealed that three homologous genes on subgenomes A, B, and D often have similar expression characteristics but different expression levels. Generally, homologous trihelix genes on subgenomes A and B, or subgenomes A and D have the same expression levels, and the rest one had relatively higher or lower expression level. For instance, TaGT-5, TaGT-36, and TaGT-68 are homologous genes, and they have similar expression characteristics. TaGT-5 and TaGT-68 belong to group I, and their transcript levels were very similar in all the tissues. By contrast, TaGT-36 belongs to group III, which showed approximately twice the expression levels of TaGT-5 and TaGT-68 in all tested tissues.
In this study, we selected ten wheat trihelix genes to analyze their expression profiles under different stress treatments, including PEG, NaCl, cold, H2O2, and exogenous ABA (Figs. 6 and 7). The results of expression analysis of selected wheat trihelix genes were essentially consistent with the results of RNA-seq data and published literatures. TaGT-10 was reported to be significantly up-regulated in leaves under PEG stress for 12 h [19]. Our qRT-PCR results showed that the transcript level of TaGT-10 reached the first peak under PEG stress at 12 h and was up-regulated again 2.4-fold at 24 h compared with the mock group. TaGT-10 revealed different expression profiles in leaves and roots under PEG stress. The transcript level of TaGT-10 in roots constantly decreased within 24 h under PEG stress. TaGT-27 is homologous with AtGT2L, which was reported to be up-regulated under salt, cold, and ABA stresses [17]. The similar result of TaGT-27 was validated via qRT-PCR in our research. In addition, TaGT-27 was found to be up-regulated in roots under PEG stress. OsGTγ-1 (Os02g33770), OsGTγ-2 (Os11g06410), and OsGTγ-3 (Os12g06640) were significantly up-regulated in rice under salt stress, but their expression data were only analyzed at 0–6 h [15]. We found that the GTγ clade genes TaGT-5 and TaGT-23 were down-regulated under salt stress, whereas TaGT-20 (homologous with OsGTγ-2) was significantly up-regulated in roots. TaGT-20 and OsGTγ-2 had similar expression profiles under salt stress, exhibited no significant change at 0–3 h and started to be significantly up-regulated at 6 h. Further findings indicated that the expression levels of TaGT-20 were 7- and 9-fold those of the mock group at 12 and 24 h. ShCIGT responded to cold and drought stresses, and AtGT-3b responded to salt stress [20, 42]. Both of these genes are homologous with TaGT-42. The expression level of TaGT-42 increased in roots under cold, PEG, and NaCl stresses in our study; it was also increased under H2O2 stress. The expression profile of TaGT-42 in roots under NaCl stress was similar to those of TaGT-8, TaGT-20, TaGT-26, and TaGT-27. These genes showed nearly no change at the initial stage of stress and began to be up-regulated at approximately 6 h. TaGT-42 was continually up-regulated by more than 7-fold at 12 and 24 h. The result indicated that TaGT-42 and TaGT-20 may indirectly participate in response to salt stress and directly respond to secondary stresses, such as oxidative damage.
Conclusions
In summary, our study is the first genome-wide analysis of trihelix genes in wheat and its close relatives. By integrating ortholog identification, phylogenetic analysis, tandem and segmental duplication identification, and conserved motif and structural analysis, comparative analysis with the available genomic information of wheat and its relatives was conducted to enable exploration of the evolution process of the identified trihelix genes. Some trihelix genes were confirmed to participate in response to multiple abiotic stresses, based on the tissue-specific expression patterns and the results of differential expression analysis under abiotic stresses. The results of our study build the foundation for stress-resistant breeding of wheat and its relatives.
Methods
Identification and characterization of trihelix genes
The wheat genome and protein database (IWGSC RefSeq v1.0, hexaploid bread wheat variety Chinese Spring) was downloaded from URGI (https://wheat-urgi.versailles.inra.fr/Seq-Repository/Assemblies) [43]. The genome and protein databases of T. urartu, Ae. tauschii, and B. distachyon were downloaded from MBKbase (http://www.mbkbase.org/Tu/), Sequencing the Aegilops tauschii Genome (http://aegilops.wheat.ucdavis.edu/ATGSP/annotation/), and JGI Phytozome 12 (http://phytozome.jgi.doe.gov/), respectively [39,40,41]. The HMM profile (PF13837) was downloaded from Pfam (http://pfam.xfam.org/family/pf13837) and used to search trihelix domains through HMMER3.0 software (http://hmmer.org/download.html) [44, 45]. The Python script was used to eliminate redundant sequences, and the sequence with the longest length was chosen as the representative of each gene. The SMART (http://smart.embl-heidelberg.de) and HMMER (https://www.ebi.ac.uk/Tools/hmmer/) websites were used to confirm the trihelix domain in all the identified trihelix genes [46, 47].
