
Abstract
Background
The ability of bacteria to acquire resistance to antibiotics relies to a large extent on their capacity for genome modification. Prokaryotic genomes are highly plastic and can utilize horizontal gene transfer, point mutations, and gene deletions or amplifications to realize genome expansion and rearrangements. The contribution of point mutations to de novo acquisition of antibiotic resistance is well-established. In this study, the internal genome rearrangement of Escherichia coli during to de novo acquisition of antibiotic resistance was investigated using whole-genome sequencing.
Results
Cells were made resistant to one of the four antibiotics and subsequently to one of the three remaining. This way the initial genetic rearrangements could be documented together with the effects of an altered genetic background on subsequent development of resistance. A DNA fragment including ampC was amplified by a factor sometimes exceeding 100 as a result of exposure to amoxicillin. Excision of prophage e14 was observed in many samples with a double exposure history, but not in cells exposed to a single antibiotic, indicating that the activation of the SOS stress response alone, normally the trigger for excision, was not sufficient to cause excision of prophage e14. Partial deletion of clpS and clpA occurred in strains exposed to enrofloxacin and tetracycline. Other deletions were observed in some strains, but not in replicates with the exact same exposure history. Various insertion sequence transpositions correlated with exposure to specific antibiotics.
Conclusions
Many of the genome rearrangements have not been reported before to occur during resistance development. The observed correlation between genome rearrangements and specific antibiotic pressure, as well as their presence in independent replicates indicates that these events do not occur randomly. Taken together, the observed genome rearrangements illustrate the plasticity of the E. coli genome when exposed to antibiotic stress.
Similar content being viewed by others


Background
The ability of bacteria to acquire resistance to antibiotics relies to a large extent on their capacity for genome modification, including intracellular mobility of mobile genetic elements [1]. Prokaryotic genomes can utilize horizontal gene transfer, point mutations, and gene deletions or amplifications to realize genome expansion and rearrangements [2, 3] and are considered to be highly plastic as a result.
Prokaryotic genome content can be divided into the core genome, containing all essential and house-keeping genes, supplemented by the mobilome, composed of mobile genetic elements (MGEs) [4]. MGEs can be intercellular, such as plasmids, integrative and conjugative elements (ICEs), and extracellular in the form of bacteriophages. Bacteriophages are major drivers of horizontal transfer of virulence factors [5] and antibiotic resistance genes [6]. While lytic phages ultimately induce bacterial cell lysis, lysogenic or temperate phages integrate into the bacterial genome and replicate with the host genome as prophages [7]. After integration, prophages can undergo a complex decay process involving point mutations, genome rearrangements, deletions, and invasion by other mobile DNA elements [8], resulting in cryptic prophages that are metabolically and genetically inert. In Escherichia coli (E. coli) K-12, nine cryptic prophages remain, accounting for 3.6% of all genomic DNA [9], which contribute to survival in adverse environments such as exposure to antibiotics, oxidative stress, heat stress, and acid stress [10].
Intracellular MGEs are not by themselves transmissible to other cells, but can change location within the genome. Transposons, introns, and insertion sequences (ISs) belong to this category. ISs constitute an important part of most prokaryotic and eukaryotic genomes, occurring in a wide range of copy numbers [11]. ISs, which vary in size from 0.7–2.5 bp, only carry genes involved in their transposition but can induce duplications, deletions, and genome arrangements [12]. Because of the coding density of the prokaryotic genome, most insertions are expected to cause frame-shifting and thus deleterious alterations, but some may confer a selective advantage by providing new regulatory sequences [13,14,15].
The contribution of point mutations to de novo acquisition of antibiotic resistance is well-established [16, 17]. Gene duplication and amplification plays an important role in creating genomic variability, enabling adaptation to modified growth conditions [18]. Gene amplification in response to antibiotic stress has been reported with duplications ranging from a few bp [19] to 300 kb [20]. Gene deletions also contribute to development of antibiotic resistance [21,22,23]. Several questions are unanswered at present: is de novo development of resistance accompanied by genomic rearrangements? If yes, do the same rearrangements occur during to induced resistance against different antibiotics? Do the same genetic events always occur during to exposure to the same drug?
Here, we provide an overview of genome rearrangements that occur in populations of E. coli cells exposed to increasing subinhibitory concentrations of amoxicillin, enrofloxacin, kanamycin, or tetracycline.
Results
The main objective of this study was to investigate whether genome rearrangements occur in E. coli during de novo acquisition of resistance to antibiotics. Whole genome population sequencing was applied to compare wildtype E. coli to populations (app. 109 cells) derived from that wildtype with acquired resistance to either one, or to two antibiotics sequentially [24] (Fig. 1). Genomic DNA was isolated from strains developed in four independent rounds of experiments inducing resistance to specific antibiotics by growing the cells at step-wise increasing concentrations. An overview of all identified genome rearrangements is presented in Table 1. These elements will be discussed separately below.

