
Abstract
Background
Copy Number Variations (CNVs) have becoming very significant variants, representing a major source of genomic variation. CNVs involvement in phenotypic expression and different diseases has been widely demonstrated in humans as well as in many domestic animals. However, genome wide investigation on these structural variations is still missing in Felis catus. The present work is the first CNV mapping from a large data set of Next Generation Sequencing (NGS) data in the domestic cat, performed within the 99 Lives Consortium.
Results
Reads have been mapped on the reference assembly_6.2 by Maverix Biomics. CNV detection with cn.MOPS and CNVnator detected 592 CNVs. These CNVs were used to obtain 154 CNV Regions (CNVRs) with BedTools, including 62 singletons. CNVRs covered 0.26% of the total cat genome with 129 losses, 19 gains and 6 complexes. Cluster Analysis and Principal Component Analysis of the detected CNVRs showed that breeds tend to cluster together as well as cats sharing the same geographical origins. The 46 genes identified within the CNVRs were annotated.
Conclusion
This study has improved the genomic characterization of 14 cat breeds and has provided CNVs information that can be used for studies of traits in cats. It can be considered a sound starting point for genomic CNVs identification in this species.
Similar content being viewed by others

Background
Short Tandem Repeats and Single Nucleotide Variants (Single Nucleotide Polymorphism – SNPs, as they are known) have been widely used in the study of the genome diversity and inherited diseases for a long time. Other structural and more complex variants - like Copy Number Variants (CNV) - were also known [1], and the recent advances in genome technologies, especially the development of array platforms and next generation sequencing, has allowed more global analyses of CNVs at a genome-wide level. These variants consist of changes in copy number of DNA sequences in comparison to a reference genome, including duplications (gain state) and deletions (loss state). The absence of variations is defined as normal state [2]. Conventionally, CNVs are defined as 1 Kb to several Mb in length, therefore being distinct from the smaller structural variants as indels and are more variable than SNPs, which vary by a single base pair [3].
An early study of the completed human genome for large-scale copy-number variants (LCVs) identified approximately 200 polymorphisms within the genomes of 20 normal individuals [4]. In another study, the regions including LCVs overlapped with genes involved in human disease syndromes and cancer, such as CMT4B2 gene (Charcot-Marie-Tooth disease type 4B2) and DPY, LRP12, FOG2 genes (squamous cell carcinoma) [5]. CNVs not only influence human phenotypes but are also related to genome evolution. The location and frequencies of the human and chimpanzee CNVs have been compared and duplications and deletions of genes with functions related to cell proliferation and inflammatory response have been found. In particular, a loss of the chimpanzee TBC1D3 gene, compared to the eight paralog copies of this gene in the human reference individual, may reflect a positive selection and adaptive phenotypic differentiation during primate evolution [6]. These studies used array-based comparative genomic hybridization methods (array CGH, aCGH), which lacked sensitivity to detect a wide range of CNVs [7].
The introduction of SNP high density genotyping and Next Generation Sequencing (NGS) approaches, together with the development of new bioinformatics tools, has led to better strategies for CNV detection [8, 9]. The 1000 Genomes Project Consortium (2010), using 179 low coverage whole genome human sequences, identified 28,025 structural variants, suggesting that CNVs represent 4.8–9.5% of the human genome [9]. NGS is also an alternative tool for genotyping CNVs associated to diseases, such as inherited kidney diseases [10].
Genome-wide studies to detect CNVs have been extended to domestic animals. CNVs have been shown to affect phenotypes such as pigmentation, morphology and production traits [11, 12]. CNVs have also been suggested to be responsible for complex disease traits such as periodic fever syndrome in Shar-Pei dogs [13] and canine squamous cell carcinoma of the digit [14].
NGS has supported the discovery of CNVs in animals too, providing higher sensitivity and allowing their identification at a genome-wide level in cattle [15], chickens [16], mice [17] and dogs [18]. Recently, CNV detection with NGS data has been used for association studies on production traits in livestock, such as fatty acids dynamics in beef cattle [19].
The genome assembly of the domestic cat is based on approximately 3× Sanger sequencing and ~ 14× short-read Illumina-based NGS. This assembly_6.2 and the re-sequencing of additional breed individuals have identified a wealth of genetic variation within the cat genome which led in turn to the development of a successful DNA 63 K array [20]. However, an analysis of large structural variants as CNV in the domestic cat across breeds and individuals has not been attempted. New cat genomes are now available from the 99 Lives cat genome sequencing project, which has produced high quality NGS data of 30× coverage, using similar techniques and technologies [21,22,23,24]. In such a context, the present study is the first genome-wide CNV detection in the domestic cat and is based on the sequence data from the cat 99 Lives Project. Ultimately this work is meant to assist the evaluation of cat breeds and to be used for the association of CNVs to breed-specific phenotypes, including disease phenotypes.
Materials and methods
Samples
Forty-two whole genome sequences representing 14 different cat breeds were available from the 99 Lives Cat Genome Sequencing Initiative (http://felinegenetics.missouri.edu/99lives) [21]. The genome sequences were produced as previously described [22] and sequencing data are available at NCBI BioProject PRJNA308208. The represented breeds, the number of individuals per breed and additional sequencing information are reported in Table 1.