The theoretical pI and Mw of the genes were analyzed by uploading protein sequences to ExPASy (Compute pI/Mw tool, https://web.expasy.org/compute_pi/) [48]. WOLF PSORT (https://wolfpsort.hgc.jp/) was utilized to predict subcellular localization [49].
Identification of orthologs
OrthoGNC software was used to precisely predict the pairwise orthology relations among T. aestivum, T. urartu, Ae. tauschii, B. distachyon, and Oryza sativa following the gene neighborhood conservation method [50].
Phylogenetic analysis and gene synteny analysis
MEGA7.0 was utilized to align the full-length sequences of trihelix proteins in wheat, B. distachyon, rice, and Arabidopsis, and we used the neighbor-joining method with a bootstrap value of 1000 replicates and default parameters to construct the unrooted phylogenetic tree [51]. To analyze synteny relationships among the T. aestivum and B. distachyon and O. sativa genomes, MCScanX was used with default settings [52, 53], and the figures were drawn using Circos 0.69 [54].
Motif and structural analysis
To analyze the conservative motifs of trihelix TFs, the MEME tool (http://meme-suite.org/tools/meme) was used [55]. The limits of the maximum number of motifs are specified as 13. To visualize the exon/intron structures of trihelix gene family members, coding sequences (CDS) and genomic sequences were uploaded to Gene Structure Display Server 2.0 (http://gsds.cbi.pku.edu.cn/) [56].
Expression profile analysis
To analyze tissue-specific expression patterns of trihelix genes in wheat, we downloaded the RNA-seq data from the expVIP website (http://www.wheat-expression.com/) [57, 58]. The study title was “choulet_URGI”, and 15 types of tissues from hexaploid wheat were involved. Gplots package of R program (https://www.R-project.org/) was used to draw the heatmap.
We also downloaded the RNA-seq data titled “SRP043554” and “SRP045409” to analyze the expression profiles of wheat trihelix genes under cold and drought stresses. Using the DESeq2 package of R, we analyzed the differential expression of wheat trihelix genes and generated the MA plots [59]. Points highlighted in red represent significantly differentially expressed trihelix genes (padj < 0.05). Open triangles pointing either up or down indicate the points falling out of the window.
Plant materials
Seeds of T. aestivum Chinese Spring were stored in our laboratory. The seeds were germinated in distilled water under the dark condition and cultured in the greenhouse with a 12-h light/12-h dark cycle at 22 °C. To conduct the qRT-PCR analysis of wheat trihelix genes in response to photoperiod, the leaves of two-week-old wheat seedlings were sampled every 3 h for nine continuous time points (0, 3, 6, 9, 12, 15, 18, 21 and 24 h). To enable the expression profile analysis of wheat trihelix genes under abiotic stresses, leaves and roots of two-week-old seedlings were sampled at 0, 1, 3, 6, 12, and 24 h after treatment with 20% (v/v) PEG6000, 200 mM NaCl, 10 mM H2O2, 100 mM ABA, and cold condition (4 °C). All of the samples were collected and stored at − 80 °C. The experiments were conducted with three independent biological replicates.
Total RNA isolation and qRT-PCR analysis
A total RNA extraction kit (Zomanbio, China) was used to extract total RNA from wheat. A PrimeScript™ RT reagent kit with gDNA Eraser (Perfect Real Time; Takara, Japan) was used to synthesize the first cDNA chain. qPCR SYBR Green Mix (Vazyme, China) was used to conduct qRT-PCR analysis in a CFX™ real-time PCR detection system (Bio-Rad, USA). The primer sequences used in this study are indicated in Additional file 1: Table S7. TaActin (GenBank ID: AB181991.1) was used as the internal control. Three technical replicates were used to analyze each sample, and the expression data were analyzed via the 2-ΔΔCT method [60].