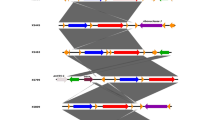
Experimental scheme. Wild-type E. coli MG1655 was exposed to stepwise increasing concentrations of amoxicillin (AMX), enrofloxacin (ENRO), kanamycin (KAN), or tetracycline (TET), resulting in strains with a de novo acquired resistance to a single antibiotic. The resistant strains were subsequently made resistant to the three other antibiotics. Each induction of resistance was performed in duplicate, resulting in four replicates with an identical exposure history of the double resistant strains
In all cells with acquired resistance to amoxicillin, either primary or secondary, (Table 1), ampC was amplified (Fig. 2). Three amplicons were identified varying in size from 3.5 to 10.5 kb (Fig. 2, a-c). Amplicon B-C were present in tetracycline resistant cells exposed to amoxicillin, all other cells contained amplicon A. In addition to ampC, 9 other genes were present in all three amplicons. Because population sequencing only provides the average copy number for the entire population, qPCR was used to quantify the number of repeats for one set of evolution experiments (Fig. 3). Strains with low levels of induced amoxicillin resistance carry on average 3–25 copies of the ampC gene. In strains that developed resistance to 1280 μg/mL amoxicillin, the average copy number ranges from 48 to 65. Within single populations, the ampC copy number varied strongly, with copy numbers ranging from single digits to a few hundred, suggesting high amplicon instability.

Amplification of three different fragments, all including ampC, upon acquisition of resistance to amoxicillin. Fragment B and C were detected in tetracycline resistant strains exposed to amoxicillin, fragment A was detected in all strains with an ampC amplification. See Table 1 for detailed information on prevalence of shown amplifications. The figure depicts genomic organization at point of deletion. The genes involved and the resulting gene products are displayed under the figure. Genes in bold are amplified in all three fragments

ampC copy number for different strains carrying an ampC amplification. The ampC copy number was determined with qPCR, using untreated wild-type E. coli as a reference. With exception of wild-type (which only acquired resistance to amoxicillin), all strains carried a previous resistance to enrofloxacin (ENROR), kanamycin (KANR), or tetracycline (TETR), resulting in a secondary resistance to amoxicillin The number displayed under the strain indicates the concentration amoxicillin used for resistance development. Bars indicate the average copy number from 25 colonies
In addition to the ampC amplification, deletions were also identified in various strains. A 14.4 kb deletion (Fig. 4a) was detected in 17 samples. No correlation with exposure to a specific antibiotic could be identified, but excision only occurred when cells were exposed to a second antibiotic (Table 1). All deleted genes were identified as part of prophage e14. A 312 bp deletion in clpS and clpA (Fig. 4b) was identified in one of the tetracycline resistant strains exposed to enrofloxacin (Table 1). The reading frame is not disturbed, but the 312 bp deletion includes the clpS stop codon and the clpA start codon, resulting in a fusion protein containing 28 N-terminal amino acids from clpS and 743 C-terminal amino acids from clpA.

Deletions detected in strains with de novo acquired antibiotic resistance a: Deletion of prophage e14 associated genes in strains exposed to any of the four antibiotics. b: Partial deletion of clpS and clpA in strains exposed to enrofloxacin and tetracycline. Figures depict genomic organization at point of deletion. The genes involved and the resulting gene products are displayed under the figure. Prophage associated genes are not shown because the resulting gene products are mostly not characterized. See Table 1 for detailed information on prevalence of the deletions
In a de novo enrofloxacin resistant E. coli subsequently treated with kanamycin, a 5.4 kb deletion was observed, containing 4 full-length and one partial gene deletion (Fig. 5a). Most notably, sbmA, encoding the peptide antibiotic transporter associated with kanamycin resistance, is located within this deletion. Point mutations in this gene were also found in many strains with acquired resistance to kanamycin (accompanying article). A 6.1 kb deletion, composed of 7 full-length and 2 truncated genes, was detected in a strain exposed to tetracycline after acquisition of resistance to amoxicillin (Fig. 5b). Two partially deleted genes, slyA and nemA, as well as 6 full-length genes are included in this deletion.

Deletions detected in resistant strains made resistant to kanamycin and amoxicillin. a: Deletion of sbmA and surrounding genes in enrofloxacin resistant E. coli exposed to kanamycin. b: Partial or full deletion of 8 genes upon induction of amoxicillin resistance in tetracycline resistant E. coli. Figures depicts genomic organization at point of deletion. The genes involved and the resulting gene products are displayed under the figure. See Table 1 for detailed information on prevalence of the deletions
In addition to genome amplifications and deletions, the role of transposable elements in acquisition of antibiotic resistance was also investigated. Two different insertion sequences, IS186 and IS1, were detected in four different genes (Fig. 6). Insertion of IS186 in fimA and yeaR correlates with exposure to amoxicillin or enrofloxacin, respectively (Table 1). Transposition of IS186 into oppB or IS1 into cyoA is much rarer, in comparison. In all four cases, the reading frame is disturbed, resulting in C-terminal deletions.