The genome data for all the cats were produced by Illumina-based short-read technology using PCR-free libraries of ~ 350 bp and/or ~ 550 bp. Most genomes are at ~30× coverage, with 100–150 bp paired end reads. All the reads were mapped to the cat reference assembly_6.2 by Maverix Biomics (http://www.ncbi.nlm.nih.gov/assembly/320798) [25].
CNVs detection
Two programs using Read Depth (RD) based methods, cn.MOPS [26] and CNVnator [27], were employed to detect CNVs in the cat genomic data. Cn.MOPS and CNVnator software support the readily available bam files for the analysis and are considered the most suitable tools for Illumina sequencing data [28].
Cn.MOPS
The CNVs were firstly identified using the R “cn.MOPS” routine. The output was filtered to exclude false calls following the software manual indications, so only losses with median in the expected log fold change < − 1 and gains with median > 0.6 were considered, as described in detailes by the authors [26]. Window Length (WL) was set at 5 Kb.
CNVnator
The second CNV detection method utilized CNVnator. The filtering was carried out considering only CNVs with size ≥1 Kb, zero mapping quality (q0) < 0.5 and Pval1 < 0.001. Since the sequences used for the analysis had ~20× - 30× coverage, the WL was set at 100 bp as suggested by the authors [27].
Consensus mapping of CNV, CNVR and data analysis
To reduce the false positive calls from each of the detection method, the CNVs obtained from cn.MOPS and CNVnator were compared using the -intersectBed command of Bedtools software [29]. Only CNVs that overlap for at least 80% (consensus_CNVs) were considered true calls and included in further analyses. When a different loss/gain state was identified by the two software for a specific CNV, a visual inspection of the sequence read depth was carried out for that CNV to identify the true state. Particularly, samtool -view option (http://samtools.sourceforge.net/) was used to extract the CNV sequence, including 50 K bp of the flanking regions, and the read depth was visually inspected with the Golden Helix GenomeBrowse. This allowed to identify false and true positive calls and assign the true CNVs state.
CNV regions (CNVRs) for all the cats were obtained by merging consensus_CNVs using the -mergeBed command of Bedtools [29]. In order to validate the CNVRs identified and to exclude any possible false positive call, all the identified CNVRs were validated by the visual inspection. The sequence read depths of all the cats pertaining to a CNVR, and thus showing a CNV in that region, were compared to the ones of two cats showing CNV normal state for that region. Only losses or gains with clear boundaries were considered true CNV calls, contributing to CNVRs. All singleton CNV calls were also visually validated to exclude false positive calls and then considered as CNVRs. All the CNVRs boundaries were validated and re-assigned with visual inspection: for both sides, the initial descending bp and the initial ascending bp positions were identified as boundaries for losses and gains respectively.
Genes within the CNVRs were identified using the annotation of the NCBI Felis catus assembly_6.2 gene dataset and the Bedtools -intersectBed command was used to catalogue genes in the corresponding regions. Gene Ontology Terms (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed with the DAVID Bioinformatic Database (https://david.ncifcrf.gov/tools.jsp). GeneCards database (www.genecards.org) was consulted to obtain information on the function of the identified genes. Imprinted Gene database (www.geneimprint.com) was used to identify imprinted genes among the ones located within the identified CNVRs.
Cat population analyses
Structure and genomic diversity among all the cats were examined using Principal Component Analysis (PCA) and a clustering analysis, grouping the individuals according to their CNVR similarities [30]. A scoring matrix of the CNVRs was developed by encoding a value of ‘0’ or ‘1’ according to the presence or absence of any mapped CNV in the corresponding CNVR. The matrix considered only the 92 CNVRs shared by two or more individuals. A hierarchical agglomerative clustering, based on Unweighted Pair-Group Average method (UPGMA), was applied to the scoring matrix using the pvclust function from the pvclust R package [31]. Multiscale bootstrap resampling was performed to calculate the Approximately Unbiased P-value (AU-P) using 10,000 bootstraps to assess the robustness of branches. The AU-P and Bootstrap Probability value (BP-P) are presented for each node, as well as the edge numbers. The PCA was performed with Past3 software using the same matrix of the clustering analysis [32].
Results
CNVs detection with cn.MOPS
After filtering, 2282 CNVs were identified using cn.MOPS software. A mean of 23 CNVs/cat was observed. The Siamese cat sample SIA_4 showed 585 CNVs and was excluded as an outlier. Therefore, 1697 CNVs were considered for downstream analyses.
CNVs detection with CNVnator
After editing, CNVnator detected 285,533 CNVs. A mean of 5827 CNVs/cat was reported and the Siamese cat sample SIA_4 was also confirmed as an outlier. The Siamese cat sample SIA_2 was the cat with the highest number of CNVs (9413) while 4405 CNVs were counted in the Birman cat sample BIR_2, which was the one with the lowest number of CNVs. After the SIA_2 outlier exclusion, 234,484 CNVs were considered in downstream analyses.
Consensus mapping of CNV, CNVR and data analysis
Comparing cn.MOPS and CNVnator detections, a total of 999 consensus_CNVs was obtained, representing 59% of the 1697 CNVs identified by cn.MOPS. Out of the 147 consensus_CNVs with different state, 78 were identified by visual inspection as true call and their state was assigned according to their read depth respect to the flanking regions (Additional file 1: Table S1).
