
Abstract
Background
Orchids produce a colorless protocorm by symbiosis with fungi upon seed germination. For mass production of orchids, the prevailing approaches are both generation of protocorm-like bodies (PLBs) from callus and multiplication of adventitious buds on inflorescence. However, somaclonal variations occur during micropropagation.
Results
We isolated the two most expressed transposable elements belonging to P Instability Factor (PIF)-like transposons. Among them, a potential autonomous element was identified by similarity analysis against the whole-genome sequence of Phalaenopsis equestris and named PePIF1. It contains a 19-bp terminal inverted repeat flanked by a 3-bp target site duplication and two coding regions encoding ORF1- and transposase-like proteins. Phylogenetic analysis revealed that PePIF1 belongs to a new P-lineage of PIF. Furthermore, two distinct families, PePIF1a and PePIF1b, with 29 and 37 putative autonomous elements, respectively, were isolated, along with more than 3000 non-autonomous and miniature inverted-repeat transposable element (MITE)-like elements. Among them, 828 PePIF1-related elements were inserted in 771 predicted genes. Intriguingly, PePIF1 was transposed in the somaclonal variants of Phalaenopsis cultivars, as revealed by transposon display, and the newly inserted genes were identified and sequenced.
Conclusion
A PIF-like element, PePIF1, was identified in the Phalaenopsis genome and actively transposed during micropropagation. With the identification of PePIF1, we have more understanding of the Phalaenopsis genome structure and somaclonal variations during micropropagation for use in orchid breeding and production.
Similar content being viewed by others
Background
Orchidaceae, containing more than 25,000 species, is one of the largest angiosperm families and is distributed in most land areas. The capsules contain hundreds of thousands of dust-like seeds that germinate and produce colorless protocorms by symbiosis with fungi in nature. The most popular orchid, the genus Phalaenopsis, comprises approximately 66 species [1], and more than 30,000 hybrid cultivars are registered in the Royal Horticultural Society [2]. P. equestris is a model orchid plant for genomic study that relies on the groundwork of basic genomics information [3]. OrchidBase (http://orchidbase.itps.ncku.edu.tw) has been established with collected transcriptome libraries from 11 Phalaenopsis orchids [4, 5], and Orchidstra (http://orchidstra.abrc.sinica.edu.tw) has been constructed for tissue-specific expression profiles in P. aphrodite [6]. The whole-genome sequence of P. equestris was published [3] and is available in OrchidBase 3.0.
In the orchid nursery, micropropagation is performed to maintain an elite orchid variety derived from a cross between two parents with desirable traits. Multiplication of adventitious buds or induction of protocorm-like bodies (PLBs) from callus are the two major approaches. Mass propagation produces plantlets with uniform growth and flowering time and eases the management of the orchid nursery. However, among the thousands of plantlets, some mutant phenotypes due to somaclonal variation are found during the vegetative or reproductive stage [7]. Usually, the occurrence rates of somaclonal variation are higher with induction of PLBs than multiplication of adventitious buds and continuous multiplication of consecutive generations of PLBs. Thus, orchid growers usually use induction of PLBs or multiplication of adventitious buds with fewer than three generations to avoid a high rate of somaclonal variation.
Somaclonal variation studies have focused on the activation of transposable elements (TEs), DNA methylation status, and histone modifications in several crops, such as oil palm, rice, and tobacco [8,9,10,11,12,13]. TEs are defined as DNA fragments that can transpose to new locations of chromosomes and are responsible for chromosomal rearrangements [14,15,16], fragmental gene movements [17, 18], and the evolution of gene regulation and function [19, 20]. Most main TE groups are ancient and are present in all kingdoms, and thousands to tens of thousands of TE families are found in plants [18, 21,22,23]. Two major classes of TEs were classified and distinguished by their transposition intermediate: class-I RNA retrotransposons or class-II DNA transposons. Class-I retrotransposons use the transposition mechanism of “copy-and-paste”, whereas class-II transposons use “cut-and-paste” [24]. However, miniature inverted TEs (MITEs) are considered non-autonomous DNA transposons but have multitudinous copy numbers within a genome [18, 24,25,26]. MITEs lack coding sequences, so their transposition is considered to be activated by autonomous elements of class-II DNA transposons, which share similar terminal inverted repeats (TIRs) with MITEs.
Two major MITEs have been identified in plants: Tourist-like, with 3-bp target-site duplications (TSDs), usually TTA/TAA, and Stowaway-like, with 2-bp TSDs, usually TA [22]. Two pairs of active Tourist-like MITE families with their related autonomous class-II DNA transposons have been identified: a 364-bp miniature P Instability Factor (mPIF) with transposase (TPase)-encoding elements, PIF in maize [27, 28], and a 430-bp mPing with autonomous Ping and Pong in rice [29,30,31]. PIF and Pong elements share several features, including conserved amino acid sequences in their open reading frames (ORFs) and nucleotide sequence homology in their TIRs and TSDs [29, 32].
The autonomous PIF element contains two ORFs: ORF1, coding for a Myb/SANT-containing protein with unknown function, and TPase, with a catalytic DDE motif for transposition [33]. mPIF shares features with PIF elements, including identical 14-bp TIRs, similar sub-terminal sequences, and an extended 9-bp target site preference [28]. Several PIF-like elements that have been identified with no analysis of their transposition activities include an autonomous 4.4-kb DcMaster-a and a 2.5-kb DcMaster1 in carrot (Daucus carota L.), a 5.14-kb MtMaster-a in Medicago truncatula, and a full-length 5.9-kb PpPIF-1 in bamboo (Phyllostachys pubescens) [34,35,36,37]. Recently, the genome-wide identification of PIF-like elements was analyzed in 21 species of Triticeae genera by using PCR-based approaches for conserved TPase sequences in genomes and transcriptomes [32, 37].
Previously, a homemade Orchid Oligo Array (4 × 44 K) was developed and contained 14,732 gene-specific oligonucleotides (45–60 mers in length) derived from 84,617 unigenes in OrchidBase [38]. The Orchid Array was used to analyze the gene expression profiles for the consecutive PLB generations of the somaclonal variant P. Brother Spring Dancer ‘KHM487’, which has crystal-like PLBs [38]. In this study, we successfully identified an active TE, PePIF1, in a Phalaenopsis genome by combining microarray data with in silico analysis and transposon display experiments.