Intragenic IS transpositions identified in strains with acquired antibiotic resistance. IS186 insertion was detected in fimA in cells with acquired amoxicillin resistance (a), in yeaR in cells with acquired amoxicillin resistance (b), and in oppB in cells with secondary kanamycin resistance (c). IS1 insertion was found in cyoA in a single kanamycin resistant strain exposed to amoxicillin (d). See Table 1 for detailed information on prevalence of shown IS transpositions
Along with intragenic, intergenic transposition of IS5 into the 5’ UTR of dcuC and pagP, and the 5’ UTR of mgrB and yobH was detected as well (Fig. 7a/b). IS5 insertion into the 5’ UTR of dcuC and pagP was associated with kanamycin resistance, while transposition of IS5 into the 5’ UTR of mgrB and yobH only occurred upon exposure to tetracycline (Table 1). Finally, IS186 was detected in the 5’ UTR of lon (Fig. 7c), most likely associated with exposure to enrofloxacin (Table 1).

Intergenic IS transpositions identified in strains with acquired antibiotic resistance. IS5 was found in the 5’ UTR of dcuC and pagP when cells were exposed to kanamycin (a), and mgrB and yobH upon acquisition of resistance to tetracycline (b). IS186 transposition was detected in the 5’ UTR of lon in enrofloxacin resistant cells exposed to tetracycline (c)
Discussion
In E. coli, genome plasticity is a main source of functional diversity on a genomic level, enabling adaptation to diverse environments. Here, we show that several genome rearrangements occur when E. coli acquires resistance to different antibiotics. The data presented here, combined with information available on point mutations acquired during development of resistance (accompanying article), suggest that the organism uses several strategies to deal with antibiotic stress. In contrast, Pseudomonas aeruginosa exposed to a similar regimen of increasing antibiotic concentrations, only acquired point mutations [17], highlighting the ability of E. coli to adapt to antibiotic stress using several different approaches.
Gene amplification in response to antibiotic treatment has been reported before, and can result in antibiotic resistance through overproduction of target molecules [20, 25], efflux pumps [26], target modification [27], or antibiotic-modifying enzymes such as B-lactamases [28]. The level of chromosomal B-lactamase, and therefore the level of amoxicillin resistance, is gene-dose dependent [29]. Moreover, promoter mutations can result in a 6 to 21-fold increase in promoter strength [30, 31]. A wild-type strain adapted to 40 μg/ml does not yet carry any ampC repeats (data not shown) but does contain promoter mutations (accompanying article), indicating that intermediate level resistance does not require additional gene copies. In contrast, B-lactam resistance in Salmonella typhimurium is initiated by beta-lactamase gene amplification, followed by stabilizing point mutations [32].
Considering the size of the E. coli genome and the amplicon size, the number of amplifications carried by a single cell (Fig. 3) implies that, on average, strains with high levels of amoxicillin resistance increase their genome size by 5–10%. The cost of carrying amplifications has been shown to be determined mainly by the metabolic costs of the encoded enzyme rather than the cost of synthesizing additional DNA [33]. The resulting increased protein activity is therefore likely to require some kind of compensation. No difference in maintenance energy between wild-type and amoxicillin-adapted E. coli was detected, but amoxicillin-resistant E. coli showed a narrowing of the ecological range in the form of reduced pH- and salt-tolerance [34].
Gene amplifications are considered to be intrinsically unstable as homologous recombination can occur between identical repeats [18]. Beta-lactamases are secreted into the periplasm [35], hence cells that do not produce any beta-lactamase can still be protected by the enzymes produced by neighboring cells [36, 37]. Together with the metabolic costs of producing enzyme, these factors could be driving the loss of copies and explain the observed variation in copy number (Fig. 3).
Most of the genome rearrangements observed only occur in strains with a secondary acquired resistance, and not during primary exposure (Table 1). This includes the deletion of cryptic prophage e14 (Fig. 7), and transposition of insertion sequences (Figs. 6 and 7). Prophage e14 is excised after induction of the SOS response [38] and has been shown to follow norfloxacin exposure [39]. Likewise, IS transposition has been shown to occur after activation of the SOS response [40]. Although SOS response activation has been reported to follow exposure to beta-lactams [41, 42] and quinolones [43, 44], we do not observe the expected genome rearrangements during primary exposure. This suggests that, in our experimental conditions, either SOS response activation is not enough to trigger excision or transposition, or the SOS response itself is not sufficiently activated.
