
Abstract
Background
Capacitation, a prerequisite for oocyte fertilization, is a complex process involving series of structural and functional changes in sperms such as membrane modifications, modulation of enzyme activities, and protein phosphorylation. In order to penetrate and fertilize an oocyte, mammalian sperms must undergo capacitation. Nevertheless, the process of sperm capacitation remains poorly understood and requires further elucidation. In the current study, via high throughput sequencing, we identified and explored the differentially expressed microRNAs (miRNAs) and mRNAs involved in boar sperm capacitation.
Results
We identified a total of 5342 mRNAs and 204 miRNAs that were differentially expressed in fresh and capacitated boar sperms. From these, 12 miRNAs (8 known and 4 newly identified miRNAs) and their differentially expressed target mRNAs were found to be involved in sperm capacitation-related PI3K-Akt, MAPK, cAMP-PKA and Ca2+signaling pathways.
Conclusions
Our study is first to provide the complete miRNA and transcriptome profiles of boar sperm. Our findings provide important insights for the understanding of the RNA profile in boar sperm and future elucidation of the underlying molecular mechanism relevant to mammalian sperm capacitation.
Similar content being viewed by others

Background
Capacitation, a prerequisite for oocyte fertilization, is a complex process involving a series of structural and functional changes in sperms such as membrane modifications, modulation of enzyme activities, and protein phosphorylation. In order to penetrate and fertilize an oocyte, mammalian sperms must undergo hours of in vivo (in female reproductive tract) or in vitro capacitation process immediately after ejaculation [1, 2]. Multiple physiological and biochemical changes are involved in sperm capacitation [3], including protein tyrosine phosphorylation [4], membrane cholesterol efflux [5], production of reactive oxygen species (ROS) [6], membrane hyper-polarization [5, 7], as well as increase in intracellular pH [8], Ca2+, cyclic adenosine monophosphate(cAMP) [9], superoxide anion levels [10], and HCO3− concentration.
Furthermore, during cryopreservation, sperms are reported to experience capacitation-like changes [8] or “cryo-capacitation” [11, 12]. These changes include increased sperm capacitation, plasma membrane reorganization, ROS generation, increased intracellular Ca2+ and protein tyrosine phosphorylation (PTP) [13]. Although cryo-capacitated sperms exhibit striking differences in patterns of PTP compared to normal in vitro capacitated sperms [14], cryo-capacitation should not be regarded as true capacitation [15]. It has been reported that various protein kinases and protein phosphatases are present in mammalian sperm and are implicated in sperm motility acquisition, capacitation and acrosome reaction [16, 17]. For example, tyrosine phosphorylation of sperm flagellar proteins is related to acquisition of hyperactive motility [18, 19]. Protein tyrosine phosphorylation involves three pathways: cAMP-dependent protein kinase A (cAMP-PKA) signaling pathway [20], phosphatidylinositol-3-hydroxycarboxylase (PI3K) signal Pathway [21] and mitogen-activated protein kinase (MAPK) signaling pathway [22].
In addition, optimal concentration of Ca2+ can promote protein phosphorylation and sperm motility [23], therefore, Ca2+ signaling pathway is essential for the regulation of capacitation. Furthermore, many important tyrosine phosphorylated proteins are reportedly associated with capacitation, such as protein A-kinase anchoring proteins (AKAPs) in human spermatozoa [24, 25], A-kinase anchoring protein 4 (AKAP4) in hamster sperm [26], proacrosin binding protein/p32 in boar [17, 27] and calcium-binding and tyrosine phosphorylation-regulated protein (CABYR) in mouse sperm [28].
Sperms are highly differentiated and specialized cells, their main function is to transmit paternal genetic information and coding, noncoding RNAs to the oocyte [29]. Sperm contains an array of RNAs, including messenger RNAs (mRNAs), ribosomal RNAs (rRNAs) and small RNAs [30], which are residues from the process of spermatogenesis [31,32,33]. Sperm RNAs may contribute to sperm movement, capacitation, fertilization and early embryogenesis [34].
In 2006, Gur and Breitbart have demonstrated that labeled amino acids are incorporated into polypeptides during sperm capacitation, a process that is entirely inhibited by mitochondrial translation inhibitors, but not by cytoplasmic translation inhibitors. They further reported that, unlike 80S cytoplasmic ribosomes, 55S mitochondrial ribosomes are present in polysomal fractions and are actively involved in protein translation in sperm. Furthermore, inhibition of protein translation could lead to significant reduction in sperm motility, capacitation and in vitro fertilization rate. Therefore, contrary to the accepted dogma, nuclear genes are expressed in sperm while in the female reproductive tract until fertilization [35].
Small RNAs are a class of short non-coding RNAs (approximately 19–23 nucleotides) including miRNAs [29]. MiRNAs can regulate gene expression and participate in the regulation of biological processes, such as development, cell proliferation and differentiation, apoptosis and metabolism [36,37,38], via inhibition/suppression of translation or degradationof mRNA [39]. Severe dysregulation in expression patterns of miRNAs has been observed in different types of reproduction abnormalities [30, 40, 41].
For many decades, the understanding of capacitation was limited to macroscopic observation and description. By and large, the process of sperm capacitation and its underlying molecular mechanisms are poorly understood and require further elucidation [42]. In this study, as the first to utilize next generation sequencing for the study of sperm capacitation,we identified and reported differentially expressed mRNA and miRNA profiles in fresh and capacitated boar sperm. Deep sequencing information was obtained to explore the interaction of miRNA and mRNA and to further understand the underlying mechanism of sperm capacitation.