Results
Transcriptional activation of Phalaenopsis TEs during PLB micropropagation
We searched for enhanced expression of transposon- or retrotransposon-like elements from microarray data for various PLB generations of KHM487 (Fig. 1a, b). It is plausible that an active TE with enhanced transcription level during micropropagation may cause somaclonal variations. Two unigenes, EICPS_047 and EFCP_7972, annotated as PIF-like TPases with a significant increase in expression in the 6th PLB generation (Table 1) were chosen as the TE candidates in this study. Quantitative real-time PCR was performed to confirm the increased transcription of the TE candidates with RNA from consecutive PLB generations (N1-N8) of both P. Sogo Berry ‘KHM1219’ and P. I-Hsin The Big Bang ‘KHM2180’ (abbreviated as KHM1219 and KHM2180, respectively), which feature peloric flowers and several floral color patterns in the somaclonal variants, respectively (Fig. 1c-h). The TE candidates expressed in the eight consecutive generations to various extents (Fig. 2; Additional file 1: Figure S1). Among them, EFCP_7972 highly expressed in the 5th PLB generation (N5) of KHM1219 and KHM2180 and in the 2nd generation (N2) of KHM1219 (Fig. 2). These results suggest that the TPase was expressed during Phalaenopsis tissue culture and reached high expression level at least in one PLB generation. However, the differential transcription activities among PLB generations reflected that not all PLBs showed EFCP_7972 expression in every PLB generation during tissue culture, which concerns the silencing effects on TEs in plant genome.

Three pairs of normal and somaclonal variants of Phalaenopsis, including P. Brother Spring Dancer ‘KHM487’ (KHM487, a and b), P. Sogo Berry ‘KHM1219’ (KHM1219, c and d), and P. I-Hsin The Big Bang ‘KHM2180’ (KHM2180, e-h). These somaclonal variants contain different phenotypes: KHM487 with the crystal-like protocorm-like bodies (PLBs) (b), KHM1219 having peloric flowers (d) and different flower color patterns for KHM2180 (e-h)

Transcription activity of EFCP_7972 during tissue culture of KHM1219 (a) and KHM2180 (b). Data are mean ± SEM from experiments performed in triplicate
Genome structure analysis reveals EFCP_7972 as a potential autonomous element
The TE candidates were mapped to the whole-genome sequence of P. equestris [3] to identify their genome structures in Phalaenopsis. Within the 1.1-Gb assembled P. equestris genome, EICPS_047 was detected with one hit and EFCP_7972 was detected with 369 hits (Additional file 2: Table S1). The top hit in the BLAST results for P. equestris was used to identify the genome structures for each TE candidate. EFCP_7972 appeared as an intact element with the presence of TIRs in both ends (Fig. 3), but EICPS_047 was not surrounded by repeat sequences. We then focused on EFCP_7972 for the following analysis.

The transposable element (TE) structures of PePIF1 in the Phalaenopsis genome and other PIF-like elements. The triangles indicate the identified terminal inverted repeats (TIRs). Black and hatched rectangles represent the open reading frames for TPase and ORF1, respectively, and blocked rectangles indicate the presence of introns
EFCP_7972 was annotated as a PIF-like transposon and mapped in Scaffold000759 (Additional file 2: Table S1), containing a 19-bp TIR with the sequence GGGYCYGTTTGGGGCAGCT (Y represents C/T) and flanked by the 3-bp TSD, TTA (Fig. 3; Table 2). Two ORFs with a head-to-tail direction were identified within the TIRs, coding for an ORF1- and a TPase-like protein, respectively (Fig. 3). This sequence was then renamed P. equestris PIF1 (PePIF1).
ORF1 of PePIF1, containing two exons encoding a 272-amino acid protein, shows 36% identity to the ORF1 of DcMaster-a [34] (Fig. 3; Table 2) and contains a conserved MYB/SANT motif with uncharacterized function (Additional file 3: Figure S2a). ORF2 is a 427-aa TPase-like protein encoded by three exons (Fig. 3) and a catalytic DDE motif within the conserved residues; N2, N3 and C1 regions; and a helix-turn-helix (HTH) domain involved in DNA binding (Additional file 3: Figure S2b, underlined regions) [33].
PePIF1 belongs to a new P lineage of PIF-like elements separate from other plants
A phylogenetic tree of PePIF1 was constructed with the conserved catalytic DDE motif of the PIF-like TPase and compared to the Pong-like TPase by the maximum likelihood method. PePIF1 was separate from the Pong-like elements and belongs to a PIF-like element (Fig. 4). PePIF1 was well separated from the previous identified lineages, including A1~A6, B, C, D, E and M, and thus grouped into a whole new lineage, named P lineage, for Phalaenopsis (Fig. 4).

Phylogenetic analysis of TPase from PIF- and Pong-like elements. The other PIF-like elements are named according to the species initials followed by their GenBank accession number. The plant names are At: Arabidopsis thaliana, Bo: Brassica oleracea, Dc: Daucus carota, Lj: Lotus japonicus, Mt.: Medicago truncatula, Os: Oryza sativa, Pe: Phalaenopsis equestris, Sb: Sorghum bicolor, Zm: Zea mays
To investigate whether PePIF1 was amplified within the Phalaenopsis genome, we searched for other copies of PePIF1 based on the presence of TIRs, TPase and ORF1 within the whole-genome sequence of P. equestris. A total of 66 putative autonomous elements was isolated based on the presence of the coding sequences of TPase and ORF1 within two TIRs and could be divided into PePIF1a and PePIF1b with 29 and 37 elements, respectively (Fig. 4; Table 3; Additional file 4: Table S2). PePIF1a and PePIF1b shared the same 19-bp TIR and 3-bp TSD but contained only 51.6 and 37.4% amino acid identity with TPase and ORF1, respectively. Thus, PePIF1a and PePIF1b were considered two distinct families according to the 80–80-80 rule [24]. The PePIF1a family contains the EFCP_7972 sequence located on Scaffold000759, with its TPase encoded by three exons, whereas PePIF1b was identified in Scaffold000402, with its TPase encoded by two exons (Table 3). Most of these predicted autonomous elements showed 5~6- and 6~8-kp sequences for PePIF1a and PePIF1b, respectively (Table 3), and the shorter or longer elements might result from the deletion or insertion events, respectively.
