
Abstract
Background
Pectobacterium parmentieri is a newly established species within the plant pathogenic family Pectobacteriaceae. Bacteria belonging to this species are causative agents of diseases in economically important crops (e.g. potato) in a wide range of different environmental conditions, encountered in Europe, North America, Africa, and New Zealand. Severe disease symptoms result from the activity of P. parmentieri virulence factors, such as plant cell wall degrading enzymes. Interestingly, we observe significant phenotypic differences among P. parmentieri isolates regarding virulence factors production and the abilities to macerate plants. To establish the possible genomic basis of these differences, we sequenced 12 genomes of P. parmentieri strains (10 isolated in Poland, 2 in Belgium) with the combined use of Illumina and PacBio approaches. De novo genome assembly was performed with the use of SPAdes software, while annotation was conducted by NCBI Prokaryotic Genome Annotation Pipeline.
Results
The pan-genome study was performed on 15 genomes (12 de novo assembled and three reference strains: P. parmentieri CFBP 8475T, P. parmentieri SCC3193, P. parmentieri WPP163). The pan-genome includes 3706 core genes, a high number of accessory (1468) genes, and numerous unique (1847) genes. We identified the presence of well-known genes encoding virulence factors in the core genome fraction, but some of them were located in the dispensable genome. A significant fraction of horizontally transferred genes, virulence-related gene duplications, as well as different CRISPR arrays were found, which can explain the observed phenotypic differences. Finally, we found also, for the first time, the presence of a plasmid in one of the tested P. parmentieri strains isolated in Poland.
Conclusions
We can hypothesize that a large number of the genes in the dispensable genome and significant genomic variation among P. parmentieri strains could be the basis of the potential wide host range and widespread diffusion of P. parmentieri. The obtained data on the structure and gene content of P. parmentieri strains enabled us to speculate on the importance of high genomic plasticity for P. parmentieri adaptation to different environments.
Similar content being viewed by others


Background
Genomes of a given bacterial species can show considerable variation in gene content, distribution, and presence of mobile elements. Therefore, conducting systematic analyses of the entire gene repertoire of several strains from a specific species, termed the pan-genome study, is vital for understanding bacterial intraspecies diversity, population genetics, and bacterial evolution [1]. The core pan-genome constitutes of highly-conserved genes present in all the analyzed genomes and usually encodes genes related to fundamental aspects of the bacterial biology contributing to significant phenotypic traits [2]. It has been reported previously that some genes of the dispensable (accessory and unique) pan-genome might play a role in bacterial adaptation to specific growth conditions, such as colonization of new ecological niches, symbiosis, host-cell interaction, and pathogenicity. In other words, the plasticity of this pan-genome fraction contributes to bacterial evolution [3]. The diversity/plasticity mentioned above is especially crucial regarding opportunistic bacterial pathogens, such as human, animal and also plant pathogens spreading in new hosts and/or a new environment.
Specifically, the genomes of a plant quarantine pathogen Ralstonia solanacearum include chromosomal rearrangements and several genes recently acquired via a horizontal gene transfer (HGT) [4,5,6]. R. solanacearum strains exhibit an unusually broad host range as they can infect more than 250 plant species in monocot and dicot botanical families [7]. The vast host range of R. solanacearum may be correlated with its high genomic plasticity [8,9,10] attributed to the occurrence of Mobile Genetic Elements (MGE), such as phages and plasmids [11].
Amongst plant pathogenic bacteria that trigger economically important losses, the causative agents of soft rot and potato blackleg should be listed. This subgroup of Gram-negative Gammaproteobacteria, classified to the Pectobacteriaceae family currently encloses two genera: Dickeya and Pectobacterium [12]. The diseases caused by Dickeya spp. and Pectobacterium spp. result from the activity of Plant Cell Wall Degrading Enzymes (PCWDE) such as pectinases, cellulases, and proteases secreted via Type I or II secretion systems [13]. The Broad host range of these phytopathogens can be exemplified by the fact that Pectobacteriaceae have been reported to cause soft-rotting symptoms in a large number of plants, including 16 dicot plant families in 11 orders and 11 monocot families in 6 orders [14].
The development of novel diagnostic methods resulted in several reclassifications within both genera. High genomic heterogeneity was attributed to Pectobacterium carotovorum strains (exhibiting about 20 different recA PCR-RFLP patterns) in comparison to Pectobacterium atrosepticum (with just two recA PCR-RFLP patterns [15]). The application of molecular techniques: genomic sequences comparison, DNA:DNA hybridization and average nucleotide identity (ANI) analysis resulted in the reevaluation of the taxonomic position of P. carotovorum strains deposited in different collections and/or isolated in the recent years. Finally, some strains of P. carotovorum, including the most frequently studied P. carotovorum SCC3193, were reclassified to Pectobacterium wasabiae [16] and later on to Pectobacterium parmentieri species [17].
In this study, we focused on P. parmentieri infecting plants worldwide and detected in Europe, North America, Africa, Asia and New Zealand [17,18,19,20,21,22,23,24,25]. P. parmentieri has been proven to cause disease symptoms on potato plants and tubers and also to survive in unfavourable environmental conditions (such as soil or surface waters). By now, their host range in the natural environment has not been fully understood. However, there is some information about the isolation of P. parmentieri (there P. wasabiae) strains from cabbage, eggplants, sweet potato and tomato in Malaysia [26].
Currently, there are five publicly available genomes of P. parmentieri (NCBI servers, June 2018), whereas only 3 of them are closed to a full chromosome. The genome (GenBank accession: NZ_CP015749.1) of the type strain P. parmentieri CFBP 8475T (previously P. wasabiae RNS 08.42.1A) is 5.03 Mb in size with GC content of 50.4% [27]. Among the 4462 proteins it encodes, there have been described some proteins speculated to ensure specific phenotypic traits that are important for P. parmentieri virulence and adaptation to its primary host - potato, such as PCWDE, components of secretion systems and AHL-dependent quorum sensing system [27].
The aim of the here presented study is to elucidate the genomic basis of P. parmentieri spread and rapid adaptation to different climate conditions (temperature, humidity). It was achieved by completing genome sequencing of 12 P. parmentieri strains isolated from potato plants in various environments, followed by comparative genomic analyses conducted on these twelve strains and three other sequences available in GenBank. The obtained results point to a high abundance of MGE characterizing the reported P. parmentieri pan-genome, which may likely be linked with adaptation to different environmental niches and be the reason for the worldwide spreading of this species.
Results and discussion
Phenotypic characterization of P. parmentieri strains
Phenotypic characterization of the available P. parmentieri strains, isolated in Poland (10), Belgium (2), Finland (1) and France (1) (Table 1), was performed. We observed high variability between the analyzed strains regarding the potato maceration efficacy. It was interesting that the lowest and the highest maceration ability was noted for the strains isolated in Poland. Besides, IFB5408 indicates 4-fold lower maceration than IFB5626 (Table 2).
The investigation of other P. parmentieri phenotypic features, such as the activity of PCDWEs revealed statistically significant differences between strains. The tested strains indicated significant differences in pectinases, cellulases and proteases but not lipases activities (Table 2). Strains IFB5597, IFB5604, IFB5619, IFB5623 produced significantly higher amounts of pectinases. At the same time, none of them produced high levels of cellulases. However, IFB5597 as the only one of them indicate high activity of proteases. On the other hand strains IFB5408, IFB5441, IFB5485 and IFB5486 possessed the low activity of pectinases, but IFB5485 and IFB5486 produced significant amounts of cellulases.
The studied P. parmentieri strains exhibit significant differences regarding both swimming and swarming motility (Table 2). Conserning biofilm formation, IFB5408, IFB5604 and IFB5605 isolates were as efficient in this trait as P. aeruginosa PAO1, a strain commonly applied as a positive control in biofilm formation assessment (Table 2).