Results
Evaluation of sperm quality parameters
Semen quality parameters were divided into two fractions: FS and CS. Statistically significant difference was detected in FS and CS after induction of sperm capacitation in vitro, as well as insperm acrosome statuses (Table 1). Additionally, the capacitated sperms showed higher motility, viability and acrosome reaction rate compared to fresh sperms (P < 0.01).
Analysis of RNA sequencing
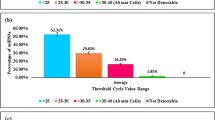
After performing transcriptome sequencing quality control, we obtained a total of 53,686,904 and 59,851,746 raw reads and 26,843,452 and 29,925,873 clean reads in fresh and capacitated sperm, respectively. The uniquely mapped reads to reference genome in fresh and capacitated sperm were 28,565,403 (53.21%) and 30,691,568 (51.28%), respectively. Additionally, in small RNA sequencing, we obtained 18,956,444 and 16,209,736 raw reads, 12,561,033 and 11,222,990 clean reads, and 3,027,230 and 2,944,033 mapped reads to reference genome in fresh and capacitated sperm, respectively. In total, Mirdeep2 detected 1092 unique miRNAs in fresh and capacitated sperm. From these, 259 and 238 were known miRNAs, 769 and 782 were new (novel) candidate miRNAs in fresh and capacitated sperm, respectively (Table 2).
GO and KEGG analysis of mRNAs and miRNAs
Log2 (Fold change, expressed in base-2 log ratio of transcript abundance – intensity, ‘log2’) > 1 and FDR (−log10) < 0.01 were selected as standard. Figure 1a and b depict the differential expression between FS and CS. In brief, we identified 5342 differentially expressed mRNAs. From these, 3716 and 1626 were known and novel mRNAs, respectively. Furthermore, 503 mRNAs were upregulated and 4839 were downregulated in FS and CS (Fig. 1b). Results of clustered differentially expressed genes are shown in Fig. 2b. In brief, 69 and 4554 mRNAs were specifically expressed in capacitated and fresh sperm, respectively (Additional file 1: Table S3–1, 2, 3).

Volcano plot of differentially expressed miRNAs and mRNAs between fresh and capacitated boar sperm. (a) miRNAs; (b) mRNAs. Each point in the volcanic figure (Volcano plot) represents a gene, numerical value of genes expression in FS and CS as the abscissa, and the negative logarithm of P-value-FDR as the ordinate. The red and green dots represent up-regulated and down-regulated differentially expressed genes, respectively. Blue or black dots represent genes that were not differentially expressed

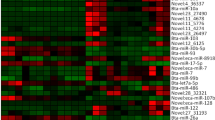
Hierarchical cluster analysis of significantly differentially expressed miRNAs and mRNA in fresh and capacitated boar sperm. (a) miRNAs; (b) mRNAs. The color represents the level of gene expression, log2 (FPKM+ 1). Chartreuse and blue color denotes high and low expression of genes, respectively. Axes x- and y- represent Euclidean distances and Pearson’s correlation
Moreover, 5342 differentially expressed genes (DEGs) were analyzed by KEGG. These mRNAs were enriched in 283 pathways, and each enriched pathway contained numbers of differentially expressed mRNAs ranging from 1 to 115 (Fig. 2). From these, 41 mRNAs were enriched in Wnt, MAPK, PI3K-Akt signaling pathways, and energy metabolism and sperm capacitation-related pathways such as Ca2+ and cAMP signaling pathways (Fig. 3b).

KEGG annotation of differentially expressed target genes of miRNAs and mRNAs. (a) miRNAs; (b) mRNAs. The vertical axis indicates the name of metabolic pathway of KEGG, and horizontal axis represents the number of genes annotated to the pathway, accounting for the proportion of the total number of genes
The enrichment analysis of KEGG pathway in differentially expressed mRNAs was depicted in Fig. 4b. We identified a total of 204 differentially expressed miRNAs (DEM) between FS and CS. Among these, 60 and 141 were known and novel miRNAs, respectively. Whereas 86 and 118 miRNAs were upregulated and downregulated, respectively (Fig. 1a). Hierarchical cluster analysis of differentially expressed miRNAs in fresh and capacitated sperm was shown in Fig. 2a. We further observed that 9 miRNAs (miR-148a-3p, miR-151-3p, miR-425-5p, miR-132, miR-451, miR7136-5p, miR-489, miR-1343, miR-1306-3p) and 49 miRNAs exhibited higher expression in CS and FS, respectively (Additional file 1: Table S4). Furthermore, 5 miRNAs (miR-378b-3p, miR493-5p, miR-133a-3p, miR-362, and miR-214) were uniquely expressed in fresh sperm.

Enrichment analysis of KEGG pathway in differentially expressed target genes of miRNAs and mRNAs. (a) miRNAs; (b) mRNAs. Each graph in the figure represents a KEGG path, and the accession name is shown in the illustration on the right. The abscissa represents enrichment factor and the ordinate represents Log10 (Q, value)
Protein internetwork of differentially expressed genes
Based on the protein interaction network, we observed a distinct interactive relationship between the differentially expressed genes. The nodes, degrees, aggregation coefficients and edges in the interaction networks reflect the strength of the interactions between differentially expressed genes. Based on these parameters, we speculated that differentially expressed genes, such as MAPK1, PGK1, PPM1B, and PGAM1, may play an important role in the regulation of fresh and capacitated boar sperm (Fig. 5).

