
Abstract
Background
Fusarium head blight (FHB) of wheat in North America is caused mostly by the fungal pathogen Fusarium graminearum (Fg). Upon exposure to Fg, wheat initiates a series of cellular responses involving massive transcriptional reprogramming. In this study, we analyzed transcriptomics data of four wheat genotypes (Nyubai, Wuhan 1, HC374, and Shaw), at 2 and 4 days post inoculation (dpi) with Fg, using RNA-seq technology.
Results
A total of 37,772 differentially expressed genes (DEGs) were identified, 28,961 from wheat and 8811 from the pathogen. The susceptible genotype Shaw exhibited the highest number of host and pathogen DEGs, including 2270 DEGs associating with FHB susceptibility. Protein serine/threonine kinases and LRR-RK were associated with susceptibility at 2 dpi, while several ethylene-responsive, WRKY, Myb, bZIP and NAC-domain containing transcription factors were associated with susceptibility at 4 dpi. In the three resistant genotypes, 220 DEGs were associated with resistance. Glutathione S-transferase (GST), membrane proteins and distinct LRR-RKs were associated with FHB resistance across the three genotypes. Genes with unique, high up-regulation by Fg in Wuhan 1 were mostly transiently expressed at 2 dpi, while many defense-associated genes were up-regulated at both 2 and 4 dpi in Nyubai; the majority of unique genes up-regulated in HC374 were detected at 4 dpi only. In the pathogen, most genes showed increased expression between 2 and 4 dpi in all genotypes, with stronger levels in the susceptible host; however two pectate lyases and a hydrolase were expressed higher at 2 dpi, and acetyltransferase activity was highly enriched at 4 dpi.
Conclusions
There was an early up-regulation of LRR-RKs, different between susceptible and resistant genotypes; subsequently, distinct sets of genes associated with defense response were up-regulated. Differences in expression profiles among the resistant genotypes indicate genotype-specific defense mechanisms. This study also shows a greater resemblance in transcriptomics of HC374 to Nyubai, consistent with their sharing of two FHB resistance QTLs on 3BS and 5AS, compared to Wuhan 1 which carries one QTL on 2DL in common with HC374.
Similar content being viewed by others
Background
Wheat yield is severely limited by diseases caused by microbial pathogens. One of the prevalent wheat diseases, fusarium head blight (FHB), is caused by ascomycetous fungi of the genus Fusarium. The most common Fusarium species causing FHB in North America is F. graminearum [1]. Fusarium graminearum (Fg) produces deoxynivalenol (DON, also known as vomitoxin), the most prevalent trichothecene in Canadian wheat. Mycotoxin-contaminated grain is sold at lower prices or is completely rejected. Wheat resistance to FHB is categorized into five types: resistance to initial infection (type I), resistance to spread (type II), resistance to DON accumulation (type III), resistance to kernel infection (type IV), and tolerance (type V) [2].
Several FHB-resistant wheat cultivars have been identified and a large number of quantitative trait loci (QTLs) conferring resistance to FHB in wheat have been discovered. At least 22 chromosomal regions have been identified as contributing consistently to FHB resistance in multiple studies (reviewed in [3]). One of the most effective and best characterized sources of resistance against FHB is the Chinese cultivar Sumai 3 and its derivatives. These harbor a major QTL for type II resistance (up to 20–25% reduction of disease severity), named Fhb1, that has been mapped to chromosome 3BS as well as a minor QTL associated with type 1 resistance on chromosome 5AS, Qfhs.ifa-5A. QTLs in the same chromosomal regions of 3BS and 5AS have been detected in a range of FHB-resistant material, including the genotype Nyubai [4]. A minor QTL associated with type II resistance was also identified by the same authors in the Chinese genotype Wuhan 1 on chromosome 2DL. They also identified 3 QTLs (2DL, 3BS, and 5A) in the double haploid progeny HC374 after crossing Nyubai with Wuhan 1.