Furthermore, we calculated correlation coefficients between different phenotypic traits by applying Pearson statistical method (p < 0.05). Weak positive linear relationships between the strain’s ability to macerate potato tissue and their cellulases activity in addition to biofilm formation capacity (0.118 and 0.085) were observed. Strains possessing the overall moderate activity of pectinases, cellulases and proteases showed the highest ability to macerate potato tissue. In other words, the observed effects of different PCDWEs are synergistic, and the individual activity of each enzyme tends to complement each other (Table 2).
The here-described P. parmentieri intraspecies variation is in agreement with several previous studies, in which P. parmentieri isolates differed in the plant maceration efficacy and PCWDE activities [20, 21, 23, 24, 28].
Genome structure of the P. parmentieri strains
Comparative genomics on 15 P. parmentieri strains (12 newly sequenced and three available from the GenBank) was performed. The number of scaffolds, GC content and the amount of N bases for all of the analyzed genomes were determined (Table 3). Ten of the newly assembled genomes were closed to a full chromosome, but in two cases we were not able to close specific genomes: strain IFB5626 consists of 2, and IFB5597 of 5 scaffolds. The length of the obtained P. parmentieri genomes varied from 4,877,201 bp (IFB5604) to 5,125,304 bp (IFB5427), which equals approx. 5% difference. GC content varied between 50.29 and 50.6%, where the strain IFB5441 and IFB5619 had the lowest and the highest values, respectively. These results are in agreement with those obtained earlier for P. parmentieri CFBP 8475T and P. parmentieri SCC3193 regarding the genome length (5,030,841 bp and 5,164,411 bp) and the GC content (50.4% and 50.37%) [16, 17].
Obtaining complete genome sequences allowed us to investigate deeply structural variations within the P. parmentieri genomes. The synteny of the closed genomes is presented in Fig. 1. In general, genomes were highly syntenic with minor structural differences. We detected a sizeable chromosomal rearrangement in one of the strains that have been isolated in Belgium (IFB5486). This deviation might have appeared during the long-time period of evolution, which could have subsequently undergone selective pressure under specific environmental conditions during the lengthy process of speciation, as it was reported before in other members of Enterobacteriales [29]. Moreover, highly variable regions in almost all of the analyzed strains were detected (Fig. 1). In these highly variable regions, genes encoding phage-related proteins were found (Fig. 1) and a more precise description of these MGE shall be provided later.

P. parmentieri genomes shape generated with the use of BRIG software
Interestingly, during de novo genome assembly of IFB5427 strain, the presence of a large plasmid of about 100 kb has been discovered. This plasmid, for the first time described in P. parmentieri species, was named pPAR01. Regarding the Pectobacteriaceae genus, the presence of other plasmids was reported only in P. carotovorum SCC1 (5 kb), D. solani PPO9091 (43 kb) and D. fangzhongdai DSM 101947 (5 kb) [30,31,32]. Notably, the above-listed plasmids are much smaller than pPAR01. A more detailed analysis of this plasmid is presented in ‘the mobilome’ paragraph below.
P. parmentieri has an open pan-genome
With the NCBI Prokaryotic Genome Annotation Pipeline, the number of total predicted genes varied from 4842 to 4534 for strains IFB5427 and IFB5485, respectively. The total number of genes encoding proteins ranged from 4561 to 4252 (Table 3) and was attributed to the strains mentioned above.
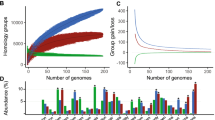
The pan-genome shape of the 15 analyzed P. parmentieri genomes is presented in Fig. 2A. A total of 7021 gene clusters (orthologs) were found, 3706 of which comprised the core genome (52.8%), 1468 the accessory genome (20.9%) and 1847 (26.3%) the unique genome fraction (Fig. 2a, Table 3). The quantity of the determined accessory genes varied from 486 to 822 for the most diverse strains SCC3193 and IFB5486, respectively. Interestingly, P. parmentieri SCC3193 indicated the highest quantity of unique genes (346 genes), while the strain IFB5485 the lowest (7 genes).

The pan-genome shape of P. parmentieri. a The pan-genome pie chart showing gene content visualised with the use of Roary software. b Heap’s law chart representation regarding conserved genes vs total genes in 15 genomes
To investigate the openness of P. parmentieri pan-genome Heap’s Law was utilized [33, 34]. The openness of pan-genome reflects the diversity of the gene pool within bacterial species. For species with an open pan-genome, an addition of a newly sequenced genome significantly changes the pan-genome size [2], contrarily to what is observed in the case of strains exhibiting a closed pan-genome. The calculated alpha value of 0.81 indicated that the P. parmentieri pan-genome is open (Fig. 2b) and that could be the result of high genomic plasticity within this species [1]. This finding is in agreement with the level of openness of the pan-genome for other representatives of Gammaproteobacteria as it was reported before [35,36,37,38]. In contrast to P. parmentieri pan-genome, closely related and very homogenous D. solani species have a closed pan-genome [39].
Genomic relatedness of P. parmentieri strains
Genetic relatedness among the P. parmentieri strains was studied by both computing the ANI values and by establishing genomic similarities with a pan-genome based analysis. As expected for strains belonging to the same species, high ANI values ranging from 98.98 to 99.97 were observed. Phylogenetic analyses derived from the comparison of all protein sequences encoded within the genomes is presented in Fig. 3. The genomes of P. parmentieri strains formed two main clades, from which the first (Clade I) is further divided into two subclades (IA and IB). Clade IA comprises genomes of P. parmentieri reference strain WPP0163 (isolated in the USA), three strains isolated in Poland in the year 2014 (IFB5597, IFB5604, and IFB5619) and also one strain isolated in Belgium (IFB5485) (Fig. 3). Clade IB, on the other hand, encloses genomes of three strains isolated in Poland, two in the year 2013 (IFB5432, IFB5441) and one strain from 2014 (IFB5623). The second clade (Clade II) consists of genomes of P. parmentieri CFBP 8475T, two strains isolated in 2013 (IFB5408, IFB5427), two in 2014 in Poland (IFB5605, IFB5626) and one strain isolated in Belgium (IFB5486). Interestingly, P. parmentieri SCC3193 being the model strain used for studying P. parmentieri molecular biology for many years (previously P. c. subsp. carotovorum SCC3193/P. wasabiae SCC3193 [40,41,42]) is distinctively separated from the clades mentioned above (has the highest number of unique genes). Furthermore, strains constituting Clade II, share the highest quantity of accessory genes (on average 794) in contrast to strains grouped within Clade I, for which an average number of accessory genes is 645) (Fig. 3, Table 3). The above-presented grouping corresponds with the ANI-based clustering as ANI Cluster I reflects Clade II, while ANI Cluster II is consistent with Clade IB (Additional file 1: Figure S1A).

Phylogenetic tree constructed with the method blast all against all using protein sequences. The tree was rooted on P. wasabiae CFBP 3304. The color codes: blue – strains isolated in 2013 (Poland), green – strains isolated in 2014 (Poland), brown – strains isolated in Belgium, pink – reference strains
In general, P. parmentieri strains did not group accordingly to geographical site of isolation (Fig. 3), which may suggest their high geographical mobility. However, to fully understand the biogeographical pattern of genomic variation in P. parmentieri, a more significant number of strains from different regions should be sequenced and analyzed.