Protein interaction network analysis for the differentially expressed genes between fresh and capacitated sperm. The circle represents the node (differentially expressed protein), the line represents the edge, the red node represents the high aggregation coefficient, and the green node represents the low aggregation coefficient
Target mRNA prediction and pathway analysis of DE miRNAs
In total, we predicted 19,788 target mRNAs using the miRnada and RNAhybrid tools. Canonical pathway analysis further revealed that these genes are annotated to 276 signaling pathways (Fig. 3b). Similarly, these predicted target mRNAs are annotated and associated with energy metabolism and sperm capacitation signaling pathways, including phosphatidylinositol-signaling system, glycolysis, MAPK, calcium, and PI3K-Akt signaling pathway (Fig. 6).

The pie chart of annotated target genes associated with energy metabolism and sperm capacitation signaling pathways. The number represents the number of target genes, and the annotations on the right represent different signaling pathways
qRT-PCR validation
The identified miRNAs and mRNAs (n = 8 each) were randomly selected for verifying their expression level in fresh and capacitated sperm via qRT-PCR. The results showed that, except for conservative-1-2721 and conservative-7-221178, the expression levels of all mRNAs and miRNAs in fresh and capacitated sperm are consistent with the results of high-throughput sequencing (Fig. 7c, d).

qRT-PCR validation of differentially expressed miRNAs and mRNAs. (a) and (c): qRT-PCR results of eight differentially expressed mRNAs and miRNAs between fresh and capacitated boar sperm; (b) and (d): The small RNA and transcriptome sequencing results of eight differentially expressed mRNAs and miRNAs between fresh and capacitated boar sperm
Discussion
In recent past, a number of studies have demonstrated that sperm RNAs contribute to spermatogenesis, sperm movement, capacitation, fertilization, and early embryogenesis [34]. The commonly shared characteristics of miRNAs and mRNAs in mammals indicate their important roles in regulation, control and guidance of sperm functions. Building on extensive scruitiny of scientific literature, our study reported, for the very first time, the comprehensive and systemic investigation of the miRNA and mRNA profiles in fresh and capacitated boar sperm using high throughput sequencing. In current investigation, we identified a total of 204 DE miRNAs and 5342 DE mRNAs between fresh and capacitated boar sperm.
Substantial past evidences have indicated that multiple physiological and biochemical changes are involved in the process of capacitation, such as protein tyrosine phosphorylation [4], sperm membrane cholesterol efflux [5], and increase in intracellular pH [8], Ca2+ and bicarbonate (HCO3−) concentration [9]. Generally, Ca2+ and HCO3− are considered as two important biological elements required for sperm capacitation and are believed to promote protein tyrosine phosphorylation. In this study, heparin induces boar sperm capacitation and affects the expression of miRNAs and their target mRNAs. Some of these miRNAs and mRNAs contribute to protein tyrosine phosphorylation and are mainly involved in membrane-related activities such as G-protein coupled receptor activity, signal transducer activity, transmembrane signaling receptor activity in mitochondria.
In the present study, we analyzed the KEGG pathway of DE miRNAs and their target mRNAs. The results showed that these DE miRNAs and their target mRNAs were mainly enriched in the PI3K-Akt, MAPK, cAMP-PKA, and calcium signaling pathways, which are thought to be important for protein tyrosine phosphorylation and sperm capacitation. It has been reported that PI3K-Akt signaling pathway plays an important role in cell cycle growth, development, apoptosis and cancer [43]. In sperms, the actin polymerization and depolymerization processes can mimic sperm capacitation and acrosome reaction [21]. MAPK signaling pathway is reportedly involved in physiological processes, such as cell proliferation, differentiation, variation and apoptosis, and plays an important role in regulating sperm flagellar activity, hyperactivation and acrosome reaction, especially via the ERK (Ras/Raf/MEK/ERK) signaling pathway [22]. Furthermore, activated ion channels, such as Ca2+ channel (CatSper), can trigger signal transduction factors that are generally required for intiating the cAMP-PKA signaling pathway and subsequent steps in sperm capacitation [44].
In our study, miR-1343 was upregulated in capacitated sperm compared to fresh sperm. The target mRNAs of miR-1343,COL11A1 and PDE4A, can participate in PI3K-Akt and cAMP-PKA signaling pathways. AKAP3, as a target of miR-1285, can combine with PKA and PDE4A to function as skeletal protein in sperm and regulates the concentration of local cAMP and sperm capacitation [45]. VDAC1 and HSPA2 are targets of miR-127. They are involved in calcium signaling pathway and MAPK signaling pathway. It has been reported that VDAC1 is mainly located in sperm mitochondrial membrane and in outer dense fibers of sperm flagella, which affects sperm motility, survival rate, acrosome reaction, capacitation, tyrosine phosphorylation, fertilization and embryo development [46, 47].
Furthermore, some newly identified miRNAs and their targets may be associated with sperm capacitation. CATSPER4, the target of miR-151-3p, is a sperm-specific calcium channel. CatSper controls the concentration of intracellular calcium and forward movement of sperm [48]. The CatSper channel has been identified in human [49], murine [50], and equine sperm [51]. Recently, it has also been identified in ovine sperm [52]. CABYR, a target of novel miRNA (unconservative_7_234335), is a calcium binding tyrosine phosphorylated chemical fiber sheath protein involved in sperm capacitation [28]. The binding protein ACRBP, a target of new miRNA (unconservative_11_42222), promotes the maturation and tyrosine phosphorylation of acrosinthat are closely related to sperm capacitation [53]. One previous study has also demonstrated that the bovine sperm capacitation process requires AKAP3-degradation; and the degree of such was regulated by the level of AKAP3 tyrosine phosphorylation [54]. In our investigation, we found that a number of mRNAs related to sperm capacitation in other species were also differentially expressed in boar sperm. Some important target mRNAs and proteins of miRNAs associated with tyrosine phosphorylation during in vitro capacitationare listed in Table 3. Nevertheless, despite the novel and fascinating findings of our current study, new scientific questions, such as how these DE miRNAs and mRNAs interact with each other to regulate sperm capacitation, remains unanswered and warrants further investigations.