In addition to the intact autonomous elements, abundant defective copies that lost their TIRs, coding sequences of TPase or ORF1 were detected (Table 3). The length distributions of these remnant elements decreased in accordance with the defective TIR or coding sequences. For example, the remnant elements without ORF1 were 3~4-kb and 5~6-kb long from PePIF1a and PePIF1b, respectively (Table 3); 3~5-kb long for defective elements with the loss of one TIR, and 2~3-kb long with the loss of two TIRs (Table 3). Further deletion of one TIR and one coding sequence resulted in the shortest elements, only with 1~2-kb sequences (Table 3). In addition, 1303 copies of MITE-like elements with both TIRs were identified without any coding sequences, most being 200 to 300 bp long (Table 3). In total, more than 3000 copies of PePIF1 were present in the Phalaenopsis genome, yet most were defective copies or MITE-like elements.
Abundant PePIF1 insertions in the coding sequences of the Phalaenopsis genome
In analyzing the P. equestris genome, we noted a high proportion of TEs located in gene regions [3]. To investigate whether the PIF-like elements were inserted in the coding sequences, we analyzed flanking sequences of PePIF1 in the Phalaenopsis genome. A total of 828 PePIF1-related elements inserted into 771 predicted proteins were identified in the genome sequence of P. equestris (Table 3). A rough view for 100 predicted genes with PePIF1 insertions, there were 76 intronic regions and 24 genes containing partial coding sequences of PePIF1, concomitant with our previous observation that P. equestris genome contains abundant intronic TE, and that alters the expression of coding genes [3]. Similarly, intronic insertion of Alu elements causes alternative splicing [39]. Among them were 6 and 14 putative autonomous elements of PePIF1a and PePIF1b families, respectively; 409 defective elements of PePIF1; and 399 MITEs identified as insertions within the predicted gene regions (Table 3). In addition, within the 771 predicted proteins in the P. equestris genome, 594 predicted genes encode non-transposon genes, 6 encode transposable elements, and the other 171 annotated to uncharacterized proteins.
Transpositional activity of PePIF1 during consecutive PLBs analyzed by transposon display
To assess the transposition activity of PePIF1, transposon display analysis was adopted to analyze the layout of the PePIF1 sequence in the genomic DNAs of seedlings derived from consecutive PLB generations of KHM1219 and KHM2180, the normal and crystal PLBs of KHM487 (Fig. 1a, b), and individual flowers of KHM2180 with distinct pigmentation patterning (Fig. 1e-h). For transposon display analysis, PePIF1a- and PePIF1b-specific primers were designed from the weak conserved sub-terminal regions with the addition of 2-bp selection primers (see Methods, Fig. 3). Most amplified fragments of PePIF1a and PePIF1b were identical for the consecutive generations of KHM2180 as well as the normal and crystal PLBs of KHM487 (Additional file 5: Figure S3). The amplification of PePIF1b revealed several differential bands in the consecutive generations of KHM1219 and the individual flowers of KHM2180 with distinct pigmentation patterning (Fig. 5, red arrowhead). Considering the two somaclonal variants (Fig. 1f, g) of KHM2180 were resulted from the mother plants (Fig. 1e) with independent mutation events, the differential bands were identified for the loss of PePIF1b insertion, which means most bands were found in WT and m1, but not in m2 for the loss of PePIF1b insertion (Fig. 5b). The results suggest that PePIF1b could be transposed and resulted in a change of amplified fragment patterns in the genome of KHM1219 during PLB micropropagation and also in the mutant flowers of KHM2180. Therefore, PePIF1b could be an active transposon in the Phalaenopsis genome. In contrast, the banding pattern for PePIF1a was not changed, thus excluding the possibility of contamination or genome rearrangement (Additional file 5: Figure S3).

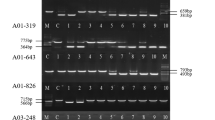
Transposon display profile of PePIF1 in various PLB generations of KHM1219 (a) and the wild-type and somaclonal variants of KHM2180 with various pigmentation patterning (b). a Seedlings from various PLB generations of KHM1219 (N1, N2, N3 and N6) are labeled 1, 2, 3, and 6. b Wild type and somaclonal variants of KHM2180 are labeled W, m1, and m2. The DNA size markers are indicated as “M” on the right and left side of the gel. AA, AT, AC, AG, CA, CT, CC, and CG indicate the second selective primers used for PCR amplification. The red arrowheads indicate the differentially amplified fragments with a new band
The differentially amplified fragments (Fig. 5, red arrowhead) were recovered and sequenced to confirm the presence of PePIF1 insertion, then mapped to the P. equestris genome to identify the genes inserted with or near PePIF1 during consecutive generations of KHM1219 or the somaclonal variants of KHM2180. Among them, 27 genes were identified with PePIF1 insertion, and only two, PEQU_19321 and PEQU_41063, were inserted in the coding sequences and the others were located in the intron regions (Additional file 6: Table S3). In addition, 52 predicted genes were found with 26 PePIF1s locating in the intergenic region, such as genes encoding DEMETER and arginine N-methyltransferase (Additional file 6: Table S3). We designed primers for five candidate insertion sites, PEQU_09928, PEQU_19321, PEQU_26837, PEQU_34751, and PEQU_41063, for PCR amplification of the flanking sequences to confirm the PePIF1 insertions. PCR amplification with both gene-specific and PePIF1b-specific primers provided more precise result for the gene and TE structures than transposon display, which used both PePIF1b-specific primer and adaptor primer. PEQU_26837, encoding serine/threonine-protein kinase GRIK2-like protein [40], produced differentially amplified fragments in both KHM1219 and KHM2180 (Additional file 7: Figure S4a), where the amplified 3-kb fragments of PEQU_26837 were the same in m2 and wt of KHM2180, suggesting the situation for loss of insertions in the mutants. However, m1 contained a large fragment with 6-kb sequence, so the PCR amplification with gene specific primers revealed detail information that the lost 3-kb fragment in m1 became a new insertion with 6-kb sequence and that was not detected in transposon display analysis. Similar situation was also found in PEQU_19321, encoding U-box domain-containing protein 43-like protein [41], where the amplified DNA fragment was larger than 3-kb sequence, and was another copy lost in wild type but present in m1 and m2, but they were not detected in transposon display analysis (Additional file 7: Figure S4b). However, the amplified patterns for PEQU_41063, PEQU_09928, and PEQU_34751, encoding disease resistance protein RGA3, histone-lysine N-methyltransferase, and RNA demethylase ALKBH5-like protein, respectively, were not distinct among all the samples tested (Additional file 7: Figure S4c-e). These results suggest that there were false positives present within these differential bands in transposon display or DNA samples prepared for PCR testing did not include cells where transposition occurred.