Abbreviations
- FAO:
-
Food and Agriculture Organization
- HMM:
-
Hidden Markov Model
- IWGSC:
-
The International Wheat Genome Sequencing Consortium
- MW:
-
Molecular weight
- padj:
-
adjusted p value
- pI:
-
Isoelectric point
- TF:
-
Transcription factor
- TPM:
-
transcript per million
References
Spitz F, Furlong EEM. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–26.
Jin J, Tian F, Yang D, Meng Y, Kong L, Luo J, et al. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017;45:D1040–5.
Riechmann JL, Heard J, Martin G, Reuber L, Jiang C, Keddie J, et al. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science. 2000;290:2105–10.
Kaplan-Levy RN, Brewer PB, Quon T, Smyth DR. The trihelix family of transcription factors - light, stress and development. Trends Plant Sci. 2012;17:163–71.
Nagano Y. Several features of the GT-factor trihelix domain resemble those of the Myb DNA-binding domain. Plant Physiol. 2000;124:491–3.
Hiratsuka K, Wu X, Fukuzawa H, Chua NH. Molecular dissection of GT-1 from Arabidopsis. Plant Cell. 1994;6:1805–13.
Perisic O, Lam E. A tobacco DNA binding protein that interacts with a light-responsive box II element. Plant Cell. 1992;4:831–8.
Green PJ, Kay SA, Chua NH. Sequence-specific interactions of a pea nuclear factor with light-responsive elements upstream of the rbcS-3A gene. EMBO J. 1987;6:2543–9.
Le Gourrierec J, Li YF, Zhou DX. Transcriptional activation by Arabidopsis GT-1 may be through interaction with TFIIA-TBP-TATA complex. Plant J. 1999;18:663–8.
Tzafrir I. Identification of genes required for embryo development in Arabidopsis. Plant Physiol. 2004;135:1206–20.
Gao MJ, Lydiate DJ, Li X, Lui H, Gjetvaj B, Hegedus DD, et al. Repression of seed maturation genes by a trihelix transcriptional repressor in Arabidopsis seedlings. Plant Cell. 2009;21:54–71.
Li X, Qin G, Chen Z, Gu H, Qu L. A gain-of-function mutation of transcriptional factor PTL results in curly leaves, dwarfism and male sterility by affecting auxin homeostasis. Plant Mol Biol. 2008;66:315–27.
Breuer C, Kawamura A, Ichikawa T, Tominaga-Wada R, Wada T, Kondou Y, et al. The trihelix transcription factor GTL1 regulates ploidy-dependent cell growth in the Arabidopsis trichome. Plant Cell. 2009;21:2307–22.
Xie ZM, Zou HF, Lei G, Wei W, Zhou QY, Niu CF, et al. Soybean trihelix transcription factors GmGT-2A and GmGT-2B improve plant tolerance to abiotic stresses in transgenic Arabidopsis. PLoS One. 2009;4:e6898.
Fang Y, Xie K, Hou X, Hu H, Xiong L. Systematic analysis of GT factor family of rice reveals a novel subfamily involved in stress responses. Mol Gen Genomics. 2010;283:157–69.
Yoo CY, Pence HE, Jin JB, Miura K, Gosney MJ, Hasegawa PM, et al. The Arabidopsis GTL1 transcription factor regulates water use efficiency and drought tolerance by modulating stomatal density via transrepression of SDD1. Plant Cell. 2011;22:4128–41.
Xi J, Qiu Y, Du L, Poovaiah BW. Plant-specific trihelix transcription factor AtGT2L interacts with calcium/calmodulin and responds to cold and salt stresses. Plant Sci. 2012;185-186:274–80.
Wang X, Li Q, Chen H, Zhang W, Ma B, Chen S, et al. Trihelix transcription factor GT-4 mediates salt tolerance via interaction with TEM2 in Arabidopsis. BMC Plant Biol. 2014;14:339.
Zheng X, Liu H, Ji H, Wang Y, Dong B, Qiao Y, et al. The wheat GT factor TaGT2L1D negatively regulates drought tolerance and plant development. Sci Rep. 2016;6:27042.
Yu C, Song L, Song J, Ouyang B, Guo L, Shang L, et al. ShCIGT, a Trihelix family gene, mediates cold and drought tolerance by interacting with SnRK1 in tomato. Plant Sci. 2018;270:140–9.