Conclusion
To conclude, our study is first to provide evidence that in vitro capacitation induces comprehensive changes in expression patterns of miRNAs and mRNAs in boar sperm. We also generated valuable data on novel boar miRNAs and genomic clusters expressed in fresh and capacitated boar sperm. Our findings, along with existing evidences, support the notion that differentially expressed miRNAs and their target mRNAs in fresh and capacitated boar sperm play considerable role in the regulation of sperm apoptosis, mitochondrial membrane potential and spermatogenesis alteration. These discoveries indicate a functional redundancy of these genes in controlling sperm capacitation and thereby, fertility. Our findings provide important insights for the understanding of the RNA profile in boar sperm and future elucidation of the underlying molecular mechanism relevant to mammalian sperm capacitation.
Methods
Sperm collection and induction of sperm capacitation
Fresh ejaculates were harvested from 11 sexually mature Landrace boars using manual collection method as previously described [55]. Collection of samples was performed in accordance with the regulations for the Administration of Affairs Concerning Experimental Animals (Ministry of Science and Technology, China, revised in June 2004) and was approved by the Institutional Animal Care and Use Committee in the College of Animal Science and Technology, Sichuan Agricultural University, Sichuan, China, under permit No. DKYB20081003 [56]. The sperm quality parameters were determined with SQA-V (MES, Israel). Only fresh ejaculates with sperm motility greater than 0.8, normal morphology, and sperm concentrations higher than 1 × 108 mL− 1 were used in this study. In order to eliminate individual differences in our analysis, five sample pools (n = 5) were generated by mixing the fresh ejaculates 2–3 boars for each pool. Then, these ejaculate pools were equally divided into two aliquots. One aliquot (Fresh sperm, FS) was immediately frozen in liquid nitrogen and stored at − 80 °C, and another aliquot (Capacitated sperm, CS) was immediately prepared for capacitation.
Before sperm capacitation, fresh sperms were washed thrice with BTS solution and centrifugated at 600 g for 5 min. Then, fresh sperms were incubated in Tris-buffered medium (TBM, 113.1 mM NaCl, 3 mM KCl, 7.5 mM CaCl2·2H2O, 5 mM Sodium pyruvate, 11 mM Glucose, 1 mM Caffine, 20 mM Tris, 1 mg mL− 1 BSA, and with a final pH 7.6~ 7.8) supplemented with 10 mM heparin at 38.5 °C, 5% CO2, 100% humidified incubator for 30 min [57]. Finally, capacitated sperms were immediately frozen in liquid nitrogen and stored at − 80 °C until RNA extraction.
Sperm viability and acrosome reaction assay
Sperm viability was assessed according to Kovacs and Foote [58] with some modifications. Briefly, 100 μL fresh and capacitated sperms were mixed with isovolumetric and pre-incubated Trypan blue (0.4% in distilled water) and placed in incubator at 37 °C for 2 min. Then, 20 μL of mixture was placed on a glass slide with a cover slip to observe and calculate the percentage of non-colored sperm (at least 500 sperms) under an optical microscope (400×). The capacitated sperms were evaluated based on the changes in pattern of 0.05% Coomassie brilliant blue (CBB) according to the method described by Zhuo et al. with some modifications [59]. Finally, 20 μL-capacitated sperms were smeared on glass slide, air-dried and then soaked in pre-incubated CBB and placed in an incubator at 37 °C for 5 min, then washed with distilled water three times and air-dried. The percentage of capacitated sperms (acrosome reaction rate) was observed (at least 200 sperm) using a phase contrast microscope (1000×) [60].
Total RNA extraction, library preparation and sequencing
For each sample (n = 5), total RNA extraction of fresh and capacitated sperms was performed with Trizol LS Reagent (Ambion, USA) [61]. Briefly, the straws were thawed by plunging into a 37~ 38 °C waterbath for 1 min. The sperms in three straws were collected in a 1.5 mL tube. The sperm suspension was centrifuged at 3400 g and 4 °C for 5 min. The pellets were resuspended with 1 mL of hypotonic solution with 0.5% of Triton X-100 (Roche, Germany). The samples were incubated for 10 min on ice for lysis of the somatic cells. After centrifugation at 5000 g for 5 min, the hypotonic/triton X-100 solution was discarded. Then, 0.75 mL of TRIzol LS reagent was added. The sperm pellets were washed three times with RNase-free PBS, and then resuspended in 0.25 mL of RNase-free water. The concentration and quality of total RNA were measured using NanoDrop ND1000 spectrophotometer (NanoDrop Technologies, USA). The purity (OD 260/280 ≥ 1.8; OD260/230 ≥ 1.0) and concentration (≥250 ng μL− 1) of total RNA were qualified for library preparation. Then, small RNA libraries were generated using small RNA Sample Kit Prekit (NEB, USA) according to manufacturer’s instructions. The quality and yield after sample (n = 5) preparation were measured with Agilent 2100 Tape Station and Qubit 2.0, and libraries were sequenced on Illumina Hiseq 2500 platform.