The described phylogenetic pattern was further supported by inspection of the presence/absence of genes in the dispensable (accessory and unique) genome fraction (Additional file 1: Figure S1B), which also showed agreement with the groupings presented in the phylogenetic analysis of all protein sequences (Fig. 3). We observe that all the strains from Clade II (Fig. 3) form a monophyletic group in Additional file 1: Figure S1B. Interestingly, P. parmentieri SCC3193 (distinctively separated from other strains in the protein phylogenetic tree), in accessory genome fraction phylogenesis, is grouping with P. parmentieri WPP163 and IFB5597.
Functional annotation of the dispensable genome fraction
To describe the strain-specific genomic differences, which are usually represented within the dispensable pan-genome fraction, we performed: class of genes (COG) annotation. It was done for every protein belonging to the dispensable pan-genome fraction to obtain results biased towards genes found in all the analyzed genomes (Fig. 4). We successfully annotated 900 protein sequences. A significant fraction of the proteins was attributed to cell wall and membranes biogenesis (130 genes) and replication and recombination (108 genes). In particular, among these 108 genes, there were quite a few proteins related to MGE, such as transposases, integrases and also DNA repair proteins and the CRISPR-associated ones.

COG classes annotation of the accessory pan-genome fraction
Proteins related to transcription were also abundant (93 genes), such as LysR family transcriptional regulators, controlling numerous genes involved in virulence, metabolism, quorum sensing and motility [43], but also other regulators responsible for plasmid maintenance (Xre), initiation of expression of phage-related genes (Ogr/delta) or resistance to multiple antibiotics (MarR) [44]. Also, 26 proteins were found to be involved in carbohydrate transport and metabolism with the notable presence of pectate lyase, glucosidases, transporters or outer membrane porins (Additional file 2: Table S1). Almost the same number of proteins (24) were attributed to intracellular trafficking, secretion, and vesicular transport, including conjugal transfer proteins, filamentous hemagglutinin, Flp/Tad pilus component, especially crucial for the establishment of virulence [45] in addition to transfer proteins. Moreover, 18 proteins were designated to inorganic ion transport and metabolism, and in this COG group, we found, among others, TonB-dependent siderophore receptors capable of binding siderophore-iron complexes with high specificity and carrying out active transport across the outer membrane [46].
Besides, we have discovered that a significant part of COG class K is related to phage-related transcription regulators and phage-related proteins. Prophages, as genetic elements, may account for a significant fraction of the bacterial genome and pass to the chromosome many genes in a single event of integration [47].
The variability within the pathogenome of P. parmentieri
Genes encoding virulence factors, motility features, chemotaxis, secretion system and quorum sensing components were investigated to describe the possible genomic variation related to pathogenesis comprehensively. Focusing on PCWDE, we have assessed the presence of genes encoding: pectinases, polygalacturonases, cellulases, and proteases. In more detail, genes encoding nine pectate lyases (pel1, pel2, pel3, pelA, pelL, pelW_1, pelW_2, pelX and putative pelC), and also three genes coding for pectin esterases (pemA, pemB, and rhgT) are noted in the core genome fraction (Additional file 2: Table S1).
Interestingly, only one IFB5486 strain having sizeable chromosomal rearrangement (Fig. 1) did not possess the gene coding for a pectin lyase (pnl). However, even with the lack of pnl, this strain is still able to effectively destroy plant tissue components (Table 2) probably by producing all other well-known pectate lyases, pectate disaccharides hydrolases and/or pectinesterases. These results are in agreement with the previously performed study on Dickeya dadantii 3937 where mutations in single genes related to pectinolytic activity did not influence the overall maceration ability [48].
Regarding other PCWDE, we found 4 genes encoding polygalacturonases: pehA_1, pehA_2, rhiE, and pehX, 3 genes encoding cellulases (celV, celS, bcsZ), and 6 genes coding for proteases (prtS, prtC, prlC, pepT, htpX_1, and pcp_1) in the P. parmentieri core genome fraction (Additional file 2: Table S1). Additionally, we confirmed the presence of a protease-encoding gene (prtB_2) in all the genomes except P. parmentieri SCC3193 strain. Also, genes encoding proteins of type 1 and 2 secretion systems, important for export of proteases, cellulases and pectinases are present in the core genome.
Focusing on the genes related to oligogalacturonides degradation, we determined the occurrence of two genes encoding 2-dehydro-3-deoxygluconokinase: kdgK_1 and kdgK_2 in the core pan-genome. Besides, genes coding for other essential enzymes involved in oligogalacturonide degradation, namely: kduD, two types of the kduI gene (kduI_2, kduI_1), kdgT, kdsC, kdsD, kdgM_3 and ogl were proven to be present in all the tested genomes.
Importantly, among the investigated 15 P. parmentieri genomes we discovered peculiar differences in the quantities and types of kdgT genes encoding a 2-keto-3-deoxygluconate permease and kdgM coding for oligogalacturonate-specific porin KdgM [49]. Two different paralogs of kdgT, i.e. kdgT and kdgT_1, were identified. kdgT is present in core genomes, while the paralogous copy kdgT_1 is absent in the genomic sequences of IFB5432, IFB5441, and IFB5623 strains. A dendrogram based on KdgT and KdgT_1 protein sequences, available in GenBank, showed branching out of KdgT_1 far away from the other KdgT orthologs originating from different Pectobacteriaceae spp. (Fig. 5). Moreover, KdgT_1 protein is more closely related to KdgT from Bacillus spp.

The evolution of KdgT protein among the members of Pectobacteriaceae family. The tree was rotted on KdgT of Dickeya dadantii 3937 (Dda 3937) and Dickeya chrysanthemi NCPPB 516 (Ddh NCPPB 516). Pcc – P. carotovorum subsp. carotovorum, Pcbr - P. carotovorum subsp. brasiliense, Pba – P. atrosepticum, P. c. actinidiae - P. carotovorum subsp. actinidiae
Additionally, three paralogs of the kgdM gene were detected among the studied P. parmentieri genomes, where kgdM encodes oligogalacturonate-specific porin KdgM which expression is strongly induced by pectin derivatives [49]. Precisely, kdgM_3 is present among all the analyzed genomes, whereas kdgM_1 and kdgM_2 are observed among the analyzed genomes in different combinations. Usually, one strain possesses two paralogs of this gene (except for IFB5408, IFB5427, IFB5626 – these strains only have kdgM_3, while IFB5486 and IFB5605 have either kdgM_1 or kdgM_2). The herein reported gene duplication events might undergo a long-term selection pressure ressulting in P. parmentieri genome plasticity. Similar duplications were observed in the genomes of Escherichia coli [29].
All the studied P. parmentieri genomes possess genes encoding AHL-dependent quorum sensing system and the components of type 4 and type 6 secretion systems. In contrast, none of the tested genomes showed the presence of genes encoding the type 3 secretion system, which were usually detected in the Dickeya and Pectobacterium genomes [50]. However, we did notice the presence of genes fliR and flhB coding for effector proteins of the type 3 secretion system in the core genome fraction. These results are in agreement with some earlier data showing the absence of type 3 secretion system in the genome of P. parmentieri, but revealing at the same time the presence of genes encoding its effector proteins [25, 51, 52].
Analysis of the presence of the structural genes within the genomes of P. parmentieri strains did not adequately explain differences in the abilities of the tested strains to degrade plant cell wall components. It can be hypothesized that the variation mentioned above might have resulted from differential regulation of expression of virulence-related genes. There have been some indications that regulatory data are more useful for the explanation of phenotypic differences than the pure genomic information [39, 53].
However, all the tested genomes possess regulatory elements affecting the expression of the genes related to virulence: components of AHL-dependent quorum sensing system, 3 functional copies of kdgR (gene encoding a global negative regulator of pectin degradation), crp (encoding cAMP-activated global regulator), fur (coding for ferric iron uptake regulator), cheZ (encoding a chemotaxis regulator) and rcsF (coding for a stress response system protein) (Additional file 2: Table S1). It can be therefore concluded that future work should be directed towards a better understanding of the regulatory network controlling maceration abilities of different P. parmentieri strains.