As result of exposure to pathogenic microorganisms, such as Fg, plants have evolved intricate mechanisms to recognize and defend themselves against potential infection. One of these responses is the down-regulation of photosynthesis and other processes associated with primary metabolisms that are essential for plant growth. It has been suggested that the energy saved by down-regulation of primary metabolism is diverted and used for defense responses [5, 6]. Nevertheless, up-regulation of primary metabolism also occurs during plant-pathogen interactions and is believed to be associated with signal transduction cascades that lead to plant defense responses [7].
Pathogen-triggered cellular responses involve massive transcriptional reprogramming within the host. Hormone signaling and transcription factors (TFs) are the two major facilitators of downstream defense responses in plants [8, 9]. Plant hormones as cellular signal molecules play key roles in regulating immune responses to invasion by microbial pathogens. Their signaling pathways are interconnected in a complex network providing plants with an enormous regulatory potential to rapidly respond to biotic stress while limiting the use of resources essential for basic metabolism [8]. According to their lifestyle, plant pathogens are generally divided into necrotrophs and biotrophs [10]. Necrotrophic pathogens first destroy host cells, often through the production of phytotoxins and cell-wall degrading enzymes, and then feed on dead tissues. Biotrophic pathogens feed on live tissues. Some plant pathogens, displaying both lifestyles depending on their life stage, are called hemi-biotrophs. Fg has been described as displaying a hemi-biotrophic life style in wheat [11]. Major plant hormones that regulate defense responses include salicylic acid (SA), jasmonic acid (JA) and ethylene (ET). Generally, SA plays a key role in defense against biotrophic pathogens, while JA and ET are critical to defense against necrotrophic pathogens [10]. The effective defense against biotrophic pathogens is largely due to programmed cell death in the host, and to the associated activation of defense responses regulated by SA–dependent pathways. Defense responses against necrotrophic pathogens are activated by JA and ET signaling.
Transcriptional reprogramming is governed by TFs and co-regulatory proteins organized in discrete transcriptional complexes [12]. Transcription factors are often sites of signal convergence and signal-regulated TFs act in concert with other context-specific TFs and transcriptional co-regulators to establish sensory transcription-regulatory networks required for plant immunity [9]. The TF families involved in plant immunity include AP2/ERF, bHLH, bZIP, MADS box, MYB, NAC, and WRKY [9, 13, 14]; their respective roles are reviewed in [9].
RNA sequencing (RNA-seq) technology has been very informative for transcriptomics studies. The recent release of the complete wheat genome sequence (pseudomolecules) and detailed annotations allowed exploratory analysis of DEGs associated with resistance and susceptibility against FHB, specifically in known FHB resistance QTL regions. We applied RNA-seq technology to study the transcriptomics response of four wheat genotypes (the FHB resistant Nyubai, Wuhan 1 and their progeny line HC374, and the FHB-susceptible Shaw) after inoculation with Fg.
Results
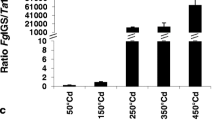
RNA-seq data were acquired for Nyubai, Wuhan 1, HC374 and Shaw from water-treated (control) and Fg-inoculated spikelets at 2 and 4 days post inoculation (dpi). Paired-end reads only were considered in the mapping to the reference genome (average mapping rate of 96%). Results show that the proportion of pathogen transcripts in the wheat samples was highest in the susceptible genotype Shaw and lower in the three resistant genotypes, HC374, Nyubai and Wuhan 1, and increased from 2 to 4 dpi across all wheat genotypes; these results were confirmed by estimation of the fungal biomass, based on expression level of Fg-GAPDH, and by measurement of DON concentration (correlations of 0.99 and 0.98, respectively, Fig. 1).