Quality analysis and mapping
Clean data (clean reads) were obtained by removing reads containing adapter, reads containing ploy-A/T/C/G and low qualities reads from raw data. All the downstream analyses were performed on high quality clean data.
The sequence alignment and subsequent analysis using a reference genome were performed using the designated reference genome of Sus scrofa (ftp://ftp.ensembl.org/pub/release-75/fasta/sus_scrofa/). Then, clean reads of small RNA were mapped to the Sus scrofa genome sequence with miRDeep2 [62]. Clean reads of transcriptome were compared with reference genome by TopHat2 [63]. Information of the location in reference genome or gene, as well as peculiarity of sequence characteristics of the sequenced samples were also obtained.
MiRNA identification and differential expression analysis
Bowtie [64] software was used to compare clean reads with Silva database, GtRNAdb database, Rfam database and Repbase. Filtered non-coding RNA, including ribosome RNA (rRNA), transport RNA (tRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), and repeat sequences, and miRNAs of unannotated reads were obtained. Then, Mirdeep2 [62] was used to identify the known miRNA and novel miRNA, and to predict the function of miRNAs. Differential expression of miRNA and mRNA between the FS and CS fractions were analyzed with TPM and FPKM algorithm [65] using the DESeq R package (1.10.1). miRNA and mRNA with an adjusted P < 0.01 and absolute value of log2 (Fold change) > 1 were assigned as differentially expressed.
MiRNA target prediction
For prediction of the potential targets of differentially expressed miRNAs, miRnada [66] and RNAhybrid [67] were used to compare the gene sequence information of corresponding species to the known miRNA and novel miRNA identified in boar sperm.
GO and KEGG enrichment analyses
GO enrichment analysis was performed on target genes of miRNAs and differentially expressed mRNA usingthe GOseqR package [68]. In addition, the differentially expressed protein coding genes were also analyzed using GO. The enrichment of miRNA target genes and differentially expressed protein-coding genes in KEGG pathways were analyzed by the KOBAS [67] software.
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) validation
In order to verify the accuracy of highthroughput sequencing results, we randomly selected and confirmed the expression of 8 miRNAs and 8 mRNAs in fresh and capacitated sperms by qRT-PCR. Fresh and capacitated sperm samples were prepared for resampling by mixing sperms from 2 to 3 individual boars. All primers were either designed based on homologous counterparts in the GenBank database using Primer Premier 5.0 software or adopted from previous literatures (Table 4). U6 [69] and PPIA [70] were used as reference genes, respectively. qRT-PCR was performed using SYBR PrimeScript miRNA RT-PCR Kit (Takara Biotech, China) on a StepOnePlus real-time PCR system (Applied BioSystems, USA) according to our laboratory protocol [56].
Statistical analysis
All results are shown as adjusted least squares means ± standard error means (LSM ± SEM). The mean cycle threshold (Ct) value was converted to relative expression level using the 2−△△Ct method [61]. Statistical analyses of the expression levels of the miRNA and mRNA were performed using unpaired t test (SPSS software version 18.0, IBM). Differences of P < 0.05 were considered as statistically significant, and differences of P < 0.01 were considered as highly statistically significant.
Abbreviations
- DEGs:
-
Differentially expressed genes
- DEM:
-
Differentially expressed miRNAs
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PTP:
-
Protein tyrosine phosphorylation
- qRT-PCR:
-
Quantitative reverse-transcription polymerase chain reaction
- ROS:
-
Reactive oxygen species
- RPKM:
-
Reads per kilobases of exon region per million mapped reads
References
Chang MC. Fertilizing capacity of spermatozoa deposited into the fallopian tubes. Nature. 1951;168(4277):697.
Austin CR. The capacitation of the mammalian sperm. Nature. 1952;170(4321):326.
Hernándezgonzález EO, Sosnik J, Edwards J, Acevedo JJ, Mendozalujambio I, Lópezgonzález I, Demarco I, Wertheimer E, Darszon A, Visconti PE. Sodium and epithelial sodium channels participate in the regulation of the capacitation-associated hyperpolarization in mouse sperm. J Biol Chem. 2006;281(9):5623.
Baker MA, Hetherington L, Ecroyd H, Roman SD, Aitken RJ. Analysis of the mechanism by which calcium negatively regulates the tyrosine phosphorylation cascade associated with sperm capacitation. J Cell Sci. 2004;117(Pt 2):211.
Thérien I, Manjunath P. Effect of progesterone on bovine sperm capacitation and acrosome reaction. Biol Reprod. 2003;69(4):1408–15.
Huang YH, Chu ST, Chen YH. A seminal vesicle autoantigen of mouse is able to suppress sperm capacitation-related events stimulated by serum albumin. Biol Reprod. 2000;63(5):1562–6.
Momozawa K, Fukuda Y. Caffeine in fertilization medium is not essential for bovine IVF by fully capacitated spermatozoa. J. Reprod. Dev. 2003;49(6):507.
Fraser LR, Abeydeera LR, Niwa K. Ca(2+)-regulating mechanisms that modulate bull sperm capacitation and acrosomal exocytosis as determined by chlortetracycline analysis. Mol. Reprod. Dev. 1995;40(2):233.
Baldi E, Casano R, Falsetti C, Krausz C, Maggi M, Forti G. Intracellular calcium accumulation and responsiveness to progesterone in capacitating human spermatozoa. J Androl. 1991;12(5):323–30.