Unfortunately, we cannot a priori exclude that random drift may be related to the lack of correlation between genomic differences and phenotypic differences, as observed in various host-associated microorganisms [54]; still, the number of sequenced genomes of P. parmentieri is too limited to draw a definitive conclusion on such hypothesis.
Furthermore, we have analyzed the genes encoding proteins involved in LPS biosynthesis, motility, chemotaxis, iron uptake and utilization in addition to resistance to oxidative stress (Additional file 2: Table S1). Several genes involved in LPS biosynthesis are present in the accessory pan-genome fraction, which correlated with the observed different degrees of pathogenicity-related traits of the P. parmentieri strains. In more detail, superb variation among the strains regarding motility and ability to form biofilm was presented (Table 2). For example, strain IFB5408 showed almost the same ability to form biofilm as P. aeruginosa PAO1, and also indicated the most efficient swimming motility (Table 2). Moreover, this strain possesses additional, strain-specific copies of genes encoding lipopolysaccharide ABC transporter substrate-binding protein LptA, LPS export ABC transporter periplasmic protein LptC, and also an additional copy of lipopolysaccharide ABC transporter ATP-binding protein.
The occurrence of two genes of possible eukaryotic origin, for example, plant ferredoxin-like protein and benzoic acid/salicylic acid carboxyl methyltransferase, was confirmed in all the tested genomes.
In the previous studies, P. parmentieri SCC3193 was stated to harbour an arsenic resistance cluster [16] common among bacteria that possess it either on the chromosome or on the plasmid. [55]. However, in the core pangenome of P. parmentieri, there was only one gene found encoding an arsenate reductase, namely arsC. The other members of the arsenic-resistant cluster were enclosed within the unique pan-genome fraction specific to P. parmentieri SCC3193 strain (Additional file 2: Table S1). Notably, none of the arsC genes present in the P. parmentieri genomes analyzed here was located within any MGE as it was the case of P. parmentieri SCC3193 genome [16].
The large mobilome of P. parmentieri
A high number of phage-related genes among the analyzed P. parmentieri strains was found, constituting especially the accessory and the unique pan-genome fraction. Therefore, a comprehensive evaluation of unique pan-genome fraction content was subsequently performed. P. parmentieri SCC3193 strain has the highest number of unique genes, namely 346 genes (Table 3). Besides, six Polish strains possess an intermediate number of unique genes (from 112 to 177).
Regarding unique pan-genome fraction almost half of the genes codes for phage-like proteins (e.g. transposases, phage structural proteins) and several conjugation transfer proteins. As a consequence of these findings, the presence of prophages was evaluated, and up to 4 intact prophages per strain were found (Table 4, Additional file 3: Figure S2). These prophages harboured genes encoding different toxins (e.g. HigB, CcdB, and YafO), bacteriocins, and a few antitoxins genes. In general, the toxins mentioned above are either involved in the inhibition of the gene expression or blockage of the foreign DNA invasion into the bacterial cell.
In more detail, HigB being a ribosome-dependent toxin preferentially cleaving mRNA at adenosine-rich codons is responsible for plasmid stability [56], and have been identified in the genomes of IFB5427, IFB5432 and IFB5486 strains (Table 4). We have also found a putative antitoxin HigA present in prophage regions of the genomes of IFB5427, IFB5605, IFB5626 isolates in addition to CFBP8475T. HigB and HigA are present in the genome of IFB5427. However, it is unlikely that they interact with one another because they do not fall under the regulation of the same operon (Additional file 4: Table S2).
The second most common toxin that is known to act on DNA gyrase [57] and is encoded by the F plasmid-carried gene, namely ccdB, was found in the genomes of IFB5432, IFB5441 and IFB5623. Besides, a gene coding for YafO toxin, which is responsible for protein synthesis inhibition [58] was detected in the genomes of IFB5427 and IFB5605. Another toxin RelE participating in growth arrest and cell death by inducing mRNA degradation at the ribosomal A-site under stress conditions [59] was encoded by the prophage regions of IFB5432 and IFB5623 sequences. Notably, genes coding for various bacteriocins were identified within the prophage hot spot regions’ of IFB5408, IFB5605 and IFB5626 genomes (Additional file 4: Table S2).
It is worth to underline that only incomplete prophages were spotted in the genomes of IFB5485, IFB5604, IFB5619 and two reference genomes: SCC3193, WPP0163. Accordingly to our findings and as it has been stated before [27] P. parmentieri strains are capable of acquiring efficiently large portions of extracellular DNA. Interestingly, some additional copies of the toxins mentioned above were found outside of phage regions; for instance, the relE gene is present in each P. parmentieri genome in at least a triplicate. Besides, genomes of all the tested strains possess a colicin V coding gene (Additional file 4: Table S2). The latter discovery is especially intriguing as the resulting protein is usually produced from large low-copy plasmids and inhibits translation during amino acid starvation [60]. However, we only confirmed the presence of any plasmid in just one strain, namely P. parmentieri IFB5427.
Shedding light on phage infections
CRISPR-Cas systems provide adaptive immunity in prokaryotes and can be effective against phage infections. They are composed of CRISPR arrays consisting of short repeats separated by spacer sequences derived from invading nucleic acids in addition to the CRISPR-associated (Cas) genes [61]. Four loci of CRISPR arrays (specifically four loci of multiple spacers) were detected in each P. parmentieri genome, named here from CRISPR1 to CRISPR4 (Additional file 5: Table S3). After performing clustering based on nucleotide alignment of the extracted sequences, it turned out that almost all the P. parmentieri CRISPR are grouping into two separate clades: the first cluster includes two CRISPR repeats: CRISPR1 and CRISPR2 and the second one additional two: CRISPR3 and CRISPR4 (Additional file 6: Figure S3). The genomes of P. parmentieri CFBP 8475T (a strain isolated in France) and IFB5486 (strain originating from Belgium) also possess 4 CRISPR repeats. However, their CRISPR sequences are specific and differ from the previously described CRISPR repeats. Notably, CRISPR1 and CRISPR2 group together, similarly to CRISPR3 and CRISPR4 forming also separate clades. We have found differences among quantities of each spacer related to specific CRISPR arrays ranging from 4 spacers up to 94 per one CRISPR array (Additional file 5: Table S3). It was proven before that when a bacterial cell is infected with a new phage, a piece of its foreign DNA can be integrated at the end of the CRISPR array as a new spacer. This spacer can then target and destroy the invading DNA [61]. Therefore, CRISPR regions can reveal information on both phage sequences and order of infection they conducted. However, we cannot be sure if there is always only one new spacer per phage, or if it is possible that more than one spacer is incorporated within the same infection event. Undoubtedly, we can say that there were numerous phage infection events in the analyzed P. parmentieri strains.
P. parmentieri plasmid pPAR01 - a possible source of additional genomic variation
As mentioned before, IFB5427 strain harbours a pPAR01 plasmid of 101,998 bp with 125 CDS and GC content of 49.45%, which is approximately 1% lower than the GC content of all the analyzed P. parmentieri bacterial chromosomes. Among the encoded proteins IncFII (plasmid replication initiator protein), RepA (initiation replication protein essential for plasmid replication), ParM (plasmid segregation), relaxase TraJ (a conjugation system) and a conjugal transfer protein should be listed. Besides, we have found a gene coding for YafN antitoxin located on the pPAR01 plasmid, and a gene encoding YafO toxin on the chromosome of this strain within a ‘hot spot’, being an entire prophage region. YafO is a toxin which inhibits protein synthesis and constitutes a type II toxin-antitoxin system, in which the YafN acts as an antitoxin. However, we do not possess any evidence that this toxin-antitoxin pair complements each other.