Discussion
Identification of a putative active PIF-like TE from a Phalaenopsis genome sequence
In this study, we used a strategy combining the experimental analysis and in silico genome mining approaches and identified an active PIF-like TE, PePIF1, in a Phalaenopsis genome. First, candidate TEs were identified by their transcription activities of TPase during consecutive PLB micropropagation by using microarray assay. Second, PePIF1 was identified as a potential autonomous TE because of the presence of TIRs accompanying the coding sequences of TPase and ORF1 in a Phalaenopsis genome. Third, the transposition activities of PePIF1 were investigated in consecutive PLBs and single flowers with changed pigmentation patterns by using transposon display assay.
PIF-like elements in Phalaenopsis showed distinct evolutionary lineage
Several PIF-like elements have been identified from plant genomes and grouped into separate evolution lineages. The phylogenetic analysis of the catalytic regions of TPases of PIF-like elements from rice and other plants resulted in four lineages: A1~A5, B, C, and D [33]. Carrot contains fewer than 10 copies of putative autonomous PIF-like elements and they were grouped into a new M lineage, which contains DcMasterA and MtPIF1 [34]. In addition, nine putative autonomous PIF-like elements are present in Lotus japonicus and they were grouped into the A3 lineage [42]. For M. truncatula, 22 putative autonomous and 67 non-autonomous PIF-like elements were grouped into three lineages, A5, D and M, and two new lineages, A6 and E [35]. Recently, 240 nonredundant PIF-like TPase sequences were amplified from 21 species of Triticeae genera and grouped into four main lineages [37]. Among them, 156 cDNA fragments from 15 species showed transcriptional activities [32]. In this study, we identified 66 putative autonomous PePIF1 elements and grouped them into a novel P lineage with two families, PePIF1a and PePIF1b. These results indicate that the evolution of PIF-like elements strikingly differed among various plant species for both the phylogenetic lineages and their copy numbers, so the types and numbers of TEs from each plant species must be investigated to understand their effects on genome evolution.
Abundant PePIF1 may result from transposition and abortive gap repair (AGR) processes
PePIF1 belongs to class II DNA transposons, which use the cut-and-paste mechanism for transposition, but more than a thousand copies of PePIF1 were identified from a Phalaenopsis genome, including 66 predicted autonomous and 1221 defective elements. Length distribution analysis suggested that direct deletion of the putative autonomous elements usually occurred for generation of the defective elements, although other types of mutations and rearrangements, including nested insertions to increase the TE length, were also observed [35]. Alternatively, the production of these high-copy-defective elements may be due to the AGR model, which suggests that new defective elements are usually derived from the DNA repair system to refill the double-strand breakage in the location after autonomous-element excision [43]. Therefore, one PePIF1 might transpose to another location by a cut-and-paste strategy but produce a new error-prone copy in the original site by a DNA repair system. In M. truncatula, various copy numbers of five lineages of PIF-like elements were affected by the efficiency of the transpositional activity or the AGR process [35]. The AGR model explained the presence of many copies of PePIF1, especially the defective elements containing both TIRs without middle sequences, and the transposition frequency accompanying the AGR process could be high in the Phalaenopsis genome. In addition, while the defective elements inserted in the genome, mutations (point mutations, insertion and deletion) occurred and leaded to the loss of one TIR or certain part of the element.
Several defective copies with only one end of elements might be from the assembly limitation of the P. equestris genome, which was assembled only from short-reads, and the most repetitive sequences, TIRs, were left as gaps or Ns in the assembled sequence. Therefore, the whole genome sequences assembled with short read-based sequences were difficult for the estimation of TE copy numbers. The repetitive sequences might be assembled together due to their similarity in sequences, so the true copy numbers should be higher than what was predicted from genomic sequences. In addition, the TIRs, as the most similar region, might be assembled together and then lost from the original copies. So the number of defective copies without one or two TIRs might be over estimated.
Relationship of PePIF1 and its MITE-like mPePIF1
Class-II DNA transposons are considered the ancestors of certain groups of MITEs and activate the transposition of these MITEs [44]. For example, mPing is suggested to be activated by its autonomous class-II DNA transposons, Ping and Pong, on the basis of their sequence similarities of TIRs and experimental analysis [29, 45]. In addition, several PIF-like elements show TIR sequence similarities to those of MITE families, including Heartbreaker from maize [46], Kiddo from rice [47], and Krak from carrot [34]. Therefore, we considered that MITE-like miniature PePIF1 (mPePIF1) was derived from PePIF1 and contained similar TIRs, TSDs, and sub-terminal sequences as PePIF1 but without the coding capacity.
In addition, most transposition activities of TEs occurred via MITE-like elements but not autonomous elements [28, 29]. The primers used for transposon display were designed for the sub-terminal regions of PePIF1a or PePIF1b, so the transposition activities of PePIF1a or PePIF1b could be distinguished. However, the sub-terminal sequences of PePIF1a or PePIF1b were similar to their derived MITE-like mPePIF1, so transposition of PePIF1 could be produced by the MITE-like mPePIF1, derived from PePIF1a or PePIF1b.