O'Flaherty C, Beorlegui N, Beconi MT. Participation of superoxide anion in the capacitation of cryopreserved bovine sperm. Int J Androl. 2003;26(2):109–14.
Bravo MM, Aparicio IM, Garciaherreros M, Gil MC, Peña FJ, Garciamarin LJ. Changes in tyrosine phosphorylation associated with true capacitation and capacitation-like state in boar spermatozoa. Mol. Reprod. Dev. 2005;71(1):88–96.
Kaneto M, Harayama H, Miyake M, Kato S. Capacitation-like alterations in cooled boar spermatozoa: assessment by the chlortetracycline staining assay and immunodetection of tyrosine-phosphorylated sperm proteins. Anim Reprod Sci. 2002;73(3–4):197.
Bailey JL, Bilodeau JF, Cormier N. Semen cryopreservation in domestic animals: a damaging and capacitating phenomenon. J Androl. 2000;21(1):1.
Naresh S, Atreja SK. The protein tyrosine phosphorylation during in vitro capacitation and cryopreservation of mammalian spermatozoa. Cryobiology. 2015;70(3):211–6.
Green CE, Watson PF. Comparison of the capacitation-like state of cooled boar spermatozoa with true capacitation. Reproduction. 2001;122(6):889–98.
Hunter T. Signaling--2000 and beyond. Cell. 2000;100(1):113–27.
Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116(2):191.
Mahony MC, Gwathmey TY. Protein tyrosine phosphorylation during Hyperactivated motility of Cynomolgus monkey (Macaca fascicularis) spermatozoa. Biol Reprod. 1999;60(5):1239.
Nassar A, Mahony M, Morshedi M, Lin MH, Srisombut C, Oehninger S. Modulation of sperm tail protein tyrosine phosphorylation by pentoxifylline and its correlation with hyperactivated motility. Fertility & Sterility. 1999;71(5):919–23.
Signorelli J, Diaz ES, Morales P. Kinases, phosphatases and proteases during sperm capacitation. Cell & Tissue Research. 2012;349(3):765–82.
Breitbart H, Rotman T, Rubinstein S, Etkovitz N. Role and regulation of PI3K in sperm capacitation and the acrosome reaction. Mol. Cell. Endocrinol. 2010;314(2):234–8.
Osaki LH, Gama P. MAPKs and signal transduction in the control of gastrointestinal epithelial cell proliferation and differentiation. Int J Mol Sci. 2013;14(5):10143–61.
Bhoumik A, Saha S, Majumder GC, Dungdung SR. Optimum calcium concentration: a crucial factor in regulating sperm motility in vitro. Cell Biochem. Biophys. 2014;70(2):1177–83.
Carrera A, Moos J, Ning XP, Gerton GL, Tesarik J, Kopf GS, Moss SB. Regulation of protein tyrosine phosphorylation in human sperm by a calcium/calmodulin-dependent mechanism: identification of a kinase anchor proteins as major substrates for tyrosine phosphorylation. Dev Biol. 1996;180(1):284–96.
Mandal A, Naaby-Hansen S, Wolkowicz MJ, Klotz K, Shetty J, Retief JD, Coonrod SA, Kinter M, Sherman N, Cesar F. FSP95, a testis-specific 95-kilodalton fibrous sheath antigen that undergoes tyrosine phosphorylation in capacitated human spermatozoa. Biol Reprod. 1999;61(5):1184–97.
Jha KN, Shivaji S. Identification of the major tyrosine phosphorylated protein of capacitated hamster spermatozoa as a homologue of mammalian sperm a kinase anchoring protein. Mol. Reprod. Dev. 2002;61(2):258.
Dubé C, Leclerc P, Baba T, Reyes-Moreno C, Bailey JL. The Proacrosin binding protein, sp32, is tyrosine phosphorylated during capacitation of pig sperm. J Androl. 2005;26(4):519.
Naabyhansen S, Mandal A, Wolkowicz MJ, Sen B, Westbrook VA, Shetty J, Coonrod SA, Klotz KL, Kim YH, Bush LA. CABYR, a novel calcium-binding tyrosine phosphorylation-regulated fibrous sheath protein involved in capacitation. Dev Biol. 2002;242(2):236–54.
Capra E, Turri F, Lazzari B, Cremonesi P, Gliozzi TM, Fojadelli I, Stella A, Pizzi F. Small RNA sequencing of cryopreserved semen from single bull revealed altered miRNAs and piRNAs expression between high- and low-motile sperm populations. BMC Genomics. 2017;18(1):14.
Abu-Halima M, Hammadeh M, Schmitt J, Leidinger P, Keller A, Meese E, Backes C. Altered microRNA expression profilesof human spermatozoa inpatients with different spermatogenic impairments. Fertility & Sterility. 2013;99(5):1249–55.
Pessot CA, Brito M, Figueroa J, Concha II, Yañez A, Burzio LO. Presence of RNA in the sperm nucleus. Biochem. Biophys. Res. Commun. 1989;158(1):272–8.
Dadoune JP, Siffroi JP, Alfonsi MF. Transcription in haploid male germ cells. Int Rev Cytol. 2004;237(237):1–56.
Chalmel F, Rolland AD, Niederhauser-Wiederkehr C, Chung SSW, Demougin P, Gattiker A, Moore J, Patard JJ, Wolgemuth DJ, Jégou B. The conserved transcriptome in human and rodent male gametogenesis. Proc Natl Acad Sci U S A. 2007;104(20):8346–51.