Level of Fg infection as estimated by proportion of Fg RNAs in RNA-seq reads (a), by accumulated level of FG-GAPDH RNA measured by RT-qPCR (b), and by DON concentration (c) across the samples. Error bar = one standard error of mean
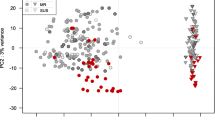
A total of 37,772 DEGs were identified: 28,961 from the wheat host and 8811 from the pathogen, with highest numbers in the susceptible host Shaw (Figs. 2, 3). Control samples were excluded for the differential analysis of Fg genes, because these samples theoretically didn’t contain any Fg mRNA. Clustering, correlation, differential expression feature extraction (DEFE) pattern and network analyses were done for wheat and pathogen genes separately. The principal component (PC) analysis of wheat DEGs (Fig. 4) revealed that differential expression was primarily driven by the Fg treatment (PC1), and secondly by duration of the treatment and genotype factors (PC2); these two PCs explained > 97% of variance.

Total number of differentially expressed genes (DEGs) originating from wheat (a) and the pathogen (b), combining all DEG analyses. Up: upregulated, Down: down-regulated by Fg; 2d and 4d: 2 and 4 dpi; S: Shaw; HC: HC374; N: Nyubai; W: Wuhan 1

Global view of the DEG expression profiles between genotypes and treatments. The top dendrogram on the left represents 28,961 wheat genes and the bottom one 8811 Fg genes. Fg and H2O: treatments with Fg and water (control); 2d and 4d: 2 and 4 dpi

Principal component analysis of the wheat DEGs dataset based on the top 1000 most variable DEGs. PC1 explained 89% of variance and PC2 8%. The ellipses were 90% confidence intervals highlighting treatment/time clusters. Fg and H2O: treatments with Fg and water (control); 2d and 4d: 2 and 4 dpi; S: Shaw; HC: HC374; N: Nyubai; W: Wuhan 1
For the purpose of this study, our analyses were focused on wheat genes. Using the recently developed differential expression feature extraction method (DEFE, Pan Y, Li Y, Liu Z, Surendra A, Wang L, Foroud NA, Goyal RK, Ouellet T, Fobert PR: Differential expression feature extraction and its application in wheat RNA-seq data analysis, forthcoming), four differential gene expression analyses were performed and seven DEFE pattern schemes were extracted (Table 1). For example, in comparisons between Fg-treated samples and the corresponding water-treated control samples, the pattern FW01020000 denotes up-regulation by Fg at 4 dpi in the susceptible Shaw, but down-regulation at 4 dpi in HC374, and no significant changes in Wuhan 1 or Nyubai. Each DEG was assigned a DEFE pattern ID from the seven pattern schemes (Additional file 1A, cols AV-BB).
Wheat DEGs highly correlated with Fg treatment
Forty clusters were identified among the 28,961 wheat DEGs (Additional file 1A, col AA). Pearson correlation analysis was performed between each cluster and FHB-related measurements including the percentage of RNA-seq reads from Fg (%Fg), Fg-GAPDH and DON concentration (Fig. 1) and two experimental treatments (Fg inoculation and time after Fg inoculation). We define these FHB-related measurements and experimental treatments as phenotypic traits, collectively representing the effect of Fg infection. Four clusters, collectively containing 11,848 DEGs, were significantly up-regulated by Fg treatment as evidenced by the positive correlation (p ≤ 0.05) with all five phenotypic traits (Fg inoculation, time after Fg treatment, %Fg reads, Fg-GAPDH and DON levels), while another set of 6 clusters containing 6026 genes were down-regulated as evidenced by the negative correlation with these traits (Additional file 1D). The average expression profiles of the 10 clusters are illustrated in Fig. 5.

Average values of the expression profiles for the ten DEG clusters significantly up- (a) or down- (b) regulated by Fg. The numbers in the brackets are the numbers of genes in the respective clusters. Fg and H2O: treatments with Fg and water (control); 2d and 4d: 2 and 4 dpi; S: Shaw; HC: HC374; N: Nyubai; W: Wuhan 1
Similarly, a correlation analysis was performed between each individual gene and these phenotypic traits. For a gene to be qualified as significantly correlated (p ≤ 0.05) with a trait, it needed to be a member of the 10 clusters above, and the gene itself also had to be significantly correlated with the given trait. Each of the five FHB-related phenotypic traits covered nearly 60% of the DEGs; they collectively (union) accounted for 74% and jointly 41% of them (Table 2, Additional file 1A, cols BD, BE).
Jointly considering the five phenotypic traits, there were 7769 up-regulated and 4148 down-regulated genes (Fig. 6). Gene ontology enrichment analysis indicates that the group of 7769 up-regulated genes was largely involved in gene regulation, including protein kinase activity and in particular serine/threonine kinases, serine/threonine phosphatase activity and transcription factor activity (Fig. 7). Key processes associated with biotic stress, such as regulation of cell death, response to unfolded protein and regulation of immune response, were also strongly affected. Changes associated with phytohormone pathways, including SA, JA and abscisic acid (ABA), were significant and will be discussed in more detail in a later section. Enrichment was also observed for the up-regulation of genes associated with aromatic amino acid metabolism, in particular tryptophan biosynthesis, and for genes associated with nitrogen compound transport and nitronate monooxygenase activity. Interestingly, only four genes (TraesCS1B01G250600, TraesCS1A01G235800, TraesCS1A01G238700, and TraesCS1D01G238900) in the wheat genome were annotated with nitronate monooxygenase activity; all of them were significantly associated with all five FHB-related traits (Fig. 8). More details are available in Additional file 2A.