Prophages, although remnants of defective phages, are recognized to be functional during bacterial stress [45]. Exposure of E. coli to nalidixic acid or azlocillin results in induction of expression of the prophage e14 genes ymfL and ymfM [10], both hypothesized to be cell division inhibitors [46]. Furthermore, single deletions of either ymfL or ymfM result in a reduced ability to resist oxidative stress. As reactive oxygen species (ROS) production in antibiotic resistant cells exposed to other antibiotics is lowered [24], excision of this prophage is in line with the radical-based theory, which suggests a pivotal role for ROS in the action of bactericidal antibiotics.
Insertion sequences are necessary for mediating large-scale variation during bacterial genome evolution [47]. The E. coli genome contains many insertion sequences, among which IS5 and IS186 are considered to be among the most active [48]. The point of insertion can be specifically correlated with resistance to one antibiotic; fimA for amoxicillin resistance, yeaR and lon for enrofloxacin resistance, dcuC/pagP for kanamycin resistance, and mgrB/yobH for tetracycline resistance, indicating that IS transposition is not a random event (Table 1). As a single IS element can integrate in many genomic locations [11], the observed insertions likely contribute to resistance development.
Intragenic insertion of an insertion sequence most often results in a loss of function of the resulting gene product [49, 50]. In this dataset, intragenic insertion was observed in four different genes, including fimA (Fig. 6a). fimA codes for a type-1 fimbrial protein, which is a virulence factor in pathogenic E. coli [51]. Resistance to quinolones is associated with a decrease in fimA expression, caused by an IS10 transposition into fimA [52]. In general, antibiotic resistance is correlated with lowered virulence [53, 54], but such association has yet to be established for beta-lactam resistance and fimA expression.
IS186 has previously been described to cause fluoroquinolone resistance by inserting in the coding sequence of the AcrAB repressor acrR [55]. In our data set, this insertion was not observed, but rather a transposition of IS186 into yeaR (Fig. 6b). Although yeaR expression is induced in response to nitrate and nitrite [56], or nitric oxide [57], the function of the resulting gene product is as of yet unknown.
In Pseudomonas aeruginosa, oppB is involved in pacidamycin resistance [58]. In E. coli, it is required for uptake of phaseolotoxin, but currently there is no evidence for a role in antibiotic resistance [59]. Likewise, for cyoA there is no known connection to development of antibiotic resistance. In addition, transposition of IS1 into cyoA only occurred in a single replicate and might therefore be less relevant (Fig. 6d).
Intergenic insertions may disrupt promoter function or create new promoters, thereby modifying gene expression, which has been observed in antibiotic-resistant bacterial strains [13, 55, 60,61,62]. Intergenic insertion of IS5 or IS186 took place on three different occasions. Insertion of IS5 in the 5’ UTR of dcuC and pagP (Fig. 7a) is exclusively associated with kanamycin exposure. Neither dcuC, responsible for the transport of C4-dicarboxylates during anaerobic growth, nor pagP, a lipid A palmitoyltransferase, are known targets involved in resistance to aminoglycosides. However, as aminoglycosides bind to the outer membrane during entry into the bacterial cell [63], alteration of the lipid A structure might result in a decreased affinity of aminoglycosides for the membrane. LPS changes in the outer membrane have been linked to aminoglycoside resistance [64]. In Salmonella typhimurium, deletion of pagP results in hypersensitivity to antimicrobial peptides [65].
Another IS5 transposition upstream of mgrB and yobH (Fig. 7b) is correlated with resistance to tetracycline (Table 1). MgrB negatively regulates the two-component system PhoP/PhoQ, which controls virulence and adaptation to Mg2+-limited environments [66]. Insertions of IS5 family elements within mgrB have been shown to cause polymyxin resistance [67, 68], but no information exists on the contribution of this element to tetracycline resistance. The role of lon during development of antibiotic resistance is well-established. IS186 insertions into the lon promoter have been reported before [69, 70] and contribute to low level multidrug resistance through stabilization of Lon protease substrates MarA and SoxS [71].
In general, no correlation can be found between the presence of different rearrangements as different combinations are observed in many strains (Additional file 1: Table S1). The number of genome rearrangements detected varies from 1 to 4 per sequenced strains, and this does not appear influenced by the number of acquired point mutations (accompanying article). The appearance of the same rearrangement in independent lineages is most likely a reflection of the specificity of the response to different antibiotics. Although genetic drift cannot be excluded as a driver, it is not very likely as a wildtype control after even more cell duplications had only 6 point mutations and no other modifications.
Conclusions
In general, the overview of all genomic alterations presented here illustrates the remarkable plasticity of the E. coli genome when exposed to antibiotic stress. Many of the amplifications, deletions, or insertions have not been reported before as genomic modifications occurring during resistance development. However, the appearance in all or at least several replicates indicates that these events are not likely to occur randomly and hence might play a functional role during acquisition of antibiotic resistance.