Hu G, Wang HY, Greenawalt DM, Azaro MA, Luo M, Tereshchenko IV, Cui X, Yang Q, Gao R, Shen L. AccuTyping: new algorithms for automated analysis of data from high-throughput genotyping with oligonucleotide microarrays. Nucleic Acids Res. 2006;34(17):e116.
Gur Y, Breitbart H. Mammalian sperm translate nuclear-encoded proteins by mitochondrial-type ribosomes. Genes Dev. 2006;20(4):411–6.
Li M, Liu Y, Wang T, Guan J, Luo Z, Chen H, Wang X, Chen L, Ma J, Mu Z, et al. Repertoire of porcine MicroRNAs in adult ovary and testis by deep sequencing. Int J Biol Sci. 2011;7(7):1045–55.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
Kloosterman WP, Plasterk RH. The diverse functions of MicroRNAs in animal development and disease. Dev Cell. 2006;11(4):441–50.
Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15(8):509–24.
Lian J, Zhang X, Tian H, Liang N, Wang Y, Liang C, Li X, Sun F. Altered microRNA expression in patients with non-obstructive azoospermia. Reprod. Biol. Endocrinol. 2009;7(1):1–10.
Liu T, Cheng W, Gao Y, Wang H, Liu Z. Microarray analysis of microRNA expression patterns in the semen of infertile men with semen abnormalities. Mol Med Rep. 2012;6(3):535–42.
Salicioni AM, Platt MD, Wertheimer EV, Arcelay E, Allaire A, Sosnik J, Visconti PE. Signalling pathways involved in sperm capacitation. Soc. Reprod. Fertil. Suppl. 2007;65:245–59.
Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase–AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501.
Chávez JC, Vega-Beltrán JLDL, Escoffier J, Visconti PE, Treviño CL, Darszon A, Salkoff L, Santi CM. Ion Permeabilities in mouse sperm reveal an external trigger for SLO3-dependent hyperpolarization. PLoS One. 2013;8(4):e60578.
Bajpai M, Fiedler SE, Huang Z, Vijayaraghavan S, Olson GE, Livera G, Conti M, Carr DW. AKAP3 selectively binds PDE4A isoforms in bovine spermatozoa. Biol Reprod. 2006;74(1):109–18.
Sabirov RZ, Merzlyak PG. Plasmalemmal VDAC controversies and maxi-anion channel puzzle. Biochim Biophys Acta. 2012;1818(6):1570.
De PV, Messina A, Lane DJ, Lawen A. Voltage-dependent anion-selective channel (VDAC) in the plasma membrane. FEBS Lett. 2010;584(9):1793–9.
Álvarez-Rodríguez M, Vicente-Carrillo A, Rodríguez-Martinez H. Exogenous individual lecithin- phospholipids (phosphatidylcholine and Phosphatidylglycerol) cannot prevent the oxidative stress imposed by cryopreservation of boar sperm. Journal of Veterinary Medicine and Surgery. 2017;1(2):11.
Tamburrino L, Marchiani S, Minetti F, Forti G, Muratori M, Baldi E. The CatSper calcium channel in human sperm: relation with motility and involvement in progesterone-induced acrosome reaction. Hum Reprod. 2014;29(3):418–28.
Qi H, Moran MM, Navarro B, Chong JA, Krapivinsky G, Krapivinsky L, Kirichok Y, Ramsey IS, Quill TA, Clapham DE. All four CatSper ion channel proteins are required for male fertility and sperm cell hyperactivated motility. Proc Natl Acad Sci U S A. 2007;104(4):1219–23.
Loux SC, Crawford KR, Ing NH, González-Fernández L, Macías-García B, Love CC, Varner DD, Velez IC, Choi YH, Hinrichs K. CatSper and the relationship of Hyperactivated motility to intracellular calcium and pH kinetics in equine Sperm1. Biol Reprod. 2013;89(5):123.
Luna C, Yeste M, Mm RDA, Domingo J, Casao A, Rodriguez-Gil JE, Pérez-Pé R, Cebrián-Pérez JA, Muiño-Blanco T. Effect of seminal plasma proteins on the motile sperm subpopulations in ram ejaculates. Reprod. Fertil. Dev. 2017;29(2):394–405.
Dong HT, Shi WS, Tian Y, Cao LP, Jin Y. Expression and tyrosine phosphorylation of sp32 regulate the activation of the boar proacrosin/acrosin system. Genet. Mol. Res. 2015;14(1):2374–83.
Shahar S, Hillman P, Lubart R, Ickowicz D, Breitbart H. Activation of sperm EGFR by light irradiation is mediated by reactive oxygen species. Photochem Photobiol. 2014;90(5):1077–83.
King GJ, Macpherson JW. A comparison of two methods for boar semen collection. J Anim Sci. 1973;36(3):563–5.
Zhang Y, Zeng CJ, He L, Ding L, Tang KY, Peng WP. Selection of endogenous reference microRNA genes for quantitative reverse transcription polymerase chain reaction studies of boar spermatozoa cryopreservation. Theriogenology. 2015;83(4):634–41.
Zhao Z, Jie XU, Zhao J, Zhang ZG, Gao JM. Initial Comparison of CTC Staining among Different Porcine Sperm Capacitation Impacts. Journal of Beijing University of Agriculture. 2009;24(1):33–5 In Chinese.
Kovács A, Foote RH. Viability and acrosome staining of bull, boar and rabbit spermatozoa. Stain Technol. 1992;67(3):119–24.