The CNVRs were then identified using the 930 remaining consensus_CNVs. Each of the 389 identified CNVR (including 269 singletons) was visually validated to identify false positive calls, including all detected singletons. An example for the CNVR validation is shown in Additional file 2: Figure S1 at chrD1:10,624,001-10,645,000 where the comparison with the reference genome and two normal state cats, ABY_1 and BEN_1, clearly allows to identify true CNV calls. A total of 154 CNVRs were, containing 589 validated consensus_CNVs, were confirmed. Among those, 62 were identified as singleton. Table 2 summarizes the statistics of consensus_CNVs found in each breed.
The size of the singleton regions (62 CNVRs) ranged from 5 Kb to 283 Kb, while the remaining 92 CNVRs ranged in size from 5 Kb to 529 Kb. The 154 CNVRs and are graphically represented in Fig. 1 and reported in Additional file 3: Table S2 and Additional file 4: Table S3.

Physical distribution of the Copy Number Variants Regions (CNVRs) according to states (gain, loss and complex) on the Felis catus vs 6.2 assembly
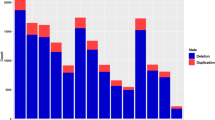
Considering the length of the cat autosomes mapped with Felix catus vs 6.2 assembly (about 2.2 Gb), the CNVRs covered about 0.26% of the cat genome. A total of 129 loss, 19 gain and 6 complex regions were identified. The statistics and the contribution of each breed in relation to singletons are reported in Fig. 2. All breeds show to have at least one singleton CNVR with the NAP showing the largest number.

Summary of the number of singletons for each breed and breed contribution to the total number of detected singletons
The contribution of every individual to each CNVR is reported in Additional file 4: Table S3. Two CNVRs were in common to the 66% of the cats: the first on chrD4:83,618,990-83,634,627, shared by 28 cats of 10 breeds and the second on chromosome chrA1:89,919,879-89,940,219, shared by 27 cats of 13 breeds. The CNVRs identified in at least 10 individuals were 13, while 26 CNVRS were present in only two cats. A total of 16 CNVRs were found in only one breed (ABY, BIR, BSH, BUR, MCO, NAP and OSH) as reported in Table 3.
In Fig. 3 the distribution of the CNVRs across the genome is reported together with the proportion of coverage within each chromosome. The number of CNVR per chromosome spans from 2 (chrE1) to 21 (chrA1) while the proportion of the total CNVR per chromosome spans from 0.05% (chrC2) to 0.69% (chrB3).

Distribution of CNVRs across the genome
In Fig. 4 the distribution of CNVRs by five size classes is reported. Only one CNVR, a gain, showed a size large than 500 Kb (chrB3:70,859,943-71,389,394, 520 Kb), while 67 are smaller than 20 Kb.

Distribution of CNVR size by class
Considering the assembly_6.2 annotation, 46 genes were located within the CNVRs (Additional file 3: Table S2 and Additional file 4: Table S3) and 13 mapped in CNVRs identified in only one breed (Table 3).
All the 46 identified genes were submitted to the David Bioinformatic Database but for only 25 genes the GO terms and KEGG metabolic pathways were available as reported in Additional file 5: Table S4.
The GO terms and the KEGG pathway clusters, resulting from the DAVID classification database, are reported in Table 4. Only two clusters have a P-Value lower than 0.05 and correspond to a biological process term (G-protein coupled receptor signaling pathway) and a KEGG pathway (Olfactory transduction).
Cat population analyses
Both the PCA and the cluster analysis depicted a similar population stratification based on different breeds (Figs. 5, 6).

Scatter plot showing groups of stratification

Clustering analysis using CNVRs. Colored rectangles correspond to geographical origin
Figure 5 shows the population stratification based on geographical origin (A) and on breeds (B). The principal components explained the 9.62 and 8.63% of the variance for PC1 and PC2 respectively. The breeds were grouped according to their geographical origin as follows: i) Asian Breeds - SIA, OSH, BUR, BIR and SPH; ii) Western Breeds - ABY, MCO, BSH, PER and RAG; iii) Mediterranean breed - EGY; iv) Mixed breeds – BEN, REX and NAP.
The clustering in Fig. 5-a shows two main groups of individuals represented by Asian breeds (green) and Western breeds (red). In Fig. 5-b a clear separate clustering is shown for BIR, BUR and OSH that are part of the Asian breeds group in Fig. 5-a. Among the Western breeds the BSH the PER and the RAG cluster very closely (Fig. 5-a).
The CNVR clustering tree also presented close associations for individuals belonging the same breed and for breeds with similar origins (Fig. 6).
The breeds clustered well according to the geographical origin, except for very few cases (RAG_2 and ABY_4 for the Western breeds; SIA_3 for the Asian breeds). For breeds with only one representative, the distribution was based on their geographical origins: Persian (PER_1) within the Western breed group while Egyptian (EGY_1) and Sphynx (SPH_1) within the Eastern breed group. Additionally, cats pertaining to the same breeds tend to cluster together as, e.g. the Burmese cats. Clustering with AU-P values > 90 were identified for several groupings of breeds, as for groups of individuals sharing the same geographical origins.
Discussion
Genome-wide mapping of CNVs has allowed new insights into genomic variation. Different techniques based on different approaches have been developed with the aim of more efficient and accurate CNV detection [28]. While aCGH has been used to detect CNVs for several years, the advent of NGS technologies has improved the global detection and analysis of CNV data. In humans, as in domestic animals, different CNV analyses have already been performed and have demonstrated the strong relationship between the presence of CNVs and phenotypic diversity, evolution and the onset of several diseases [33, 12]. Although the examination of repetitive elements was carried out in cats [25] and a preliminary annotation of the whole genome reference sequence (Felis catus assembly_6.2) has revealed that repetitive elements comprise about 55.7% of the whole genome, no formal studies focusing on CNVs have been performed [34].