In addition to YafN, pPAR01 also comprises another putative antitoxin VapB; anyhow, we did not identify a toxin corresponding to this protein either on the plasmid or the chromosome of IFB5427. Besides, pPAR01 plasmid harbours a toxin RelE, but again without any proper antitoxin gene on any genetic unit.
The here described pPAR01 plasmid retains other exciting features, which could be related to its possible transmissibility. For instance, anti-restriction proteins ArdA and KlcA, which may allow for the associated MGE to evade the ubiquitous Type I DNA restriction systems in the recipient bacterium [62] resulting in the acquisition of cognate modification. Also, the presence of the gene coding for N6 DNA methylase suggests that maintaining this plasmid may affect histone methylation pattern within the host DNA to avoid further invasion by foreign nucleic acids [63]. pPAR01 also encodes proteins involved in SOS responses, such as the DNA adenine methyltransferase YhdJ essential for DNA repair, and DNA polymerase V subunit UmuC playing a role in replication under the stress responses [64]. On the other hand, SOS inhibition proteins (PsiA and PsiB), which impede bacterial stress responses are also located on this plasmid. Given reported induction of bacterial SOS responses by conjugative DNA transfer, it can be speculated that the presence of genes coding for SOS inhibition proteins may increase plasmid transferability to the host cell [65].
Conclusions
In conclusion, the obtained results show differences in the ability of P. parmentieri strains to macerate potato tubers tissue in addition to variation in their phenotypic traits. The observed high genomic plasticity can explain these features within P. parmentieri species expressed by a high number of MGE found both in the core and particularly in the dispensable pan-genome fraction. The analysis of the genomes of 15 P. parmentieri strains indicated that the dispensable pan-genome fraction constitutes 47% of the whole pan-genome. Also, the occurrence of prophage sequences and CRISPR-Cas system elements is abundant in the dispensable genome. The widespread presence of MGE may have caused considerable genome rearrangements in addition to the gene loss, as it was noted in the case of lack of type 3 secretion system components. Moreover, we can hypothesize that such high genomic variation among P. parmentieri strains could be the basis for the widespread presence of this species and its potential wide host range. In conclusion, our comparative genomic analysis of P. parmentieri highlights the contribution of plastic genomic structure to adaptive lifestyle and ability to survive and cause disease symptoms in different climatic zones.
Methods
Strains used in the study
P. parmentieri strains used in this study are presented in Table 1. Ten of these strains originating from Poland have been partially described previously regarding their phenotypic features [21]. The additional two strains have been isolated in Belgium by Johan van Vaerenbergh (ILVO, Belgium) and are included in this type of analysis for the first time. Also, we have used P. parmentieri CFBP 8475T isolated in France, P. parmentieri SCC3193 isolated in Finland, and P. wasabiae CFBP 3304 as a closely related species and an outgroup for phylogenetic analyses. For in silico analysis only, the P. parmentieri WPP163 isolated in the US was utilized. Pseudomonas aeruginosa PAO1 was used as a control in biofilm formation test.
Phenotypic characterization of P. parmentieri strains
The maceration ability of the selected P. parmentieri strains was evaluated by performing the potato slice assay as previously described [21]. Briefly, potatoes (cv. Lord) were surface sterilized, washed in tap water and cut into slices (approx. 2 cm thick). In each slice, depending on size, two to three holes were pierced and then into each of them 45 μl of bacterial inoculum (107 cfu x ml− 1) was placed. Potatoes were incubated for 48 h at 28 °C on moistened linen within closed plastic boxes. Subsequently, maceration spots diameters were measured. The experiment was performed in three biological repetitions each including eight technical ones.
The activity of pectinases, cellulases, proteases, siderophores as well as swarming motility was performed as described before [21]. To obtain consistent results and prevent undirected bacterial movement, swimming motility was evaluated on NB medium supplemented with 0.3% agar and also 0.4% PGA (Sigma-Aldrich, USA) [66]. The activity of lipases was tested on dedicated medium supplemented with Tween 80 and rhodamine B [67], where diameters of the clear halo zones appearing around the colonies were measured. The experiments were performed four times involving two technical replicates in each. Incubations were performed for 24 h at 28 °C.
Biofilm formation assay was performed as described by [45] with following modifications: 10 μl of overnight bacterial cultures in LB medium were transferred to Eppendorf tubes with 400 μl of M9 medium supplemented with 0.4% glucose (M9-C). After 16 h of incubation at 18 °C without agitation, 70 μl of 1% (w/v) crystal violet solution was added to each tube and incubated at room temperature for 20 min. Subsequent washing and OD565 measurement were performed as described by [45].
Statistical significance of differences noted in the phenotypic assays was analyzed with the use of the agricolae package from the R 3.3.1 programming environment [68]. Leven’s test was implemented to verify whether the data variances were equal. Shapiro-Wilk test was used to check whether the data followed a normal distribution. As the requirements of ANOVA were not always fulfilled, Kruskal-Wallis test with Bonferroni correction was applied for multiple comparisons (R agricolae package). All statistical hypotheses were tested at p < 0.05. For testing the Pearson’s correlation Hmisc package with rcorr was utilized.
Genome sequencing, de novo assembly and annotation
For shotgun genome sequencing, bacterial overnight cultures were grown on LA plates and subsequently sent to GATC Biotech, Konstanz, Germany. DNA was isolated, libraries have been constructed and then subjected to Illumina MiSeq or HiSeq2000 and Pacific BioSciences sequencing methods, to provide both proper coverages, and high-quality reads. Illumina paired-end reads were trimmed with Trimmomatic [69] to remove adapter sequences and poorly sequenced read ends. De novo genome assembly was performed with the use of SPAdes [70]. In the case of 2 genomes (i.e. of IFB5604 and IFB5619 strains), we could not achieve satisfactory assembly to the level of one scaffold. Therefore, MeDuSa: a multi-draft based scaffolder was applied [71] to close scaffolds to a full chromosome on the basis of reference genomes (acquired from NCBI) and also the genomes assembled by us to a single chromosome within this study. Genome annotation was firstly achieved by utilizing Prokka v1.12 [72]. Afterwards, custom-made Python scripts were used to reorient genomes in order to make them start from the dnaA gene sequence. Finally, all reoriented genomic fasta outputs were submitted to the NCBI database, and the final annotation was performed by the NCBI Prokaryotic Genome Annotation Pipeline (PGAP, https://www.ncbi.nlm.nih.gov/genome/annotation_prok/). CRISPR spacers were found with the use of CRISPRFinder (http://crispr.i2bc.paris-saclay.fr/Server/).
Comparative genomic analysis
Average Nucleotide Identity (ANI) was calculated for 15 P. parmentieri genomes applying ChunLab’s online Average Nucleotide Identity Calculator (EzBioCloud) [73] where P. parmentieri CFBP 8475T was used as an internal reference. Synteny was evaluated with the use of Mauve [74]. Phast [75] has been implemented for computational identification and visualization of prophages.
Gene ontology and the pan-genome shape of P. parmentieri strains were established using Roary [76] with the default settings. PGAP annotated sequences were used as an input. Calculations of the Heap’s Law and construction of the phylogenetic tree (constructed by hierarchical clustering of gene families established by reciprocal all-against-all protein sequences BLAST) were performed using the micropan R package for microbial pan-genomics [34]. The Class of Genes (COG) annotation of every protein belonging to the accessory fraction was done with eggNOG 4.5 server [77].