Moreover, the transposon display result showed only PePIF1b containing the transposition activity, although the differential transcription activities among PLB generations were detected for EFCP_7972, which belongs to PePIF1a. Since PePIF1a and PePIF1b showed the same TIRs, it is plausible that PePIF1b was transposed by the expressed TPase of PePIF1a. As the situation for mPing and its autonomous class-II DNA transposons, Ping and Pong, they share the similar TIR sequences and thought to be responsible for mPing transposition [29, 45].
mPePIF1 derived from transposition and amplification of themselves
Although the transposition of MITEs was activated by other DNA transposons, MITEs have the potential to amplify to high copy numbers, whereas DNA transposons amplify to fewer than 50 copies [48]. For example, most rice cultivars contain fewer than 50 copies of mPing, but the Gimbozu EG4 strain has more than 1000 copies amplified [48]. The amplification of mPing was explained by the DNA repair system for the double-strand breaks, with mPing excision by using the other mPing copy from the sister chromatid or homologous chromosome [49]. Therefore, most mPePIF1s had similar lengths of 200~300-bp sequences and were probably produced by the transposition and amplification of themselves but not by deletion or the AGR process of PePIF1.
Genes with PePIF1 insertions might cause the somaclonal variants of KHM2180 and KHM1219
The mutations with TE insertions and excision in gene coding regions may change protein functions and enzymatic activities [50], whereas the upstream regulatory regions with TE insertions may modify tissue-specific gene expression patterns [51]. With use of transposon display, the differentially amplified DNA fragments in the genomes of KHM1219 and KHM2180 were sequenced and found to possibly cause the deranged phenotypes of the somaclonal variants. In all, 27 genes were identified with PePIF1 insertions in the gene regions, with 26 PePIF1s locating in the intergenic region between 52 genes. Among them, histone-lysine N-methyltransferase [52] and DEMETER [53] may play roles in epigenetic regulation of the somaclonal variants during micropropagation in Phalaenopsis, as was previously found for whole-genome hypomethylation in somaclonal mantled palms [13] and Phalaenopsis [54]. In addition, polyadenylate-binding proteins for binding to poly(A) RNA [55], RNA demethylase [56], and zinc finger CCCH domain-containing protein for pre-mRNA splicing [57] are responsible for the RNA process at a post-transcriptional level and may also play roles in the somaclonal variations in Phalaenopsis.
However, PePIF1 was the first identified active transposon in Phalaenopsis, and other TEs or factors such as DNA methylation may also affect the somaclonal variants. Therefore, our results show the predicted genes with PePIF1 insertions in KHM1219 and KHM2180, but more evidence is needed for elucidating their roles in somaclonal variations during micropropagation.
With the amplified and sequenced differential DNA fragments of PePIF1 in the genomes of normal and somaclonal variants of KHM1219 and KHM2180, we thought that PePIF1 was an active transposon in a Phalaenopsis genome. The various generations of PLBs from KHM1219 as well as the normal and somaclonal variants of KHM2180 were induced from individual plants of P. Sogo Berry ‘KHM1219’ and P. I-Hsin The Big Bang ‘KHM2180,’ respectively, so there were no pre-existing genomic DNA polymorphisms in the analyzed samples before the experiments. However, mitotic recombination is also a major cause of the polymorphic insertion of MITE elements, which is also possible for the differentially amplified fragments of PePIF1 [58].
Conclusions
In this study, a strategy combining experimental analysis and in silico genome mining was used for successful identification of an active PIF-like TE, PePIF1, in a Phalaenopsis genome. The identification of PePIF1 can provide more understanding of the Phalaenopsis genome structure and somaclonal variations during micropropagation, which would benefit orchid breeding and production.
Methods
Plant materials
Three pairs of normal and somaclonal variants of Phalaenopsis were used in this study, including P. Brother Spring Dancer ‘KHM487’ (Fig. 1a, b), P. Sogo Berry ‘KHM1219’ (Fig. 1c, d), and P. I-Hsin The Big Bang ‘KHM2180’ (Fig. 1e-h), abbreviated to KHM487, KHM1219, and KHM2180, respectively. These somaclonal variants contain various phenotypes, including KHM487 with crystal-like PLBs, which leads to peloric flowers in mature plants (Fig. 1b). KHM1219 has peloric flowers (Fig. 1d), and KHM2180 shows distinct flower pigmentation patterning (Fig. 1e-h). Two individual plants of KHM1219 and KHM2180 were chosen and used for inducing callus to grow into PLBs. These PLBs were used for further inducing callus and for micropropagation of consecutive generations of PLBs, and grown into seedlings, then whole plants. Pooled seedlings of PLB in the same generations of KHM1219 and KHM2180 were used for quantitative real-time PCR and transposon display analyses. Single flowers of mother plants (Fig. 1e) and somaclonal variants (Fig. 1f, g) of KHM2180 were analyzed by transposon display. All plants were provided by I-Hsin Biotechnology Inc. (Chiayi, Taiwan) and kept under natural light and controlled temperature from 23 °C to 27 °C in the greenhouse at National Cheng Kung University (Tainan, Taiwan).
Phalaenopsis transcriptome microarray
The Agilent custom Orchid Oligo array (4 × 44 K) was designed for 14,732 unigenes chosen among the 84,617 unigenes in OrchidBase [4, 38]. RNA samples extracted from various generations of Phalaenopsis tissue culture derived from KHM487 were used for analysis of differential expression profiles following the Agilent eArray 5.0 program with the manufacturer’s recommendations [38].
Quantitative real-time PCR
For RNA extraction, the PLBs of various generations or stage-3 floral buds were immersed in liquid nitrogen and stored at − 80 °C. Total RNA extraction, cDNA synthesis, and quantitative real-time PCR were as described [59]. For each TE candidate, primer pairs within the gene-specific regions were designed and are listed in Additional file 8: Table S4. After amplification, melting curve analysis was used to verify amplicon specificity and primer dimer formation. A housekeeping gene, PeActin4 (PACT4, AY134752), displayed expression stability for M = 1.02 in GeNorm analysis [60] and was used for normalization [59, 61,62,63,64]. Data are mean ± SEM calculated from triplicate data, and experiments involved three independent biological repeats.