This is the first CNV detection in the domestic cat and also the first one using NGS data. CNVnator and cn.MOPS software were selected to identify the CNVs. Both packages are based on the Read Depth (RD) method, which has been suggested as the most suitable for Illumina sequencing data [28]. Compared to other methods, the RD method does not require a reference sample and is based on coverage of the sequencing data. This represents an important advantage as, during the detection, the software compares reads within the same sample and also among all the samples. Moreover, the RD method uses a negative binomial distribution statistical model [28] and has the advantage of setting the WL depending on the number of reads. The difference between the two software programs is based on the RD method pipelines described by [35]. Cn.MOPS is based on a Bayesian approach that measures the depth of coverage at each genomic position, across multiple samples, thus increasing the statistical power and decreasing the false discovery rate in the detection. CNVs and noise identification are achieved using mixture components and Poisson distribution [26]. In contrast, CNVnator uses the mean-shift approach [36] and CNV detection is made more accurate through GC and multiple-bandwidth partitioning corrections [35]. The significant difference in the number of detected CNVs between the two packages is based on the different approach for defining a single CNV. In cn.MOPS, copy number of adjacent windows are compared and those with the same copy number sequence are joined into one single CNV. The length of the final CNV is given by the sum of the two adjacent copy number sequences. The more adjacent segments with high or low copy number call are joined, the higher will be the confidence in the detections. CNVnator does not use this overlapping approach, which explains the large discrepancy in the number of CNVs detected by the two packages. Ten or more CNVs detected with CNVnator correspond to one CNV detected with cn.MOPS. For this reason, it was decided to obtain CNVRs after the selection of CNVs in common to the two software.
The number of analyzed individuals is comparable or superior to those used in previous studies on horses (16 individuals [37]), pigs (20 individuals [38]) and dogs (34 individuals [39]). The number and the mean lengths of CNVs are in accordance to those found in dogs, where 1748 CNVs with a mean length of 194,559 bp were identified [40], and in other studies using cn.MOPS [41] and CNVnator software [17].
The present CNV detection revealed a higher number of loss regions compared to gain and complex regions. This could be explained by the difference within breeds of the same species, as already reported for Holstein cattle, where the number of loss regions is consistently higher than in other cattle breeds [42, 43]. Moreover, even though CNVnator is still the most used software for CNV detection, it is known to have a major deficiency in terms of detecting gain regions [27]. This could also explain the identification of 147 CNVs with different state when the output of the two software packages was compared. Nevertheless, the visual inspection approach used in this study to validate all the CNVRs, allows to overcome the problem of having different calling state between the cn.MOPS and CNVnator software. Additionally, it further reduces the false positive calls that may occur when using only one of the software for the CNV detection. In fact, the visual inspection leads to a full validation of the called CNV, improving the true calls obtained by comparing the results of the two calling algorithms. Visual inspection was also used to reassign the boundaries at each single CNVR, allowing to further refine the genome proportion covered by CNVR. The singletons detected in this study represent the 41% of the total CNVRs, a lower proportion compared to those reported in other studies [44,45,46].
The proportion of validated non singleton CNVRs after visual inspection was 77%. This proportion of true calls is in accordance to the findings of [47] who identified a concordance for the 80% of mapped CNV using two sequencing data runs.
Grouping the individuals according to their CNVR similarities, both the PCA and the clustering analyses showed cats belonging to the same breed tended to cluster together as well as cats sharing the same geographical origin. The Western and Eastern breeds were distinct in both of the analyses and resembled the results obtained in previous studies of cat phylogenies [20, 48]. However, the Devon Rex cats, a breed developed in the United Kingdom [49], neither cluster nor have apparent correct historical origins, which are suggested as Eastern in this study. Depending on breeding associations, the genetic contributions of some cat breeds can be varied as different associations allow for different outcrosses for a given breed. Thus, some of the variation in the breed and individual associations may be due to historical breeding differences.
The Bengal breed is a hybrid, developed from crossing spotted cats from India, Egyptian Maus or Abyssinians with the Asian leopard cat (Prionailurus bengalensis). Thus, the convoluted genetics of an individual cat of the Bengal breed could easily result in placement nearly anywhere within the cat genetic spectrum. As previously stated [50], the Abyssinian, which is one of the oldest cat breeds, has mysterious origins that are not clearly defined.
The gene annotation performed in this study showed that 19% of the CNVRs harbor genes. This proportion is lower than the ones found in studies developed in other species [8, 40, 46]. This is likely due to the more incomplete information in the cat gene annotation, compared to other species.
The LOC101085660, LOC101095519, LOC101101252 and LOC101089105 genes encode for olfactory receptors and were found in CNVRs of several breeds. As previously shown in humans [51], it may be related to significant variability in olfactory capabilities, an important sensorial attribute in predators.
The RYR3 gene in the Burmese cat was found differentially expressed in adipose tissue in cats during winter/short days and summer/long days [52].