Abbreviations
- ANI:
-
Average nucleotide identity
- COG:
-
Class of genes
- HGT:
-
Horizontal gene transfer
- MGE:
-
Mobile genetic elements
- PCWDE:
-
Plant Cell Wall Degrading Enzymes
References
Guimarães LC, Florczak-Wyspianska J, de Jesus LB, Viana MVC, Silva A, Ramos RTJ, et al. Inside the Pan-genome - methods and software overview. Curr Genomics. 2015;16:245–52. https://doi.org/10.2174/1389202916666150423002311.
Vernikos G, Medini D, Riley DR, Tettelin H. Ten years of pan-genome analyses. Curr Opin Microbiol. 2015;23:148–54. https://doi.org/10.1016/j.mib.2014.11.016.
Hacker J, Carniel E. Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep. 2001;2:376–81. https://doi.org/10.1093/embo-reports/kve097.
Salanoubat M, Genin S, Artiguenave F, Gouzy J, Mangenot S, Arlat M, et al. Genome sequence of the plant pathogen Ralstonia solanacearum. Nature. 2002;415:497–502. https://doi.org/10.1038/415497a.
Xue Q-Y, Yin Y-N, Yang W, Heuer H, Prior P, Guo J-H, et al. Genetic diversity of Ralstonia solanacearum strains from China assessed by PCR-based fingerprints to unravel host plant- and site-dependent distribution patterns. FEMS Microbiol Ecol. 2011;75:507–19. https://doi.org/10.1111/j.1574-6941.2010.01026.x.
Guarischi-Sousa R, Puigvert M, Coll NS, Siri MI, Pianzzola MJ, Valls M, et al. Complete genome sequence of the potato pathogen Ralstonia solanacearum UY031. Stand Genomic Sci. 2016;11:7. https://doi.org/10.1186/s40793-016-0131-4.
Elphinstone J, Allen C, Prior P, Hayward A. Bacterial Wilt Disease and the Ralstonia solanacearum Species Complex. American Phytopathological Society; 2005. https://my.apsnet.org/ItemDetail?iProductCode=43291
Remenant B, Coupat-Goutaland B, Guidot A, Cellier G, Wicker E, Allen C, et al. Genomes of three tomato pathogens within the Ralstonia solanacearum species complex reveal significant evolutionary divergence. BMC Genomics. 2010;11:379. https://doi.org/10.1186/1471-2164-11-379.
Bocsanczy AM, Huguet-Tapia JC, Norman DJ. Comparative genomics of Ralstonia solanacearum identifies candidate genes associated with cool virulence. Front Plant Sci. 2017;8:1565. https://doi.org/10.3389/fpls.2017.01565.
Ailloud F, Lowe T, Cellier G, Roche D, Allen C, Prior P. Comparative genomic analysis of Ralstonia solanacearum reveals candidate genes for host specificity. BMC Genomics. 2015;16:270. https://doi.org/10.1186/s12864-015-1474-8.
Guidot A, Coupat B, Fall S, Prior P, Bertolla F. Horizontal gene transfer between Ralstonia solanacearum strains detected by comparative genomic hybridization on microarrays. ISME J. 2009;3:549–62. https://doi.org/10.1038/ismej.2009.14.
Adeolu M, Seema A, Naushad S, Gupta RS. Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: proposal for Enterobacterales Ord. Nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. Nov., Pectobacteriaceae fam. Nov., Yersiniaceae fam. Nov., Hafniaceae fam. Nov., Morgane. Int J Syst Evol Microbiol. 2016;66:5575–99. https://doi.org/10.1099/ijsem.0.001485.
Motyka A, Zoledowska S, Sledz W, Lojkowska E. Molecular methods as tools to control plant diseases caused by Dickeya and Pectobacterium spp: a minireview. New Biotechnol. 2017;39. https://doi.org/10.1016/j.nbt.2017.08.010.
Ma B, Hibbing ME, Kim H-S, Reedy RM, Yedidia I, Breuer JJJ, et al. Host range and molecular phylogenies of the soft rot enterobacterial genera Pectobacterium and Dickeya. Phytopathology. 2007;97:1150–63. https://doi.org/10.1094/PHYTO-97-9-1150.
Waleron K, Waleron M, Podhajska AJ, Łojkowska E. Genotyping of bacteria belonging to the former Erwinia genus by PCR-RFLP analysis of a recA gene fragment. Microbiology. 2002;148:583–95. https://doi.org/10.1099/00221287-148-2-583.
Nykyri J, Niemi O, Koskinen P, Nokso-Koivisto J, Pasanen M, Broberg M, et al. Revised phylogeny and novel horizontally acquired virulence determinants of the model soft rot phytopathogen Pectobacterium wasabiae SCC3193. PLoS Pathog. 2012;8:e1003013. https://doi.org/10.1371/journal.ppat.1003013.
Khayi S, Cigna J, Chong TM, Quêtu-Laurent A, Chan K-G, Hélias V, et al. Transfer of the potato plant isolates of Pectobacterium wasabiae to Pectobacterium parmentieri sp. nov. Int J Syst Evol Microbiol. 2016;66:5379–83. https://doi.org/10.1099/ijsem.0.001524.
Waleron M, Waleron K, Lojkowska E. Characterization of Pectobacterium carotovorum subsp. odoriferum causing soft rot of stored vegetables. Eur J Plant Pathol. 2014;139:457–69. https://doi.org/10.1007/s10658-014-0403-z.
Suárez MB, Feria FJ, Martín-Robles MJ, del Rey FJ, Palomo JL. Pectobacterium parmentieri causing soft rot on potato tubers in southern Europe. Plant Dis 2017;101:1029–1029. doi:https://doi.org/10.1094/PDIS-01-17-0013-PDN.
Nabhan S, De Boer SH, Maiss E, Wydra K. Taxonomic relatedness between Pectobacterium carotovorum subsp. carotovorum, Pectobacterium carotovorum subsp. odoriferum and Pectobacterium carotovorum subsp. brasiliense subsp. nov. J Appl Microbiol. 2012;113:904–13. https://doi.org/10.1111/j.1365-2672.2012.05383.x.
Zoledowska S, Motyka A, Zukowska D, Sledz W, Lojkowska E. Population structure and biodiversity of Pectobacterium parmentieri isolated from potato fields in temperate climate. Plant Dis. 2018;102:154–64. https://doi.org/10.1094/PDIS-05-17-0761-RE.
De Boer SH, Li X, Ward LJ. Pectobacterium spp associated with bacterial stem rot syndrome of potato in Canada. Phytopathology. 2012;102:937–47. https://doi.org/10.1094/PHYTO-04-12-0083-R.
Moleleki LN, Onkendi EM, Mongae A, Kubheka GC. Characterisation of Pectobacterium wasabiae causing blackleg and soft rot diseases in South Africa. Eur J Plant Pathol. 2013;135:279–88. https://doi.org/10.1007/s10658-012-0084-4.
Ngadze E, Brady CL, Coutinho TA, van der Waals JE. Pectinolytic bacteria associated with potato soft rot and blackleg in South Africa and Zimbabwe. Eur J Plant Pathol. 2012;134:533–49. https://doi.org/10.1007/s10658-012-0036-z.
Pitman AR, Harrow SA, Visnovsky SB. Genetic characterisation of Pectobacterium wasabiae causing soft rot disease of potato in New Zealand. Eur J Plant Pathol. 2010;126:423–35. https://doi.org/10.1007/s10658-009-9551-y.
Golkhandan E, Sijam K, Meon S, Ahmad ZAM, Nasehi A, Nazerian E. First report of Pectobacterium wasabiae causing soft rot of cabbage in Malaysia. Plant Dis 2013;97:1110–1110. doi:https://doi.org/10.1094/PDIS-01-13-0112-PDN.