Investigation of the genome structures of TE candidates
The predicted TE candidates were first mapped to the whole-genome sequence of P. equestris by using a BLASTN algorithm with a cutoff E value of 1.0 × 10− 10. The flanking regions of 10-kb sequences of these predicted TEs were used to screen for the presence of terminal repeats by using a BLAST2 algorithm with more than 11-bp matches. Then the identified repeats were verified for their co-localization with the predicted TEs in the other regions of the P. equestris genome. The target-site duplications (TSDs) were identified from the flanking sequences of the terminal inverted repeats (TIRs).
Phylogenetic analysis of PIF-like TPases
The amino acids of the catalytic DDE domain of TPase for each PIF- and Pong-like TE were corrected for the frameshifts caused by 1–2 bp insertions or deletions and used to construct the phylogenetic tree with the maximum likelihood method by using ClustalW [65] and MUSCLE [66] implemented in MEGA v6 [67]. The 1000 bootstrapping datasets were used to estimate the confidence for each tree clade. Sequence data are available at NCBI with the accession numbers for PePIF1a and PePIF1b of MG470826 and MG470827, respectively.
Copy number estimation of PIF-like elements
The sequences for TIRs and coding sequences for ORF1 and TPase were mapped to the whole-genome sequence of P. equestris to screen for other copies of PePIF1 by using BLASTN and TBLASTX algorithms for TIR and coding sequences, respectively, with a cutoff E value of 1.0 × 10− 10. The predicted autonomous elements were identified by the presence of the coding sequences for TPase and ORF1 between two TIRs with maximum length of 30-kb sequences. All these elements of PePIF1 were mapped to the predicted proteins from the genomic sequences of P. equestris to identify the predicted proteins with PePIF1 insertions.
Transposon display for the transposition activity of PePIF1
The transposon display procedure was modified from a previous report [46]. DNA samples were digested with BfaI and ligated with adapters. The pre-selective amplifications involved use of an adapter-complementary primer and another TE-specific primer, PePIF1a_TD1 or PePIF1b_TD1, complementary to the weak conserved sub-terminal sequence of PePIF1 (Additional file 8: Table S4) with the temperature cycling parameters 94 °C for 5 min; 24 cycles of 94 °C for 30 s, 59 °C for 30 s, and 72 °C for 1 min, and a final cycle of 72 °C for 5 min. Selective amplification for detection involved use of 1/4-diluted pre-selective amplification products, adapter-complementary primer+NN and IRDye-700-labeled TE-specific PePIF1a_TD2 or PePIF1b_TD2 (Additional file 8: Table S4) (Protech Technology Enterprise Co., Taipei) with a “touchdown” protocol: 94 °C for 5 min; 35 cycles of 94 °C for 30 s, 69–61 °C for 30 s, and 72 °C for 1 min, and a final cycle of 72 °C for 5 min. The annealing temperature was reduced from 69 °C to 61 °C in 1 °C increments for each cycle. Separation of these fluorescently labeled transposon display fragments was performed with 7.5% polyacrylamide sequencing gel, with imaging by use of the Li-COR 4300 DNA Analyzer System (Li-COR Biotechnology, Lincoln, NB, USA). The differentially amplified fragments were recovered from the gel, cloned into the T-easy vector (Invitrogen), and randomly sequenced for 6–8 clones.
Abbreviations
- AGR:
-
Abortive gap repair
- MITEs:
-
Miniature inverted-repeat transposable elements
- mPIF:
-
Miniature P Instability Factor
- ORF:
-
Open reading frame
- PIF:
-
P Instability Factor
- PLBs:
-
Protocorm-like bodies
- TEs:
-
Transposable elements
- TIRs:
-
Terminal inverted repeats
- TPase:
-
Transposase
- TSDs:
-
Target-site duplications
References
Christenson EA. Phalaenopsis. Porland, OR: Timber Press; 2001.
OrchidWiz. OrchidWiz Encyclopedia Version X3.3 June 2017 Database. Ames, IA: OrchidWiz Database LLC; 2017.
Cai J, Liu X, Vanneste K, Proost S, Tsai WC, Liu KW, et al. The genome sequence of the orchid Phalaenopsis equestris. Nat Genet. 2015;47:65–72.
Fu CH, Chen YW, Hsiao YY, Pan ZJ, Liu ZJ, Huang YM, et al. OrchidBase: a collection of sequences of the transcriptome derived from orchids. Plant Cell Physiol. 2011;52:238–43.
Tsai WC, Fu CH, Hsiao YY, Huang YM, Chen LJ, Wang M, et al. OrchidBase 2.0: comprehensive collection of Orchidaceae floral transcriptomes. Plant Cell Physiol. 2013;54:e7.
Chao YT, Yen SH, Yeh JH, Chen WC, Shih MC. Orchidstra 2.0-a transcriptomics resource for the ochid family. Plant Cell Physiol. 2017;58:e9.
Chen FC, Chen WH. In: Chen WH, Chen HH, editors. Chapter 5. Somaclonal variation in orchids. Orchid Biotechnology. Singapore: World Scientific Publishing Co Pte Ltd; 2007.
Cheng C, Daigen M, Hirochika H. Epigenetic regulation of the rice retrotransposon Tos17. Mol Gen Genomics. 2006;276:378–90.
Jaligot E, Rival A, Beule T, Dussert S, Verdeil JL. Somaclonal variation in oil palm (Elaeis guineensis Jacq.): the DNA methylation hypothesis. Plant Cell Rep. 2000;19:684–90.
Jaligot E, Beule T, Rival A. Methylation-sensitive RFLPs: characterisation of two oil palm markers showing somaclonal variation-associated polymorphism. Theor Appl Genet. 2002;104:1263–9.
Jaligot E, Beule T, Baurens FC, Billotte N, Rival A. Search for methylation-sensitive amplification polymorphisms associated with the mantled variant phenotype in oil palm (Elaeis guineensis Jacq). Genome. 2004;47:224–8.
Kubis SE, Castilho AM, Vershinin AV, Heslop-Harrison JS. Retroelements, transposons and methylation status in the genome of oil palm (Elaeis guineensis) and the relationship to somaclonal variation. Plant Mol Biol. 2003;52:69–79.
Ong-Abdullah M, Ordway JM, Jiang N, Ooi SE, Kok SY, Sarpan N, et al. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature. 2015;525:533–7.