The SYDE2, PAX6, ELP4 and the CHRM5 genes, have been found in genomic regions that have been recognized to be under positive selection in cats during their domestication [53]. Interestingly, the PAX6 and the ELP4 genes were found by the same authors as genes underlying segmental duplication in the domestic cat genome, as occurring in this study.
The PTCHD3 gene was found in the CNVR (complex) at chrB2:2,291,441-2,347,122 in 5 breeds (BEN, BIR, OSH, RAG, SPH). This gene has been previously found in a rare CNV in humans [54] where the CNV homozygous deletion was not associated to an abnormal phenotype. More recently, [55] have found this gene as associated with diabetes in humans and [56] have classified the gene as a potential imprinted gene. The PTCHD3 gene is not yet included in the Imprinted Genes database (www.geneimprint.com), as no other gene found in the CNVRs here reported. The presence of imprinted genes in CNVRs has already been shown in other species such as in cattle [57]. The regulation of the gene expression is mainly determined by the genetic imprinting and it could be interesting to further investigate this aspect in cat too.
Conclusions
The CNV calling performed in this study represents the first effort for the detection of genomic structural variation in the domestic cat. The clustering among the cat breeds that was possible to obtain in this study using CNVRs, complement findings of other studies based on other type of markers, leading to a closer insight of common and proprietary functional aspects of each population. Further studies based on further resequencing and on novel NGS technologies, might disclose other insights on CNV in Felis catus species and could complement the results obtained with the mapping performed in this study. Since CNV are well known to be related to gene expression regulation, also in complex diseases, this first mapping is meant to be the first information on a class of genomic variants that can be related to recorded phenotypes in cat populations.
Abbreviations
- ABY:
-
Abyssinian breed
- aCGH:
-
Array-Based Comparative Genomic Hybridization
- AU-P:
-
Approximately Unbiased P-value
- BEN:
-
Bengal breed
- BIR:
-
Birman breed
- BP-P:
-
Bootstrap Probability value
- BSH:
-
British Short Hair breed
- BUR:
-
Burmese breed
- CNV:
-
Copy Number Variants
- CNVR:
-
Copy Number Variants Region
- EGY:
-
Egyptian breed
- GO:
-
Gene Ontology Terms
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LCVs:
-
Large-Scale Copy-Number Variants
- MCO:
-
Main Coon breed
- NAP:
-
Napoleon breed
- NGS:
-
Next Generation Sequencing
- OSH:
-
Oriental Short Hair breed
- PER:
-
Persian breed
- RAG:
-
Ragdoll breed
- RD:
-
Read Depth
- REX:
-
Devon Rex breed
- SIA:
-
Siamese breed
- SNP:
-
Single Nucleotide Polymorphism
- SPH:
-
Sphynx breed
- UPGMA:
-
Unweighted Pair-Group Average
References
Bridges CB. The bar “gene” a duplication. Science. 1936;83(2148):210–1.
Henrichsen CN, Chaignat E, Reymond A. Copy number variants, diseases and gene expression. Hum Mol Genet. 2009;18(R1):R1–8.
Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5(10):e254.
Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305(5683):525–8.
Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36(9):949–51.
Perry GH, Yang F, Marques-Bonet T, Murphy C, Fitzgerald T, Lee AS, et al. Copy number variation and evolution in humans and chimpanzees. Genome Res. 2008;18(11):1698–710.
Pinto D, Darvishi K, Shi X, Rajan D, Rigler D, Fitzgerald T, et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotechnol. 2011;29(6):512–20.
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54.
Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16(3):172–83.
Nagano C, Nozu K, Morisada N, Yazawa M, Ichikawa D, Numasawa K, et al. Detection of copy number variations by pair analysis using next-generation sequencing data in inherited kidney diseases. Clin Exp Nephrol. 2018;22(4):881–8.
Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20(2):207–11.
Clop A, Vidal O, Amills M. Copy number variation in the genomes of domestic animals. Anim Genet. 2012;43(5):503–17.
Olsson M, Meadows JRS, Truvé K, Pielberg GR, Puppo F, Mauceli E, et al. A novel unstable duplication upstream of HAS2 predisposes to a breed-defining skin phenotype and a periodic fever syndrome in Chinese Shar-Pei dogs. PLoS Genet. 2011;7(3):e1001332.
Karyadi DM, Karlins E, Decker B, vonHoldt BM, Carpintero-Ramirez G, Parker HG, et al. A copy number variant at the KITLG locus likely confers risk for canine squamous cell carcinoma of the digit. PLoS Genet. 2013;9(3):e1003409.
Bickhart DM, Hou Y, Schroeder SG, Alkan C, Cardone MF, Matukumalli LK, et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. 2012;22(4):778–90.
Sohrabi SS, Mohammadabadi M, Wu D, Esmailizadeh A. Detection of breed-specific copy number variations in domestic chicken genome. Genome. 2018;61(1):7–14.
Harr B, Karakoc E, Neme R, Teschke M, Pfeifle C, Pezer Ž, et al. Genomic esources for wild populations of the house mouse, Mus musculus and its close relative Mus spretus. Sci Data. 2016;13(3):160075.
Freedman AH, Gronau I, Schweizer RM, Ortega-Del Vecchyo D, Han E, Silva PM, et al. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 2014;10(1):e1004016.
de Lemos MVA, Peripolli E, Berton MP, Feitosa FLB, Olivieri BF, Stafuzza NB, et al. Association study between copy number variation and beef fatty acid profile of Nellore cattle. J Appl Genet. 2018;59(2):203–23.