Materials and methods
Sample description
All samples for sequencing were gathered from experiments described in [24]. Briefly, batch cultures of wild-type E. coli were adapted to increasing concentrations amoxicillin, enrofloxacin, kanamycin, or tetracycline, followed by a second round of adaptation to any of the three other antibiotics (Fig. 1). For every step, bacteria were reinoculated to an OD600 of 0.1. Each round of adaptation was performed twice, resulting in four secondary rounds of adaptation for each antibiotic. This way four strains derived from the same wildtype with an identical exposure history were obtained.
WGS
Genome isolation was carried out with the DNeasy blood and tissue kit (Qiagen). Samples were prepared for IonTorrent sequencing as described before (accompanying article). After the quality control and read mapping, the BAM files were subjected to copy number analysis using the cn.mops package in R (https://cran.r-project.org/) [72].
The copy number analysis procedure entailed: 1) segmentation of the genome in counting bins, 2) counting the reads for each bin, 3) sample normalization and GC correction, and 4) copy number detection in each sample. Loci with amplifications or deletions indicated by a ≥ 2-fold difference in copy number were selected. All genomic aberrations detected by the algorithm were checked by visual inspection of the data at each particular genomic region. In addition, stretches of single nucleotide polymorphisms identified in the TVC-generated data were found to be indicative for a suboptimal mapping result due to insertions. Insertions detected in this way were confirmed with PCR or qPCR. No genome rearrangements were detected in the sequenced wild-type strain. Deletions smaller or equal to 26 nucleotides were described in the accompanying paper.
PCR
PCR was used to verify a number of amplifications, deletions, or insertions. Primers are given in Table 2. Amplification was performed in 25 μL working volumes with DreamTaq polymerase (Thermo Scientific) with the following cycling conditions: 5′ at 95 °C, 35 cycles of 35″ at 95 °C, 55″ at given annealing temperature and 90″ at 72 °C, ending with a 90″ extension at 72 °C. PCR products were purified using the MSBSpinRapace kit (Stratec) and sequenced by Macrogen Europe using Sanger sequencing.
Quantitative PCR
Single colonies were dissolved in 10 μL TE-buffer (pH 8.0) and incubated at 95 °C for 5 min, after which the sample was diluted 105 fold in sterile MilliQ. 5 μL of diluted sample was mixed with 20 μL master mix containing 50 nM of each primer and Power SYBR Green PCR mix (Thermofisher Scientific). Quantitative PCR was performed with the Applied Biosystems 7300 real-time PCR system (Applied Biosystems) using the following cycling conditions: 10′ at 95 °C, 40 cycles of 15″ at 95 °C and 1′ at 60 °C. A wild-type sample was prepared as described above and aliquoted for use as a reference on every plate. Cycle threshold (Ct) values were determined by automated threshold analysis using the ABI Prism 1.0 software. Gene copy numbers were determined using the ∆∆Ct method using idnT as the reference gene. IdnT was chosen because no mutations or other alterations were detected in this region for any of the resistant strains. Primers used for quantification are shown in Table 3 and were validated using serial dilutions of WT sample.
Abbreviations
- Amx:
-
Amoxicillin
- Enro:
-
Enrofloxacin
- ICE:
-
Integrative and conjugative elements
- IS:
-
Insertion sequence
- Kan:
-
Kanamycin
- MGE:
-
Mobile genetic element
- ROS:
-
Reactive oxygen species
- Tet:
-
Tetracycline
References
Partridge SR, Kwong SM, Firth N, Jensen SO. Mobile genetic elements associated with antimicrobial resistance. Clin Microbiol Rev. 2018;31(4):e00088–17.
Koonin EV, Wolf YI. Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008;36(21):6688–719.
Snel B, Bork P, Huynen MA. Genomes in flux: the evolution of archaeal and proteobacterial gene content. Genome Res. 2002;12(1):17–25.
Frost LS, Leplae R, Summers AO, Toussaint A. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol. 2005;3(9):722–32.
Boyd EF. Bacteriophage-encoded bacterial virulence factors and phage-pathogenicity island interactions. Adv Virus Res. 2012;82:91–118.
Colavecchio A, Cadieux B, Lo A, Goodridge LD. Bacteriophages contribute to the spread of antibiotic resistance genes among foodborne pathogens of the Enterobacteriaceae Family - a review. Front Microbiol. 2017;8:1108.
Casjens S. Prophages and bacterial genomics: what have we learned so far? Mol Microbiol. 2003;49(2):277–300.
Canchaya C, Proux C, Fournous G, Bruttin A, Brussow H. Prophage genomics. Microbiol Mol Biol Rev. 2003;67(2):238–76 table of contents.
Lawrence JG, Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci U S A. 1998;95(16):9413–7.
Wang X, Kim Y, Ma Q, Hong SH, Pokusaeva K, Sturino JM, et al. Cryptic prophages help bacteria cope with adverse environments. Nat Commun. 2010;1:147.
Siguier P, Gourbeyre E, Chandler M. Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol Rev. 2014;38(5):865–91.