Kazazian HH Jr. Mobile elements: drivers of genome evolution. Science. 2004;303:1626–32.
Xuan YH, Peterson T, Han CD. Generation and analysis of transposon Ac/Ds-induced chromosomal rearrangements in rice plants. Methods Mol Biol. 2016;1469:49–61.
Lisch D. How important are transposons for plant evolution? Nat Rev Genet. 2013;14:49–61.
Jiang N, Bao Z, Zhang X, Eddy SR, Wessler SR. Pack-MULE transposable elements mediate gene evolution in plants. Nature. 2004;431:569–73.
Morgante M, Brunner S, Pea G, Fengler K, Zuccolo A, Rafalski A. Gene duplication and exon shuffling by Helitron-like transposons generate intraspecies diversity in maize. Nat Genet. 2005;37:997–1002.
Kashkush K, Feldman M, Levy AA. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat Genet. 2003;33:102–6.
Song X, Cao X. Transposon-mediated epigenetic regulation contributes to phenotypic diversity and environmental adaptation in rice. Curr Opin Plant Biol. 2017;36:111–8.
Bennetzen JL. Transposable elements, gene creation and genome rearrangement in flowering plants. Curr Opin Genet Dev. 2005;15:621–7.
Feschotte C, Jiang N, Wessler SR. Plant transposable elements: where genetics meets genomics. Nat Rev Genet. 2002;3:329–41.
El Baidouri M, Carpentier MC, Cooke R, Gao D, Lasserre E, Llauro C, et al. Widespread and frequent horizontal transfers of transposable elements in plants. Genome Res. 2014;24:831–8.
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–82.
Wessler SR, Bureau TE, White SE. LTR-retrotransposons and MITEs: important players in the evolution of plant genomes. Curr Opin Genet Dev. 1995;5:814–21.
Naito K, Zhang F, Tsukiyama T, Saito H, Hancock CN, Richardson AO, et al. Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature. 2009;461:1130.
Walker EL, Eggleston WB, Demopulos D, Kermicle J, Dellaporta SL. Insertions of a novel class of transposable elements with a strong target site preference at the r locus of maize. Genetics. 1997;146:681–93.
Zhang X, Feschotte C, Zhang Q, Jiang N, Eggleston WB, Wessler SR. P instability factor: an active maize transposon system associated with the amplification of Tourist-like MITEs and a new superfamily of transposases. P Natl Acad Sci USA. 2001;98:12572–7.
Jiang N, Bao Z, Zhang X, Hirochika H, Eddy SR, McCouch SR, et al. An active DNA transposon family in rice. Nature. 2003;421:163–7.
Kikuchi K, Terauchi K, Wada M, Hirano HY. The plant MITE mPing is mobilized in anther culture. Nature. 2003;421:167–70.
Nakazaki T, Okumoto Y, Horibata A, Yamahira S, Teraishi M, Nishida H, et al. Mobilization of a transposon in the rice genome. Nature. 2003;421:170–2.
Markova DN, Mason-Gamer RJ. Transcriptional activity of PIF and pong-like class II transposable elements in Triticeae. BMC Evol Biol. 2017;17:178.
Zhang X, Jiang N, Feschotte C, Wessler SR. PIF- and Pong-like transposable elements: distribution, evolution and relationship with Tourist-like miniature inverted-repeat transposable elements. Genetics. 2004;166:971–86.
Grzebelus D, Yau YY, Simon PW. Master: a novel family of PIF/Harbinger-like transposable elements identified in carrot (Daucus carota L.). Mol Gen Genomics. 2006;275:450–9.
Grzebelus D, Lasota S, Gambin T, Kucherov G, Gambin A. Diversity and structure of PIF/Harbinger-like elements in the genome of Medicago truncatula. BMC Genomics. 2007;8:409.
Zhou MB, Liu XM, Tang DQ. PpPIF-1: first isolated full-length PIF-like element from the bamboo Phyllostachys pubescens. Genet Mol Res. 2012;11:810–20.
Markova DN, Mason-Gamer RJ. The role of vertical and horizontal transfer in the evolutionary dynamics of PIF-like transposable elements in Triticeae. PLoS One. 2015;10:e0137648.
Hsiao YY, Huang TH, Fu CH, Huang SC, Chen YJ, Huang YM, et al. Transcriptomic analysis of floral organs from Phalaenopsis orchid by using oligonucleotide microarray. Gene. 2013;518:91–100.
Kreahling J, Graveley BR. The origins and implications of Aluternative splicing. Trends Genet. 2004;20:1–4.
Shen W, Reyes MI, Hanley-Bowdoin L. Arabidopsis protein kinases GRIK1 and GRIK2 specifically activate SnRK1 by phosphorylating its activation loop. Plant Physiol. 2009;150:996–1005.
Kalladan R, Lasky JR, Chang TZ, Sharma S, Juenger TE, Verslues PE. Natural variation identifies genes affecting drought-induced abscisic acid accumulation in Arabidopsis thaliana. P Natl Acad Sci USA. 2017;114:11536–41.
Holligan D, Zhang X, Jiang N, Pritham EJ, Wessler SR. The transposable element landscape of the model legume Lotus japonicus. Genetics. 2006;174:2215–28.
Rubin E, Levy AA. Abortive gap repair: underlying mechanism for Ds element formation. Mol Cell Biol. 1997;17:6294–302.
Jurka J, Kapitonov VV. PIFs meet Tourists and Harbingers: a superfamily reunion. P Natl Acad Sci USA. 2001;98:12315–6.
Shan X, Liu Z, Dong Z, Wang Y, Chen Y, Lin X, et al. Mobilization of the active MITE transposons mPing and Pong in rice by introgression from wild rice (Zizania latifolia Griseb.). Mol Biol Evol. 2005;22:976–90.
Casa AM, Brouwer C, Nagel A, Wang L, Zhang Q, Kresovich S, et al. The MITE family heartbreaker (Hbr): molecular markers in maize. P Natl Acad Sci USA. 2000;97:10083–9.
Yang G, Dong J, Chandrasekharan MB, Hall TC. Kiddo, a new transposable element family closely associated with rice genes. Mol Gen Genomics. 2001;266:417–24.