Gandolfi B, Alhaddad H, Abdi M, Bach LH, Creighton EK, Davis BW, et al. Applications and efficiencies of the first cat 63K DNA array. Sci Rep. 2018;8:7024.
Mauler DA, Gandolfi B, Reinero CR, O'Brien DP, Spooner JL, Lyons LA, et al. Precision medicine in cats: novel Niemann-pick type C1 diagnosed by whole-genome sequencing. J Vet Intern Med. 2017;31(2):539–44.
Gandolfi B, Grahn RA, Creighton EK, Williams DC, Dickinson PJ, Sturges BK, et al. COLQ variant associated with Devon rex and Sphynx feline hereditary myopathy. Anim Genet. 2015;46(6):711–5.
Aberdein D, Munday JS, Gandolfi B, Dittmer KE, Malik R, Garrick DJ, et al. A FAS-ligand variant associated with autoimmune lymphoproliferative syndrome in cats. Mamm Genome. 2017;28(1–2):47–55.
Oh A, Pearce JW, Gandolfi B, Creighton EK, Suedmeyer WK, Selig M, et al. Early-onset progressive retinal atrophy associated with an IQCB1 variant in African black-footed cats (Felis nigripes). Sci Rep. 2017;7:43918.
Pontius JU, Mullikin JC, Smith DR. Agencourt sequencing team, Lindblad-Toh K, Gnerre set al. Initial sequence and comparative analysis of the cat genome. Genome Res. 2007;17(11):1675–89.
Klambauer G, Schwarzbauer K, Mayr A, Clevert DA, Mitterecker A, Bodenhofer U, et al. Cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 2012;40(9):e69.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21(6):974–84.
Pirooznia M, Goes FS, Zandi PP. Whole-genome CNV analysis: advances in computational approaches. Front Genet. 2015;6:138.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2.
Tian M, Wang Y, Gu X, Feng C, Fang S, Hu X, et al. Copy number variants in locally raised Chinese chicken genomes determined using array comparative genomic hybridization. BMC Genomics. 2013;14:262.
Ryota S, Hidetoshi S. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22(12):1540–2.
Hammer Ø, Harper DAT, Ryan PDPAST. Paleontological statistics software package for education and data analysis. Palaeontol Electron. 2001;4(1):9.
Shastry BS. Copy number variation and susceptibility to human disorders. Mol Med Rep. 2009;2(2):143–7.
Tamazian G, Simonov S, Dobrynin P, Makunin A, Logachev A, Komissarov A, et al. Annotated features of domestic cat – Felis catus genome. Gigascience. 2014;3:13.
Miller CA, Hampton O, Coarfa C, Milosavljevic A. ReadDepth: a parallel R package for detecting copy number alterations from short sequencing reads. PLoS One. 2011;6(1):e16327.
Wang LY, Abyzov A, Korbel JO, Snyder M, Gerstein MMSB. A mean-shift-based approach for the analysis of structural variation in the genome. Genome Res. 2009;19(1):106–17.
Doan R, Cohen N, Harrington J, Veazey K, Juras R, Cothran G, et al. Identification of copy number variants in horses. Genome Res. 2012;22(5):899–907.
Wang J, Jiang J, Wang H, Kang H, Zhang Q, Liu JF. Improved detection and characterization of copy number variations among diverse pig breeds by array CGH. G3 (Bethesda). 2015;5(6):1253–61.
Serres-Armero A, Povolotskaya IS, Quilez J, Ramirez O, Santpere G, Kuderna LFK, et al. Similar genomic proportions of copy number variation within gray wolves and modern dog breeds inferred from whole genome sequencing. BMC Genomics. 2017;18(1):977.
Molin AM, Berglund J, Webster MT, Lindblad-Toh K. Genome-wide copy number variant discovery in dogs using the CanineHD genotyping array. BMC Genomics. 2014;19(15):210.
McNew SM, Beck D, Sadler-Riggleman I, Knutie SA, Koop JAH, Clayton DH, et al. Epigenetic variation between urban and rural populations of Darwin’s finches. BMC Evol Biol. 2017;17(1):183.
Jiang L, Jiang J, Yang J, Liu X, Wang J, Wang H, et al. Genome-wide detection of copy number variations using high-density SNP genotyping platforms in Holsteins. BMC Genomics. 2013;14:131.
Gao Y, Jiang J, Yang S, Hou Y, Liu GE, Zhang S, et al. CNV discovery for milk composition traits in dairy cattle using whole genome resequencing. BMC Genomics. 2017;18(1):265.
Yi G, Qu L, Liu J, Yan Y, Xu G, Yang N. Genome-wide patterns of copy number variation in the diversified chicken genomes using next-generation sequencing. BMC Genomics. 2014;15:962.
Han R, Yang P, Tian Y, Wang D, Zhang Z, Wang L, et al. Identification and functional characterization of copy number variations in diverse chicken breeds. BMC Genomics. 2014;15:934.
Prinsen RTMM, Strillacci MG, Schiavini F, Santus E, Rossoni A, Maurer V, et al. A genome-wide scan of copy number variants using high-density SNPs in Brown Swiss dairy cattle. Livest Sci. 2016;191:153–60.
Letaief R, Rebours E, Grohs C, Meersseman C, Fritz S, Trouilh L, et al. Identification of copy number variation in French dairy and beef breeds using next-generation sequencing. Genet Sel Evol. 2017;49:77.