Schneider D, Duperchy E, Coursange E, Lenski RE, Blot M. Long-term experimental evolution in Escherichia coli. IX. Characterization of insertion sequence-mediated mutations and rearrangements. Genetics. 2000;156(2):477–88.
Turton JF, Ward ME, Woodford N, Kaufmann ME, Pike R, Livermore DM, et al. The role of ISAba1 in expression of OXA carbapenemase genes in Acinetobacter baumannii. FEMS Microbiol Lett. 2006;258(1):72–7.
Heritier C, Poirel L, Nordmann P. Cephalosporinase over-expression resulting from insertion of ISAba1 in Acinetobacter baumannii. Clin Microbiol Infect. 2006;12(2):123–30.
Corvec S, Caroff N, Espaze E, Giraudeau C, Drugeon H, Reynaud A. AmpC cephalosporinase hyperproduction in Acinetobacter baumannii clinical strains. J Antimicrob Chemother. 2003;52(4):629–35.
Munita JM, Arias CA. Mechanisms of antibiotic resistance. Microbiol Spectr. 2016;4(2):1–24.
Feng Y, Jonker MJ, Moustakas I, Brul S, Ter Kuile BH. Dynamics of mutations during development of resistance by Pseudomonas aeruginosa against five antibiotics. Antimicrob Agents Chemother. 2016;60(7):4229–36.
Sandegren L, Andersson DI. Bacterial gene amplification: implications for the evolution of antibiotic resistance. Nat Rev Microbiol. 2009;7(8):578–88.
Nichols BP, Guay GG. Gene amplification contributes to sulfonamide resistance in Escherichia coli. Antimicrob Agents Chemother. 1989;33(12):2042–8.
Vinella D, Cashel M, D'Ari R. Selected amplification of the cell division genes ftsQ-ftsA-ftsZ in Escherichia coli. Genetics. 2000;156(4):1483–92.
Martinez E, Holmes N, Jelfs P, Sintchenko V. Genome sequencing reveals novel deletions associated with secondary resistance to pyrazinamide in MDR Mycobacterium tuberculosis. J Antimicrob Chemother. 2015;70(9):2511–4.
Baucheron S, Monchaux I, Le Hello S, Weill FX, Cloeckaert A. Lack of efflux mediated quinolone resistance in Salmonella enterica serovars Typhi and Paratyphi A. Front Microbiol. 2014;5:12.
Zhang Y, Heym B, Allen B, Young D, Cole S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature. 1992;358(6387):591–3.
Hoeksema M, Brul S, Ter Kuile BH. Influence of reactive oxygen species on De novo Acquisition of Resistance to bactericidal antibiotics. Antimicrob Agents Chemother. 2018;62(6):e02354–17.
Brochet M, Couve E, Zouine M, Poyart C, Glaser P. A naturally occurring gene amplification leading to sulfonamide and trimethoprim resistance in Streptococcus agalactiae. J Bacteriol. 2008;190(2):672–80.
Nicoloff H, Perreten V, McMurry LM, Levy SB. Role for tandem duplication and lon protease in AcrAB-TolC- dependent multiple antibiotic resistance (mar) in an Escherichia coli mutant without mutations in marRAB or acrRAB. J Bacteriol. 2006;188(12):4413–23.
Musher DM, Dowell ME, Shortridge VD, Flamm RK, Jorgensen JH, Le Magueres P, et al. Emergence of macrolide resistance during treatment of pneumococcal pneumonia. N Engl J Med. 2002;346(8):630–1.
Seoane A, Sanchez E, Garcia-Lobo JM. Tandem amplification of a 28-kilobase region from the Yersinia enterocolitica chromosome containing the blaA gene. Antimicrob Agents Chemother. 2003;47(2):682–8.
Normark S, Edlund T, Grundstrom T, Bergstrom S, Wolf-Watz H. Escherichia coli K-12 mutants hyperproducing chromosomal beta-lactamase by gene repetitions. J Bacteriol. 1977;132(3):912–22.
Jaurin B, Grundstrom T, Normark S. Sequence elements determining ampC promoter strength in E. coli. EMBO J. 1982;1(7):875–81.
Corvec S, Caroff N, Espaze E, Marraillac J, Reynaud A. 11 mutation in the ampC promoter increasing resistance to beta-lactams in a clinical Escherichia coli strain. Antimicrob Agents Chemother. 2002;46(10):3265–7.
Sun S, Berg OG, Roth JR, Andersson DI. Contribution of gene amplification to evolution of increased antibiotic resistance in Salmonella typhimurium. Genetics. 2009;182(4):1183–95.
Pettersson ME, Sun S, Andersson DI, Berg OG. Evolution of new gene functions: simulation and analysis of the amplification model. Genetica. 2009;135(3):309–24.
Handel N, Schuurmans JM, Brul S, ter Kuile BH. Compensation of the metabolic costs of antibiotic resistance by physiological adaptation in Escherichia coli. Antimicrob Agents Chemother. 2013;57(8):3752–62.