Naito K, Cho E, Yang G, Campbell MA, Yano K, Okumoto Y, et al. Dramatic amplification of a rice transposable element during recent domestication. P Natl Acad Sci USA. 2006;103:17620–5.
Teramoto S, Tsukiyama T, Okumoto Y, Tanisaka T. Early embryogenesis-specific expression of the rice transposon Ping enhances amplification of the MITE mPing. PLoS Genet. 2014;10:e1004396.
Nordborg M, Walbot V. Estimating allelic diversity generated by excision of different transposon types. Theor Appl Genet. 1995;90:771–5.
Wicker T, Yu Y, Haberer G, Mayer KF, Marri PR, Rounsley S, et al. DNA transposon activity is associated with increased mutation rates in genes of rice and other grasses. Nat Commun. 2016;7:12790.
Li Y, Mukherjee I, Thum KE, Tanurdzic M, Katari MS, Obertello M, et al. The histone methyltransferase SDG8 mediates the epigenetic modification of light and carbon responsive genes in plants. Genome Biol. 2015;16:79.
Mok YG, Choi KY, Hong SH, Huh JH. DEMETER plant DNA demethylase induces antiviral response by interferon signalling in animal cells. Sci Rep. 2017;7:9160.
Hsu CC, Wu PS, Chen TC, Yu CW, Tsai WC, Wu K, et al. Histone acetylation accompanied with promoter sequences displaying differential expression profiles of B-class MADS-box genes for phalaenopsis floral morphogenesis. PLoS One. 2014;9:e106033.
Banerjee A, Apponi LH, Pavlath GK, Corbett AH. PABPN1: molecular function and muscle disease. FEBS J. 2013;280:4230–50.
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m6A demethylase ALKBH5 aaintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606 e596.
Chen Z, Gui B, Zhang Y, Xie G, Li W, Liu S, et al. Identification of a 35S U4/U6.U5 tri-snRNP complex intermediate in spliceosome assembly. J Biol Chem. 2017; [Epub ahead of print].
Huang CR, Burns KH, Boeke JD. Active transposition in genomes. Annu Rev Genet. 2012;46:651–75.
Hsu CC, Chen YY, Tsai WC, Chen WH, Chen HH. Three R2R3-MYB transcription factors regulate distinct floral pigmentation patterning in Phalaenopsis spp. Plant Physiol. 2015;168:175–91.
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology. 2002;3:RESEARCH0034.
Chen YH, Tsai YJ, Huang JZ, Chen FC. Transcription analysis of peloric mutants of Phalaenopsis orchids derived from tissue culture. Cell Res. 2005;15:639–57.
Hsieh M-H, Lu H-C, Pan Z-J, Yeh H-H, Wang S-S, Chen W-H, et al. Optimizing virus-induced gene silencing efficiency with Cymbidium mosaic virus in Phalaenopsis flower. Plant Sci. 2013;201:25–41.
Hsieh M-H, Pan Z-J, Lai P-H, Lu H-C, Yeh H-H, Hsu C-C, et al. Virus-induced gene silencing unravels multiple transcription factors involved in floral growth and development in Phalaenopsis orchids. J Exp Bot. 2013;64:3869–84.
Pan ZJ, Chen YY, Du JS, Chen YY, Chung MC, Tsai WC, et al. Flower development of Phalaenopsis orchid involves functionally divergent SEPALLATA-like genes. New Phytol. 2014;202:1024–42.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Acknowledgements
We thank Dr. Michel Delseny (Laboratoire Génome et développement des plantes, Université de Perpignan, France) and Dr. Kenji Kojima (Department of Life Sciences, National Cheng Kung University) for helpful discussion of this article, and Dr. I-Hsuan Chen (Graduate Institute of Biotechnology, National Chung Hsing University, Taichung, Taiwan) for assistance with the transposon display experiment.
Funding
This research was supported by the Ministry of Science and Technology, Taiwan (MOST 105–2313-B-006-002-MY3 and MOST 106–2321-B-006-009-).
Availability of data and materials
We promise that all the data supporting the set of our conclusions are included in this article and its additional files. Sequence data are available at NCBI with the accession numbers of PePIF1a and PePIF1b for MG470826 and MG470827, respectively.
Author information
Authors and Affiliations
Contributions
CCH and HHC conceived and designed the study. PHL, WCT, JLH, YYH, and WHC collected plant materials. CCH, PHL, TCC, and JLH performed the experiments. CCH, PHL, WLW, and CHT analyzed and interpreted the data. CCH and HHC wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study did not directly involve humans, animals or plants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Transcription activity of EICPS_047 during tissue culture of KHM1219 (a) and KHM2180 (b). Data are mean ± SEM from experiments performed in triplicate. (DOCX 110 kb)
Additional file 2:
Table S1. Mapping of the three transposable element candidates to the whole-genome sequence of Phalaenopsis equestris. (DOCX 13 kb)
Additional file 3:
Figure S2. Multiple alignment of the amino acid sequence of ORF1s (a) and TPases (b). (DOCX 778 kb)
Additional file 4:
Table S2. Predicted autonomous elements of PePIF1 in the genome sequence of P. equestris. (DOCX 21 kb)
Additional file 5:
Figure S3. Transposon display profile of PePIF1 in normal and crystal-like PLBs of KHM487 (a) and various PLB generations of KHM2180 (b). (DOCX 301 kb)
Additional file 6:
Table S3. The insertion sites of PePIF1 in the whole-genome sequence of P. equestris (DOCX 18 kb)
Additional file 7:
Figure S4. Confirmation of the transposon display result by PCR with the primers designed in the flanking sequences of five candidate insertion sites in various PLB generations of KHM1219 and the wild type and somaclonal variants of KHM2180 (DOCX 239 kb)
Additional file 8:
Table S4. Primers used in this study. (DOCX 13 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hsu, CC., Lai, PH., Chen, TC. et al. PePIF1, a P-lineage of PIF-like transposable element identified in protocorm-like bodies of Phalaenopsis orchids. BMC Genomics 20, 25 (2019). https://doi.org/10.1186/s12864-018-5420-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5420-4