Khayi S, Raoul des Essarts Y, Quetu-Laurent A, Moumni M, Helias V, Faure D, et al. Genomic overview of the phytopathogen Pectobacterium wasabiae strain RNS 08.42.1A suggests horizontal acquisition of quorum-sensing genes. Genetica. 2015;143:241–52. https://doi.org/10.1007/s10709-014-9793-2.
Dees MW, Lebecka R, Perminow JIS, Czajkowski R, Grupa A, Motyka A, et al. Characterization of Dickeya and Pectobacterium strains obtained from diseased potato plants in different climatic conditions of Norway and Poland. Eur J Plant Pathol. 2017;148:1–13. https://doi.org/10.1007/s10658-016-1140-2.
Raeside C, Gaffe J, Deatherage DE, Tenaillon O, Briska AM, Ptashkin RN, et al. Large Chromosomal Rearrangements during a Long-Term Evolution Experiment with Escherichia coli. MBio. 2014;5:e01377–14-e01377–14. https://doi.org/10.1128/mBio.01377-14.
Niemi O, Laine P, Koskinen P, Pasanen M, Pennanen V, Harjunpää H, et al. Genome sequence of the model plant pathogen Pectobacterium carotovorum SCC1. Stand Genomic Sci. 2017;12:87. https://doi.org/10.1186/s40793-017-0301-z.
Khayi S, Blin P, Chong TM, Chan K-G, Faure D. Complete chromosome and plasmid sequences of two plant pathogens, Dickeya solani strains D s0432-1 and PPO 9019. Genome Announc. 2018;6:e00233–18. https://doi.org/10.1128/genomeA.00233-18.
Zhao Y, Tian Y, Li X, Hu B. Complete genome sequence of a Dickeya fangzhongdai type strain causing bleeding canker of pear tree trunks. Genome Announc. 2018;6:e00177–18. https://doi.org/10.1128/genomeA.00177-18.
Tettelin H, Riley D, Cattuto C, Medini D. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol. 2008;11:472–7. https://doi.org/10.1016/j.mib.2008.09.006.
Snipen L, Liland KH. Micropan: an R-package for microbial pan-genomics. BMC Bioinformatics. 2015;16:79. https://doi.org/10.1186/s12859-015-0517-0.
Udaondo Z, Molina L, Segura A, Duque E, Ramos JL. Analysis of the core genome and pangenome of Pseudomonas putida. Environ Microbiol. 2016;18:3268–83. https://doi.org/10.1111/1462-2920.13015.
López-Pérez M, Rodriguez-Valera F. Pangenome evolution in the marine bacterium Alteromonas. Genome Biol Evol. 2016;8:1556–70. https://doi.org/10.1093/gbe/evw098.
Nourdin-Galindo G, Sánchez P, Molina CF, Espinoza-Rojas DA, Oliver C, Ruiz P, et al. Comparative Pan-genome analysis of Piscirickettsia salmonis reveals genomic divergences within Genogroups. Front Cell Infect Microbiol. 2017;7:459. https://doi.org/10.3389/fcimb.2017.00459.
Bosi E, Fondi M, Orlandini V, Perrin E, Maida I, de Pascale D, et al. The pangenome of (Antarctic) Pseudoalteromonas bacteria: evolutionary and functional insights. BMC Genomics. 2017;18:93. https://doi.org/10.1186/s12864-016-3382-y.
Golanowska M, Kabza M, Bacci G, Mengoni A, Potrykus M, Motyka A, et al. Comparison of highly and weakly virulent Dickeya solani strains, with a view on the pangenome and panregulon of this species. Front Microbiol. 2018;9:1940. https://doi.org/10.3389/FMICB.2018.01940.
Flego D, Pirhonen M, Saarilahti H, Palva TK, Palva ET. Control of virulence gene expression by plant calcium in the phytopathogen Erwinia carotovora. Mol Microbiol. 1997;25:831–8. https://doi.org/10.1111/j.1365-2958.1997.mmi501.x.
Eriksson ARB, Andersson RA, Pirhonen M, Palva ET. Two-component regulators involved in the global control of virulence in Erwinia carotovora subsp. carotovora. Mol Plant-Microbe Interact. 1998;11:743–52. https://doi.org/10.1094/MPMI.1998.11.8.743.
Andersson RA, Eriksson ARB, Heikinheimo R, Mäe A, Pirhonen M, Kõiv V, et al. Quorum sensing in the plant pathogen Erwinia carotovora subsp. carotovora : the role of expR Ecc. Mol Plant-Microbe Interact. 2000;13:384–93. https://doi.org/10.1094/MPMI.2000.13.4.384.
Maddocks SE, Oyston PCF. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology. 2008;154(Pt 12):3609–23. https://doi.org/10.1099/mic.0.2008/022772-0.
Alekshun MN, Levy SB. The mar regulon: multiple resistance to antibiotics and other toxic chemicals. Trends Microbiol. 1999;7:410–3. https://doi.org/10.1016/S0966-842X(99)01589-9.
Nykyri J, Mattinen L, Niemi O, Adhikari S, Kõiv V, Somervuo P, et al. Role and regulation of the Flp/tad pilus in the virulence of Pectobacterium atrosepticum SCRI1043 and Pectobacterium wasabiae SCC3193. PLoS One. 2013;8:e73718. https://doi.org/10.1371/journal.pone.0073718.
Chimento DP, Kadner RJ, Wiener MC. The Escherichia coli outer membrane cobalamin transporter BtuB: structural analysis of calcium and substrate binding, and identification of orthologous transporters by sequence/structure conservation. J Mol Biol. 2003;332:999–1014. https://doi.org/10.1016/j.jmb.2003.07.005.
Oliveira PH, Touchon M, Cury J, Rocha EPCC. The chromosomal organization of horizontal gene transfer in bacteria. Nat Commun. 2017;8:841. https://doi.org/10.1038/s41467-017-00808-w.
Lojkowska E, Dorel C, Reignault P, Hugouvieux-Cotte-Pattat N, Robert-Baudouy J. Use of GUS fusion to study the expression of Erwinia chrysanthemi pectinase genes during infection of potato tubers. Mol Plant-Microbe Interact. 1993;6:488–94.
Blot N, Berrier C, Hugouvieux-Cotte-Pattat N, Ghazi A, Condemine G. The oligogalacturonate-specific porin KdgM of Erwinia chrysanthemi belongs to a new porin family. J Biol Chem. 2002;277:7936–44. https://doi.org/10.1074/jbc.M109193200.
Charkowski A, Blanco C, Condemine G, Expert D, Franza T, Hayes C, et al. The role of secretion systems and small molecules in soft-rot Enterobacteriaceae pathogenicity. Annu Rev Phytopathol. 2012;50:425–49. https://doi.org/10.1146/annurev-phyto-081211-173013.
Kim H-SHS, Bing M, Perna NT, Charkowski AO, Ma B, Perna NT, et al. Phylogeny and virulence of naturally occurring type III secretion system-deficient Pectobacterium strains. Appl Environ Microbiol. 2009;75:4539–49. https://doi.org/10.1128/AEM.01336-08.
Panda P, Vanga BR, Lu A, Fiers M, Fineran PC, Butler R, et al. Pectobacterium atrosepticum and Pectobacterium carotovorum harbor distinct, independently acquired integrative and conjugative elements encoding coronafacic acid that enhance virulence on potato stems. Front Microbiol. 2016;7 MAR:397. doi:https://doi.org/10.3389/fmicb.2016.00397.
Potrykus M, Golanowska M, Hugouvieux-Cotte-Pattat N, Lojkowska E. Regulators involved in Dickeya solani virulence, genetic conservation, and functional variability. Mol Plant-Microbe Interact. 2014;27:700–11. https://doi.org/10.1094/MPMI-09-13-0270-R.