Ciofu O, Beveridge TJ, Kadurugamuwa J, Walther-Rasmussen J, Hoiby N. Chromosomal beta-lactamase is packaged into membrane vesicles and secreted from Pseudomonas aeruginosa. J Antimicrob Chemother. 2000;45(1):9–13.
Brook I. Beta-lactamase-producing bacteria in mixed infections. Clin Microbiol Infect. 2004;10(9):777–84.
Medaney F, Dimitriu T, Ellis RJ, Raymond B. Live to cheat another day: bacterial dormancy facilitates the social exploitation of beta-lactamases. ISME J. 2016;10(3):778–87.
Greener A, Hill CW. Identification of a novel genetic element in Escherichia coli K-12. J Bacteriol. 1980;144(1):312–21.
Long H, Miller SF, Strauss C, Zhao C, Cheng L, Ye Z, et al. Antibiotic treatment enhances the genome-wide mutation rate of target cells. Proc Natl Acad Sci U S A. 2016;113(18):E2498–505.
Eichenbaum Z, Livneh Z. UV light induces IS10 transposition in Escherichia coli. Genetics. 1998;149(3):1173–81.
Miller C, Thomsen LE, Gaggero C, Mosseri R, Ingmer H, Cohen SN. SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science (New York, NY). 2004;305(5690):1629–31.
Maiques E, Ubeda C, Campoy S, Salvador N, Lasa I, Novick RP, et al. Beta-lactam antibiotics induce the SOS response and horizontal transfer of virulence factors in staphylococcus aureus. J Bacteriol. 2006;188(7):2726–9.
Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130(5):797–810.
Handel N, Hoeksema M, Freijo Mata M, Brul S, ter Kuile BH. Effects of stress, reactive oxygen species, and the SOS response on De novo Acquisition of Antibiotic Resistance in Escherichia coli. Antimicrob Agents Chemother. 2015;60(3):1319–27.
Domka J, Lee J, Bansal T, Wood TK. Temporal gene-expression in Escherichia coli K-12 biofilms. Environ Microbiol. 2007;9(2):332–46.
Mehta P, Casjens S, Krishnaswamy S. Analysis of the lambdoid prophage element e14 in the E. coli K-12 genome. BMC Microbiol. 2004;4:4.
Schneider D, Lenski RE. Dynamics of insertion sequence elements during experimental evolution of bacteria. Res Microbiol. 2004;155(5):319–27.
Lee H, Doak TG, Popodi E, Foster PL, Tang H. Insertion sequence-caused large-scale rearrangements in the genome of Escherichia coli. Nucleic Acids Res. 2016;44(15):7109–19.
Gregory ST, Dahlberg AE. Transposition of an insertion sequence, ISTth7, in the genome of the extreme thermophile Thermus thermophilus HB8. FEMS Microbiol Lett. 2008;289(2):187–92.
Vincent AT, Freschi L, Jeukens J, Kukavica-Ibrulj I, Emond-Rheault JG, Leduc A, et al. Genomic characterisation of environmental Pseudomonas aeruginosa isolated from dental unit waterlines revealed the insertion sequence ISPa11 as a chaotropic element. FEMS Microbiol Ecol. 2017;93(9):fix106.
Wurpel DJ, Beatson SA, Totsika M, Petty NK, Schembri MA. Chaperone-usher fimbriae of Escherichia coli. PLoS One. 2013;8(1):e52835.
Vila J, Simon K, Ruiz J, Horcajada JP, Velasco M, Barranco M, et al. Are quinolone-resistant uropathogenic Escherichia coli less virulent? J Infect Dis. 2002;186(7):1039–42.
Velasco M, Horcajada JP, Mensa J, Moreno-Martinez A, Vila J, Martinez JA, et al. Decreased invasive capacity of quinolone-resistant Escherichia coli in patients with urinary tract infections. Clin Infect Dis. 2001;33(10):1682–6.
Johnson JR, van der Schee C, Kuskowski MA, Goessens W, van Belkum A. Phylogenetic background and virulence profiles of fluoroquinolone-resistant clinical Escherichia coli isolates from the Netherlands. J Infect Dis. 2002;186(12):1852–6.
Jellen-Ritter AS, Kern WV. Enhanced expression of the multidrug efflux pumps AcrAB and AcrEF associated with insertion element transposition in Escherichia coli mutants selected with a fluoroquinolone. Antimicrob Agents Chemother. 2001;45(5):1467–72.
Lin HY, Bledsoe PJ, Stewart V. Activation of yeaR-yoaG operon transcription by the nitrate-responsive regulator NarL is independent of oxygen- responsive regulator Fnr in Escherichia coli K-12. J Bacteriol. 2007;189(21):7539–48.
Justino MC, Vicente JB, Teixeira M, Saraiva LM. New genes implicated in the protection of anaerobically grown Escherichia coli against nitric oxide. J Biol Chem. 2005;280(4):2636–43.