Lipinski MJ, Froenicke L, Baysac KC, Billings NC, Leutenegger CM, Levy AM, et al. The ascent of cat breeds: genetic evaluations of breeds and worldwide random bred populations. Genomics. 2008;91(1):12–21.
Robinson R. The rex mutants of the domestic cat. Genetica. 1971;42:466.
Robinson R. The domestic cat, Felis catus. In: King RC, editor. Handbook of genetics. Boston: Springer; 1975.
Young JM, Endicott RM, Parghi SS, Walker M, Kidd JM, Trask BJ. Extensive copy-number variation of the human olfactory receptor gene family. Am J Hum Genet. 2008;83(2):228–42.
Mori A, Kappen KL, Dilger AC, Swanson KS. Effect of photoperiod on the feline adipose transcriptome as assessed by RNA sequencing. BMC Vet Res. 2014;10:146.
Montague MJ, Li G, Gandolfi B, Khan R, Aken BL, Searle SM, et al. Comparative analysis of the domestic cat genome reveals genetic signatures underlying feline biology and domestication. Proc Natl Acad Sci U S A. 2014;111(48):17230–5.
Seno MMG, Kwan BYM, Lee-Ng KKM, Moessner R, Lionel AC, Marshall CR, Scherer SW. Human PTCHD3 nulls: rare copy number and sequence variants suggest a non-essential gene. BMC Medical Genetics. 2011;12:45.
Yamada Y, Sakuma J, Takeuchi I, Yasukochi Y, Kato K, Oguri M, et al. Identification of five genetic variants as novel determinants of type 2 diabetes mellitus in Japanese by exome-wide association studies. Oncotarget. 2017;8(46):80492–505.
Bonaldi A, Kashiwabara A, de Araújo ÉS, Pereira LV, Paschoal AR, Andozia MB. Mining novel candidate imprinted genes using genome-wide methylation screening and literature review. Epigenomes. 2017;1(2):13.
da Silva JM, Giachetto PF, da Silva LO, Cintra LC, Paiva SR, Yamagishi MEB, et al. Genome-wide copy number variation (CNV) detection in Nelore cattle reveals highly frequent variants in genome regions harboring QTLs affecting production traits. BMC Genomics. 2016;17:454.
Acknowledgements
MFR of the 99Lives Consortium acknowledges support of state of Iowa and hatch funding.
EIG of the 99Lives Consortium acknowledges University of Massachusetts Medical School for providing funding.
EO of the 99Lives Consortium acknowledges the Intramural Program of the National Human Genome Research Institute at the National Institutes of Health, USA for providing funding.
99 lives consortium
Barbara Gandolfi1, Danielle Aberdein2, Paulo C. Alves3,4, Gregory S. Barsh5,6, Holly C. Beale7, Tomas F. Bergström8, Adam R. Boyko9, Jeffrey A. Brockman10, Marta G. Castelhano11, Patricia P. chan7, Brian W. Davis12, Ottmar Distl13, Nicholas H. Dodman14, N. Matthew Ellinwood15, Jonathan E. Fogle16, Oliver P. Forman17, Dorian J. Garrick2,17, Jens Häggström18, Christopher R. Helps19, Marjo K. Hytönen20, Daniel M. Ibrahim21, Maria Kaukonen20, Christopher B. Kaelin5,6, Emilie Leclerc22, Teri L. Lear23, Tosso Leeb24, Hannes Lohi20, Darío G Lupiáñez21, Mark A. Magnuson25,26, Richard Malik27, Michael J. Montague28, John S. Munday2, William J. Murphy29, Elaine A. Ostrander12, Niels C. Pedersen30, Simon M. Petersen-Jones31, Max F. Rothschild15, Beth Shapiro32, Joshua A. Stern30, William F. Swanson33, Karen A. Terio34, Rory J. Todhunter11, Yu Ueda30, Wesley C. Warren28, Elizabeth A. Wilcox11, Julia H. Wildschutte35, Edward I. Ginns36.
1Department of Veterinary Medicine and Surgery, College of Veterinary Medicine, University of Missouri, Columbia, Missouri, 65,211 USA.
2School of Veterinary Science, Massey University, Palmerston North 4410 New Zealand.
3CIBIO/InBIO, Centro de Investigação em Biodiversidade e Recursos Genéticos/InBIO Associate Lab & Faculdade de Ciências, Universidade do Porto, Campus e Vairão, 4485–661 Vila do Conde, Portugal.
4Wildlife Biology Program, University of Montana, Missoula, Montana, 59,812 USA.
5HudsonAlpha Institute for Biotechnology, Huntsville, Alabama, 35,806 USA.
6Department of Genetics, Stanford University, Stanford, California, 94,305 USA.
7Maverix Biomics, Inc., San Mateo, California, 94,402 USA.
8Department of Animal Breeding and Genetics, Swedish University of Agricultural Sciences, 750 07 Uppsala, Sweden.
9Department of Biomedical Sciences and Cornell Veterinary Biobank, College of Veterinary Medicine, Cornell University, Ithaca, New York, 14,853 USA.
10Hill’s Pet Nutrition Inc., Topeka, Kansas 66,601 USA.
11Department of Clinical Sciences and Cornell Veterinary Biobank, College of Veterinary Medicine, Cornell University, Ithaca, New York, 14,853 USA.
12Cancer Genetics and Comparative Genomics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20,892 USA.