Sheppard SK, Guttman DS, Fitzgerald JR. Population genomics of bacterial host adaptation. Nat Rev Genet. 2018;19:1–17. https://doi.org/10.1038/s41576-018-0032-z.
Stolz JF, Basu P, Santini JM, Oremland RS. Arsenic and selenium in microbial metabolism. Annu Rev Microbiol. 2006;60:107–30. https://doi.org/10.1146/annurev.micro.60.080805.142053.
Schureck MA, Repack A, Miles SJ, Marquez J, Dunham CM. Mechanism of endonuclease cleavage by the HigB toxin. Nucleic Acids Res. 2016;44:7944–53. https://doi.org/10.1093/nar/gkw598.
Bahassi EM, O’Dea MH, Allali N, Messens J, Gellert M, Couturier M. Interactions of CcdB with DNA gyrase. J Biol Chem. 1999;274:10936–44. https://doi.org/10.1074/jbc.274.16.10936.
Zhang Y, Yamaguchi Y, Inouye M. Characterization of YafO, an Escherichia coli toxin. J Biol Chem. 2009;284:25522–31. https://doi.org/10.1074/jbc.M109.036624.
Li G-Y, Zhang Y, Inouye M, Ikura M. Inhibitory mechanism of Escherichia coli RelE-RelB toxin-antitoxin module involves a helix displacement near an mRNA interferase active site. J Biol Chem. 2009;284:14628–36. https://doi.org/10.1074/jbc.M809656200.
Frick KK, Quackenbush RL, Konisky J. Cloning of immunity and structural genes for colicin V. J Bacteriol. 1981;148:498–507 http://www.ncbi.nlm.nih.gov/pubmed/6271732.
Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, et al. Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science (80- ). 2008;321:960–964. doi:https://doi.org/10.1126/science.1159689.
Chen K, Reuter M, Sanghvi B, Roberts GA, Cooper LP, Tilling M, et al. ArdA proteins from different mobile genetic elements can bind to the EcoKI type I DNA methyltransferase of E. coli K12. Biochim Biophys Acta - Proteins Proteomics. 2014;1844:505–11. https://doi.org/10.1016/j.bbapap.2013.12.008.
Low DA, Weyand NJ, Mahan MJ. Roles of DNA adenine methylation in regulating bacterial gene expression and virulence. Infect Immun. 2001;69:7197–204. https://doi.org/10.1128/IAI.69.12.7197-7204.2001.
Kuban W, Vaisman A, McDonald JP, Karata K, Yang W, Goodman MF, et al. Escherichia coli UmuC active site mutants: effects on translesion DNA synthesis, mutagenesis and cell survival. DNA Repair (Amst). 2012;11:726–32. https://doi.org/10.1016/j.dnarep.2012.06.005.
Baharoglu Z, Bikard D, Mazel D. Conjugative DNA transfer induces the bacterial SOS response and promotes antibiotic resistance development through integron activation. PLoS Genet. 2010;6:1–10. https://doi.org/10.1371/journal.pgen.1001165.
Bowden SD, Hale N, Chung JCS, Hodgkinson JT, Spring DR, Welch M. Surface swarming motility by Pectobacterium atrosepticum is a latent phenotype that requires O antigen and is regulated by quorum sensing. Microbiology. 2013;159(Pt 11):2375–85. https://doi.org/10.1099/mic.0.070748-0.
Samad MYA, Razak CNA, Salleh AB, Zin Wan Yunus WM, Ampon K, Basri M. A plate assay for primary screening of lipase activity. J Microbiol Methods. 1989;9:51–6. https://doi.org/10.1016/0167-7012(89)90030-4.
R Core Team. R: A language and environment for statistical computing. 2014. www.R-project.org.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. https://doi.org/10.1089/cmb.2012.0021.
Bosi E, Donati B, Galardini M, Brunetti S, Sagot M-F, Lió P, et al. MeDuSa: a multi-draft based scaffolder. Bioinformatics. 2015;31:2443–51. https://doi.org/10.1093/bioinformatics/btv171.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. https://doi.org/10.1093/bioinformatics/btu153.
Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, et al. Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol. 2017;67:1613–7. https://doi.org/10.1099/ijsem.0.001755.
Darling ACE. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–403. https://doi.org/10.1101/gr.2289704.
Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39(suppl):W347–52. https://doi.org/10.1093/nar/gkr485.
Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3. https://doi.org/10.1093/bioinformatics/btv421.
Huerta-Cepas J, Szklarczyk D, Forslund K, Cook H, Heller D, Walter MC, et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016;44:D286–93. https://doi.org/10.1093/nar/gkv1248.
Acknowledgements
We would like to acknowledge Dr. Michał Kabza from Adam Mickiewicz University in Poznan for his guidance in NGS data analysis. We want to thank Dr. George DiCenzo for critically revising the manuscript and Dr. Johan van Vaerenbergh (ILVO, Belgium) for the bacterial strains.
Availability of data and material
GenBank BioProject: This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the BioProject PRJNA433470 and link: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA433470
GenBank Accession This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accessions nos.: CP026977, CP027260, CP026979, CP026980, CP026981, CP026982, PSZH00000000, CP026983, CP026984, CP026985, CP026986, PSZG00000000.
Funding
This work was conceived and fulfilled during the realization of the project provided by the National Science Centre in Poland via grant no. 2014/14/M/NZ8/00501 awarded to EL. The founding body did not take part in the design of the study, collection, analysis, and interpretation of data and in writing of the presented manuscript.
Author information
Authors and Affiliations
Contributions
SZ performed all analyses presented in work (phenotypic characterization and bioinformatics analysis) and wrote the manuscript. AMP and WS isolated strains from the environment, read the manuscript and gave comments. EL and AM conceived the work and critically revised the final version of the manuscript. All Authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable. The P. parmentieri strains analyzed in this study are environmental isolates, that were obtained by the authors. Any other reference strains were bought or received from befriended laboratories/researchers or bought from commercial bacterial banks (P. parmentieri CFBP8475 was bought from CIRM-Plant Associated Bacteria / CIRM-CFBP).
Consent for publication
Not applicable, no humans were the object of the presented study.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Phylogenetic relatedness of the analyzed P. parmentieri strains: A. Genomic Average Nucleotide Identity (gANI) heatmap with dendrograms. B. Gene presence/absence matrix against core pan-genome generated dendrogram. The dendrogram was created basing on hierarchical clustering of the rows. (PNG 1528 kb)
Additional file 2:
Table S1. Comprehensive analysis with locus_tag information on virulence genes, virulence-related genes, motility, chemotaxis, iron metabolism, resistance to oxidative stress, quorum sensing related genes and regulators within analyzed P. parmentieri strains. Format: xlsx file. (XLSX 74 kb)
Additional file 3:
Figure S2. Synteny of intact prophages within the particular P. parmentieri strains. (PNG 1257 kb)
Additional file 4:
Table S2. Toxins and antitoxins genes encoded within the genomes of analyzed P. parmentieri strains. Format: docx. (DOCX 16 kb)
Additional file 5:
Table S3. The number of CRISPR spacers found per each CRISPR array in the analyzed P. parmentieri strains. (DOCX 12 kb)
Additional file 6:
Figure S3. Clustering of CRISPR arrays from analyzed P. parmentieri strains. Two clusters formed from CRISPR1-CRISPR2 and another two CRISPR3-CRISPR4 and two additional clusters formed only from CRISPR arrays from P. parmentieri CFBP 8475T and P. parmentieri IFB5486. (PNG 1227 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Zoledowska, S., Motyka-Pomagruk, A., Sledz, W. et al. High genomic variability in the plant pathogenic bacterium Pectobacterium parmentieri deciphered from de novo assembled complete genomes. BMC Genomics 19, 751 (2018). https://doi.org/10.1186/s12864-018-5140-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5140-9