Jiang Y, Zhuo D, Chen Y, Lin Y, Han M. Evaluation of Coomassie brilliant blue staining for detection of human sperm acrosome reaction. Acta Anatomica Sinica. 1998;29:332–6.
Dou XT, Song XC, Zhang XH, Guo D. Effects of Heparin on in vitro Sperm Capacitation in Liaoning Cashmere Goats. China Herbivores. 2011;31(1):21–3 In Chinese.
Liu Y, Ma J, Chen L, Lou P, Zhou J, Li M, Li X. Comparison of microRNA transcriptomes between immature and mature porcine testes. J. Anim. Vet. Adv. 2014;13(3):132–8.
Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36.
Sherlock G. Gene ontology: tool for the unification of biology. Canadian Institute of Food Science & Technology Journal. 2009;22(4):415.
Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, et al. High-throughput sequencing of Arabidopsis microRNAs : evidence for frequent birth and death of MIRNA genes. PLoS One. 2007;2(2):e219.
Doron B, Manda W, Aaron G, Marks DS, Chris S. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–53.
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–80.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25.
Gu Y, Li M, Zhang K, Chen L, Jiang AA, Wang J, Lv X, Li X. Identification of suitable endogenous control microRNA genes in normal pig tissues. Anim Sci J. 2011;82(6):722–8.
Zeng C, He L, Peng W, Ding L, Tang K, Fang D, Zhang Y. Selection of optimal reference genes for quantitative RT-PCR studies of boar spermatozoa cryopreservation. Cryobiology. 2014;68(1):113–21.
Vizel R, Hillman P, Ickowicz D, Breitbart H. AKAP3 degradation in sperm capacitation is regulated by its tyrosine phosphorylation[J]. Biochim Biophys Acta. 2015;1850(9):1912–20.
Brenker C, Goodwin N, Weyand I, Kashikar ND, Naruse M, Krähling M, Müller A, Kaupp UB, Strünker T. The CatSper channel: a polymodal chemosensor in human sperm[J]. EMBO J. 2012;31(7):1654–65.
Lee D, Moawad AR, Morielli T, Fernandez MC, O'Flaherty C. Peroxiredoxins prevent oxidative stress during human sperm capacitation[J]. Mol Hum Reprod. 2017;23(2):106–15.
Zhou W, Anderson AL, Turner AP, De Luliis GN, McCluskey A, McLaughlin EA, Nixon B. Characterization of a novel role for the dynamin mechanoenzymes in the regulation of human sperm acrosomal exocytosis[J]. Mol Hum Reprod. 2017;23(10):657–73.
Saewu A, Kadunganattil S, Raghupathy R, Kongmanas K, Diaz-Astudillo P, Hermo L, Tanphaichitr N. Clusterin in the mouse epididymis: possible roles in sperm maturation and capacitation[J]. Reproduction. 2017;154(6):867–80.
Ren X, Chen X, Wang Z, Wang D. Is transcription in sperm stationary or dynamic?[J]. J. Reprod. Dev. 2017;63(5):439–43.
Gao B, Qian Y, Shen D, Shi YQ, Wang SS. Effect of swine (Sus scrofa) sperm capacitation in vitro and acrosome reaction on expression of spermadhesins gene and seminal plasma motility inhibitor (SPMI) mRNA[J]. Journal of Agricultural Biotechnology. 2015;23(7):923–9.
Acknowledgements
The authors thank Chongzhou pig farm of the Sichuan Giastar Group for generously allowing semen collection.
Funding
This work was supported partly by a grant from the National Natural Science Foundation of China (No.31570533).The funding bodies do not have any role in the design of the study, the collection, analysis and interpretation of data and the authoring of the manuscript.
Availability of data and materials
The datasets used and analyzed in this study are available on reasonable request. Part of the data generated or analyzed during this study are included in this published article and additional files.
Author information
Authors and Affiliations
Contributions
YL, RHL, MXR, YZ, KL, YNR and WCH collected samples, performed the experiments, analyzed the data and drafted the manuscript. MZ contributed to samples collection, data analysis and revised the manuscript. GBZ revised the manuscript critically and given final approval to be published. IHQ revised and proof read the manuscript. CJZ granted, concept, designed the experiment and revised, given final approval version of the manuscript to be published. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All procedures for boar semen collection from Chongzhou pig farm of the Sichuan Giastar Group were approved by the Regulations for the Administration of Affairs Concerning Experimental Animals (Ministry of Science and Technology, China, revised in June 2004) and the Institutional Animal Care and Use Committee in the College of Animal Science and Technology, Sichuan Agricultural University, Sichuan, China, under permit No. DKYB20081003. Chongzhou pig farm of the Sichuan Giastar Group also understood and granted permission for Prof. Zeng’s lab to collect the boar semen for the purposes of this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Raw and collated data. Table S1. Differential expression of mRNAs between fresh and capacitated boar sperm. Table S2. Differential expression of miRNAs between fresh and capacitated boar sperm. Table S3–1. 69 specifically expressed mRNAs in capacitated boar sperm. Table S3–2. 4554 specifically expressed mRNAs in fresh boar sperm. Table S3–3. 434 co-expressed mRNAs in fresh and capacitated boar sperm. Table S4. 49 miRNAs with higher expression level in boar fresh sperm compared to capacitated sperm. (XLSX 818 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, Y., Li, RH., Ran, MX. et al. High throughput small RNA and transcriptome sequencing reveal capacitation-related microRNAs and mRNA in boar sperm. BMC Genomics 19, 736 (2018). https://doi.org/10.1186/s12864-018-5132-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5132-9