Numbers of DEGs correlated with the five FHB related traits, specific to each trait, common between 2, 3, 4, or all five traits. Values for %Fg, Fg_GAPDH and DON are from Fig. 1. a up-regulated DEGs; b down-regulated DEGs

Gene ontology enrichment analysis of DEG groups up- or down-regulated by Fg (Fg_Up and Fg_Down) and genes associated with FHB resistance (Res) and susceptibility at 2 and/or 4 dpi (Sus2, Sus4, and Sus2.4)

Gene expression profiles of nitronate monooxygenase genes. Fg and H2O: treatments with Fg and water (control); 2d and 4d: 2 and 4 dpi; S: Shaw; HC: HC374; N: Nyubai; W: Wuhan 1
The group of 4148 down-regulated genes was highly enriched with genes associated with microtubule-based processes including binding, motor activity and nucleoside-triphosphatase activity. It was also enriched in genes associated with primary metabolic processes, such as the nitrogen compound metabolic process, glucan biosynthetic process, chlorophyll biosynthetic process and photosynthesis (Fig. 7). DNA replication and repair, gene silencing by RNA, as well as translation and protein folding were also negatively impacted by Fg infection. More details are available in Additional file 2B.
The following sections examine the contribution of genes whose expression changed in response to Fg treatment, in order to gain better understanding of the molecular response of wheat to Fg infection.
DEGs potentially associated with FHB resistance
There were a very small number of genes (12) with a DEFE feature pattern FW00111111, which identifies genes up-regulated by Fg across all three resistant genotypes at both time points, but with no significant change in the susceptible Shaw. All genes in this group have the same DEFE pattern FRS111111 showing significantly higher expression in the resistant genotypes as compared with the susceptible Shaw. One gene having a feature pattern WRS111111, indicating similar transcriptional profile in the control sample, was removed from this group. The combined DEFE feature pattern FW00111111∩(FRS111111¬WRS111111) includes 11 genes consisting of three glutathione S-transferase (GST), five protein kinases, a purple acid phosphatase and two membrane proteins (Table 3, Fig. 9). Additionally, another gene (TraesCS2B01G296000, a MatE transmembrane transporter) was included in the combined DEFE feature pattern FW10111111∩(FRS111111¬WRS111111). Although it was upregulated in Shaw at 2 dpi, the extent of change (log2FC = 1.5) was minor as compared to the three resistant genotypes (log2FC > 3.5); the differences in gene expression between a resistance genotype and the susceptible Shaw (FRS111111) were at log2FC > 2 (Table 3). The two GST from chromosome 7 are possible homolog genes. The expression levels of the 12 genes were mostly correlated with pathogen inoculation in the three resistant genotypes. The expression of these genes in the susceptible Shaw was much lower, even after challenge by the pathogen, showing a highly significant negative correlation (p < 0.002) (Fig. 9, Additional file 3A).

Genes putatively associated with FHB resistance. a 12 genes upregulated by Fg treatment across the resistant Nyubai, Wuhan 1 and HC374, but not in the susceptible Shaw were identified by the joint DEFE patterns of FW00111111∩ (FRS111111¬WRS111111) or FW10111111∩(FRS111111¬WRS111111). Refer to Table 1 for the meaning of the DEFE patterns. b expression profiles of the 12 genes putatively associated with FHB resistance. c expression profile by RT-qPCR for GST (TraesCS7A01G021900). Fg and H2O: treatments with Fg and water (control); 2d and 4d: 2 and 4 dpi; S: Shaw; HC: HC374; N: Nyubai; W: Wuhan 1
The small number of commonly up-regulated genes among the resistant genotypes can be explained at least in part by the genetic relationship between wheat genotypes. Nyubai and Wuhan 1 are genetically different from each other while HC374 was derived from a cross between the two. There were more genes commonly associated with FHB resistance between Nyubai and HC374 (17) than between Wuhan 1 and HC374 (5). To study the similarity between Nyubai and HC374 and their difference with Wuhan 1, three DEFE patterns: FW00010100, FW00111100, FW00101000 and their combination with Sets 2 and 3 comparisons were investigated (Tables 4, 5, 6; Additional file 3B). For example, three sesquiterpene synthase homolog genes were found to have similar expression between Nyubai and HC374, but not Wuhan 1 (Fig. 10).