13Institute for Animal Breeding and Genetics, University of Veterinary Medicine Hannover (Foundation), 30,559, Hannover, Germany.
14Department of Clinical Sciences, Cummings School of Veterinary Medicine, Tufts University, Grafton, MA, 01536 USA.
15Department of Animal Science, College of Agriculture and Life Sciences, Iowa State University, Ames, Iowa, 50,011 USA.
16College of Veterinary Medicine, North Carolina State University, Raleigh, NC 27607.
17WALTHAM Centre for Pet Nutrition, Freeby Lane, Waltham on the Wolds, Leicestershire, LE14 4RT UK.
18Department of Clinical Sciences, Faculty of Veterinary Medicine and Animal Science, Swedish University of Agricultural Sciences, Uppsala, SE-750 07 Sweden.
19Langford Veterinary Services, University of Bristol, Langford, Bristol, BS40 5DU UK.
20Department of Veterinary Biosciences and Research Programs Unit, Molecular Neurology, University of Helsinki and Folkhälsan Research Center, Helsinki 00014 Finland.
21Max Planck Institute for Molecular Genetics, RG Development and Disease, D-14195, Berlin, Germany.
22Diana Petfood, SPF, ZA du Gohelis, 56,250 Elven, France.
23Department of Veterinary Science, University of Kentucky - Lexington, Lexington, KY, 40506 USA (In memoriam).
24Vetsuisse Faculty, Institute of Genetics, University of Bern, 3001 Bern, Switzerland.
25Department of Cell and Developmental Biology, Vanderbilt Center for Stem Cell Biology, Vanderbilt University School of Medicine, Vanderbilt University Program in Developmental Biology, Nashville, Tennessee, 37,240 USA.
26Department of Molecular Physiology and Biophysics, Vanderbilt University School of Medicine, Nashville, Tennessee, 37,240 USA.
27Centre for Veterinary Education, University of Sydney, Sydney, NSW, 2006 Australia.
28The McDonnell Genome Institute, Washington University School of Medicine, St. Louis, Missouri, 63,108 USA.
29Department of Veterinary Integrative Biosciences, College of Veterinary Medicine, Texas A&M University, College Station, Texas, 77,845 USA.
30Department of Medicine and Epidemiology, School of Veterinary Medicine, University of California at Davis, Davis, California, 95,616 USA.
31Small Animal Clinical Sciences, College of Veterinary Medicine, Michigan State University, East Lansing, Michigan, 48,824 USA.
32Department of Ecology and Evolutionary Biology, University of California, Santa Cruz, Santa Cruz, Calfornia 95,064 USA.
33Center for Conservation and Research of Endangered Wildlife (CREW), Cincinnati Zoo & Botanical Garden, Cincinnati, Ohio, 45,220 USA.
34Zoological Pathology Program, University of Illinois, Brookfield, Illinois 60,513 USA.
35Bowling Green State University, Department of Biological Sciences, Bowling Green, Ohio 43,403 USA.
36Department of Psychiatry, University of Massachusetts Medical School, Worcester, MA, 01655 USA.
Funding
Funding for the 99 Lives Cat Genome Sequencing Initiative has been provided by the Winn Feline Foundation and the George Sydney and Phyllis Redman Miller Trust (MT–13–010), the National Geographic Society Education Foundation (2P-14), the University of Missouri Gilbreath-McLorn Endowment (LAL), the Cornell Feline Health Center (ARB, MGC, RJT), contributions from the 99 Lives Consortium participants, donations from Associazione Nazionale Felina Italiana (ANFI), Zoetis, Orivet Genetic Pet Care, Langford Veterinary Services, the World Cat Federation, and public donations. Internal funds from the Università degli Studi di Milano, Italy.
Availability of data and materials
All the data used in this study were fully available without restriction. The data are fully accessible in NCBI database according to accession number as specified in Table 1. Data with no accession number are under release and can be accessed in the meantime freely upon request to the 99Lives Consortium Coordinator.
Author information
Authors and Affiliations
Consortia
Contributions
FG, MGS and AB analyzed the data and wrote the manuscript, ML, LAL, AB and MGS revised critically the results and the manuscript, 99 L provided the genomic data, MGS conceived the idea and coordinated the research. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable as all the whole genome sequence data were made available by the 99Lives Consortium.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. (XLSX). Consensus CNVs. List of the consensus list of CNVs detected by both cn.mops and CNVnator software and state confirmed with visual inspection with GenomeBrowse (XLSX 34 kb)
Additional file 2:
Figures S1. (PDF). Visual inspection of CNVR at chrD1:10624094–10,643,050. (TIF 2999 kb)
Additional file 3:
Table S2. (XLSX). Singleton CNVRs and list of annotated genes. (XLSX 14 kb)
Additional file 4:
Table S3. (XLSX). List of CNVRs. List of the CNVRs and annotated genes. Singletons were excluded. (XLSX 20 kb)
Additional file 5:
Table S4. (DOCX). David annotation of genes in all CNVRs. List of the annotation gene names GO terms (BP: biological process; CC: cellular component; MF: molecular function), and KEGG pathways. (DOCX 22 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Genova, F., Longeri, M., Lyons, L.A. et al. First genome-wide CNV mapping in FELIS CATUS using next generation sequencing data. BMC Genomics 19, 895 (2018). https://doi.org/10.1186/s12864-018-5297-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5297-2