Mistry A, Warren MS, Cusick JK, Karkhoff-Schweizer RR, Lomovskaya O, Schweizer HP. High-level pacidamycin resistance in Pseudomonas aeruginosa is mediated by an Opp oligopeptide permease encoded by the Opp-fabI operon. Antimicrob Agents Chemother. 2013;57(11):5565–71.
Staskawicz BJ, Panopoulos NJ. Phaseolotoxin transport in Escherichia coli and Salmonella typhimurium via the oligopeptide permease. J Bacteriol. 1980;142(2):474–9.
Wright MS, Mountain S, Beeri K, Adams MD. Assessment of insertion sequence mobilization as an adaptive response to oxidative stress in Acinetobacter baumannii using IS-seq. J Bacteriol. 2017;199(9):e00833–16.
Kuo HY, Chang KC, Liu CC, Tang CY, Peng JH, Lu CW, et al. Insertion sequence transposition determines imipenem resistance in Acinetobacter baumannii. Microb Drug Resist. 2014;20(5):410–5.
Olliver A, Valle M, Chaslus-Dancla E, Cloeckaert A. Overexpression of the multidrug efflux operon acrEF by insertional activation with IS1 or IS10 elements in Salmonella enterica serovar typhimurium DT204 acrB mutants selected with fluoroquinolones. Antimicrob Agents Chemother. 2005;49(1):289–301.
Hancock RE, Farmer SW, Li ZS, Poole K. Interaction of aminoglycosides with the outer membranes and purified lipopolysaccharide and OmpF porin of Escherichia coli. Antimicrob Agents Chemother. 1991;35(7):1309–14.
Bryan LE, O'Hara K, Wong S. Lipopolysaccharide changes in impermeability-type aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1984;26(2):250–5.
Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, Hackett M, et al. Lipid a acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell. 1998;95(2):189–98.
Groisman EA. The pleiotropic two-component regulatory system PhoP-PhoQ. J Bacteriol. 2001;183(6):1835–42.
Olaitan AO, Diene SM, Kempf M, Berrazeg M, Bakour S, Gupta SK, et al. Worldwide emergence of colistin resistance in Klebsiella pneumoniae from healthy humans and patients in Lao PDR, Thailand, Israel, Nigeria and France owing to inactivation of the PhoP/PhoQ regulator mgrB: an epidemiological and molecular study. Int J Antimicrob Agents. 2014;44(6):500–7.
Cannatelli A, Giani T, D'Andrea MM, Di Pilato V, Arena F, Conte V, et al. MgrB inactivation is a common mechanism of colistin resistance in KPC-producing Klebsiella pneumoniae of clinical origin. Antimicrob Agents Chemother. 2014;58(10):5696–703.
Nicoloff H, Perreten V, Levy SB. Increased genome instability in Escherichia coli lon mutants: relation to emergence of multiple-antibiotic-resistant (mar) mutants caused by insertion sequence elements and large tandem genomic amplifications. Antimicrob Agents Chemother. 2007;51(4):1293–303.
saiSree L, Reddy M, Gowrishankar J. IS186 insertion at a hot spot in the lon promoter as a basis for lon protease deficiency of Escherichia coli B: identification of a consensus target sequence for IS186 transposition. J Bacteriol. 2001;183(23):6943–6.
Nicoloff H, Andersson DI. Lon protease inactivation, or translocation of the lon gene, potentiate bacterial evolution to antibiotic resistance. Mol Microbiol. 2013;90(6):1233–48.
Klambauer G, Schwarzbauer K, Mayr A, Clevert DA, Mitterecker A, Bodenhofer U, et al. Cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 2012;40(9):e69.
Acknowledgements
Not applicable.
Funding
This study was supported by the Netherlands Food and Consumer Product Safety Authority. The funding body had no role in the design of the study, data collection, analysis, interpretation of data, or writing the manuscript.
Availability of data and materials
The binary alignment/map (bam) files of the sequenced strains are available in the NCBI Sequence Read Archive database with SRA accession number SRP159604, http://www.ncbi.nlm.nih.gov/sra.
Author information
Authors and Affiliations
Contributions
MH and BtK conceived the project. MH performed most experiments. KB performed PCR. MJ performed the bioinformatic analysis. MH and BtK wrote the manuscript. SB contributed to the final manuscript. All authors critically reviewed the manuscript and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Overview of genomic rearrangement occurring in different samples. Contains information on the combinations of genome rearrangements that appear in samples, as well as correlation with the number of point mutations also identified. (XLSX 19 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hoeksema, M., Jonker, M.J., Bel, K. et al. Genome rearrangements in Escherichia coli during de novo acquisition of resistance to a single antibiotic or two antibiotics successively. BMC Genomics 19, 973 (2018). https://doi.org/10.1186/s12864-018-5353-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5353-y