Xu H, Shi X, He L, Guo Y, Zang D, Li H, et al. Arabidopsis thaliana trihelix transcription factor AST1 mediates salt and osmotic stress tolerance by binding to a novel AGAG-box and some GT motifs. Plant Cell Physiol. 2018;59:946–65.
Wang W, Wu P, Liu T, Ren H, Li Y, Hou X. Genome-wide analysis and expression divergence of the trihelix family in Brassica rapa: insight into the evolutionary patterns in plants. Sci Rep. 2017;7:6463.
Li H, Huang W, Liu Z, Wu Z, Zhuang J. Trihelix family transcription factors in tea plant (Camellia sinensis): identification, classification, and expression profiles response to abiotic stress. Acta Physiol Plant. 2017;39:217.
Wang Z, Liu Q, Wang H, Zhang H, Xu X, Li C, et al. Comprehensive analysis of trihelix genes and their expression under biotic and abiotic stresses in Populus trichocarpa. Sci Rep. 2016;6:36274.
Yu C, Cai X, Ye Z, Li H. Genome-wide identification and expression profiling analysis of trihelix gene family in tomato. Biochem Bioph Res Co. 2015;468:653–9.
Osorio MB, Bücker-Neto L, Castilhos G, Turchetto-Zolet AC, Wiebke-Strohm B, Bodanese-Zanettini MH, et al. Identification and in silico characterization of soybean trihelix-GT and bHLH transcription factors involved in stress responses. Genet Mol Biol. 2012;35:233–46.
Song A, Wu D, Fan Q, Tian C, Chen S, Guan Z, et al. Transcriptome-wide identification and expression profiling analysis of chrysanthemum trihelix transcription factors. Int J Mol Sci. 2016;17:198.
Fedoroff NV, Battisti DS, Beachy RN, Cooper PJ, Fischhoff DA, Hodges CN, et al. Radically rethinking agriculture for the 21st century. Science. 2010;327:833–4.
Zhu J. Abiotic stress signaling and responses in plants. Cell. 2016;167:313–24.
Fitch WM. Homology: a personal view on some of the problems. Trends Genet. 2000;16:227–31.
Peterson ME, Chen F, Saven JG, Roos DS, Babbitt PC, Sali A. Evolutionary constraints on structural similarity in orthologs and paralogs. Protein Sci. 2009;18:1306–15.
Marcussen T, Sandve SR, Heier L, Spannagl M, Pfeifer M, Jakobsen KS, et al. Ancient hybridizations among the ancestral genomes of bread wheat. Science. 2014;345:1250092.
IWGSC. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;1251788:345.
Leister D. Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance gene. Trends Genet. 2004;20:116–22.
Kondrashov FA. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc R Soc B. 2012;279:5048–57.
O'Grady K, Goekjian VH, Nairn CJ, Nagao RT, Key JL. The transcript abundance of GmGT-2, a new member of the GT-2 family of transcription factors from soybean, is down-regulated by light in a phytochrome-dependent manner. Plant Mol Biol. 2001;47:367–78.
Dehesh K, Bruce WB, Quail PH. A trans-acting factor that binds to a GT-motif in a phytochrome gene promoter. Science. 1990;250:1397–9.
Zhu J. Salt and drought stress signal transduction in plants. Annu Rev Plant Biol. 2002;53:247–73.
Vogel JP, Garvin DF, Mockler TC, Schmutz J, Rokhsar D, Bevan MW, et al. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463:763–8.
Ling HQ, Ma B, Shi X, Liu H, Dong L, Sun H, et al. Genome sequence of the progenitor of wheat a subgenome Triticum urartu. Nature. 2018;557:424–8.
Luo M, Gu YQ, Puiu D, Wang H, Twardziok SO, Deal KR, et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature. 2017;551:498–502.
Park HC, Kim ML, Kang YH, Jeon JM, Yoo JH, Kim MC, et al. Pathogen- and NaCl-induced expression of the SCaM-4 promoter is mediated in part by a GT-1 box that interacts with a GT-1-like transcription factor. Plant Physiol. 2004;135:2150–61.
Appels R, Eversole K, Feuillet C, Keller B, Rogers J, Stein N, et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science. 2018;361:r7191.
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–30.
Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–63.
Letunic I, Bork P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018;46:D493–6.
Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, et al. HMMER web server: 2015 update. Nucleic Acids Res. 2015;43:W30–8.
Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, et al. Protein identification and analysis tools in the ExPASy server. In: The proteomics protocols handbook, vol. 112; 2005. p. 571–607.
Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585–7.
Jahangiri-Tazehkand S, Wong L, Eslahchi C. OrthoGNC: a software for accurate identification of orthologs based on gene neighborhood conservation. Genom Proteom Bioinf. 2017;15:361–70.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Tang H, Bowers JE, Wang X, Ming R, Alam M, Paterson AH. Synteny and collinearity in plant genomes. Science. 2008;320:486–8.
Wang Y, Tang H, DeBarry JD, Tan X, Li J, Wang X, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45.
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–8.
Hu B, Jin J, Guo A, Zhang H, Luo J, Gao G. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics. 2015;31:1296–7.
Ramírez-González RH, Borrill P, Lang D, Harrington SA, Brinton J, Venturini L, et al. The transcriptional landscape of polyploid wheat. Science. 2018;361:r6089.
Borrill P, Ramirez-Gonzalez R, Uauy C. ExpVIP: a customizable RNA-seq data analysis and visualization platform. Plant Physiol. 2016;170:2172–86.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–8.
Acknowledgements
The authors would like to thank the International Wheat Genome Sequencing Consortium (IWGSC) for providing access to the reference sequence of wheat, IWGSC RefSeq v1.0.
Funding
The work was supported by National Genetically Modified New Varieties of Major Projects of China (2016ZX08010004–004), the National Natural Science Foundation of China (No. 31771418, 31570261) and Key Project of Hubei Province (2017AHB041). The funding bodies had no role in the design of the study, collection, analysis, or interpretation of data or in the writing of the manuscript.
Availability of data and materials
The sequencing data for the genomic sequences are available in the URGI (https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Assemblies/v1.0/), MBKbase (http://www.mbkbase.org/Tu/), Sequencing the Aegilops tauschii Genome (http://aegilops.wheat.ucdavis.edu/ATGSP/annotation/), and JGI Phytozome 12 (http://phytozome.jgi.doe.gov/), respectively. The public RNA-seq data are available on the expVIP website (http://www.wheat-expression.com/).
Author information
Authors and Affiliations
Contributions
GH and GY conceived and supervised this study. JX and RH designed and conducted the experiments and analyzed the data. JH, DQ, and PS prepared the plant materials and treated the samples. TG and JF performed the qRT-PCR experiments. JC participated in the data analysis. GH, GY, JX, and RH wrote the manuscript. All of the authors have read and approved of the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Characteristic features of trihelix genes in wheat and its relatives. Table S2. Coding sequences of trihelix genes in wheat and its relatives. Table S3. Amino acid sequences of trihelix proteins in wheat and its relatives. Table S4. Syntenic relationships of trihelix genes between wheat and its relatives. Table S5. Expression data of wheat trihelix genes for tissue-specific expression profile analyses. Table S6. Differential expression of wheat trihelix genes under cold and drought stresses. Table S7. Primers for qRT-PCR.
Additional file 2:
Figure S1. Locations and duplication events of trihelix genes on T. urartu chromosomes. Red boxes indicate tandem duplications. Figure S2. Locations and duplication events of trihelix genes on Ae. tauschii chromosomes. Red boxes indicate tandem duplications, and red lines indicate segmental duplications. Figure S3. Locations and duplication events of trihelix genes on B. distachyon chromosomes. Red lines indicate segmental duplications. Figure S4. Syntenic analysis of trihelix genes between wheat and rice. Red, blue, and green bands represent subgenomes A, B, and D, respectively. Yellow bands indicate the rice genome. Figure S5. Syntenic analysis of trihelix genes between wheat and B. distachyon. Red, blue, and green bands represent subgenomes A, B, and D, respectively. Yellow bands indicate the rice genome. Figure S6. Conserved motifs of trihelix proteins. The logos of the motifs were predicted using MEME. Figure S7. MA plots of the differential expression of wheat trihelix genes under cold stress. Figure S8. MA plots of the differential expression of wheat trihelix genes under drought stress for 1 h. Figure S9. MA plots of the differential expression of wheat trihelix genes under drought stress for 6 h.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xiao, J., Hu, R., Gu, T. et al. Genome-wide identification and expression profiling of trihelix gene family under abiotic stresses in wheat. BMC Genomics 20, 287 (2019). https://doi.org/10.1186/s12864-019-5632-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5632-2