Expression profiles of sesquiterpene synthases. a RNA-seq data of the three homologs. b cumulative expression profile by RT-qPCR for the three sesquiterpene synthases (gene specific assays could not be designed due to high sequence similarity among the three genes). Fg and H2O: treatments with Fg and water (control); 2d and 4d: 2 and 4 dpi; S: Shaw; HC: HC374; N: Nyubai; W: Wuhan 1
Other genes associated with FHB resistance but with distinct expression profiles in different genotypes are listed in Additional file 3C. They illustrate some of the differences in the overall defense response to Fg infection in each genotype. For example, there were roughly similar proportions of genes annotated as involved in the regulation of gene expression, defense, secondary metabolism, protein degradation and transport among the 107 DEGs unique to Wuhan 1, while about half of the 50 DEGs unique to Nyubai were involved in gene regulation, a quarter in secondary metabolism and very few in the other 3 functional categories. Among the 107 DEGs unique to Wuhan 1, 92 were up-regulated only at 2 dpi and the other 15 only at 4 dpi, but none were up-regulated at both time points. Among the 50 Nuybai DEGs, 46 were up-regulated at 2 dpi and 19 were also up-regulated at 4 dpi. Thus, DEGs in Wuhan 1 were transiently up-regulated by Fg, while many in Nyubai were much more strongly up-regulated by the pathogen at both time points. Only 21 DEGs had a distinct expression profile for HC374, with most (18/21) showing higher up-regulation by Fg at 4 dpi only. Interestingly, nine of these genes are annotated as acid beta-fructofuranosidases (Fig. 7; Additional file 2C, Additional file 3C). These genes are part of a small protein family containing distinct members co-localized on chromosomes 6A, B and D.
RT-qPCR analysis was performed for a subset of the genes associated with resistance mentioned above. Similar expression profiles were obtained when the genes tested had no close homolog (GST, Traes7A01G021900, Fig. 9c) or when all of the homologs had a similar expression pattern (sesquiterpene synthase TraesCS6A01G183000 and homologs, Fig. 10).
DEGs potentially associated with FHB susceptibility
In comparisons between Fg-treated samples and their controls, the number of DEGs of both host and pathogen were highest in the susceptible host Shaw, consistent with the Fg biomass (estimated by Fg-GAPDH expression), mRNA abundance, and DON concentration (Figs. 1, 2). In wheat, there were 3739 genes uniquely up-regulated at 4 dpi in Shaw (FW01000000 = 3739), which was the most frequent DEFE feature pattern of the series (Additional file 1B). In Fg-treated samples, there were 2891 genes expressed unanimously lower at 4 dpi in all three resistant genotypes than in the susceptible Shaw (FRS020202 = 2891). This pattern had the highest frequency in that series as well. These two interesting feature patterns prompted us to investigate DEGs potentially associated with FHB susceptibility in Shaw. FRS020202 confirms FW01000000 in up-regulation by Fg treatment at 4 dpi only in Shaw. There were a smaller number of genes (291) with a DEFE feature pattern of WRS020202, which represents innate difference between all three FHB resistant genotypes and the susceptible Shaw without Fg treatment. Combining these three feature patterns in the formula FW01000000∩FRS020202¬WRS020202 revealed a group of 1727 genes putatively associated with susceptibility in Shaw at 4 dpi (Fig. 11). Similarly, there were 66 genes with a formula of FW10000000∩FRS202020¬WRS202020 (same as previous formula, but at 2 dpi) putatively associated with susceptibility at 2 dpi in Shaw and 477 genes of FW11000000∩FRS222222¬WRS222222 putatively associated with susceptibility in Shaw at both 2 and 4 dpi.

DEGs putatively associated with FHB susceptibility. Frequency distribution at 2 dpi (a), 4 dpi (b), and both time points (c). The highlighted numbers in the Venn diagrams are considered in the text. Refer to Table 1 for the meaning of the DEFE patterns
Gene Ontology enrichment analysis indicated that the 66 genes uniquely up-regulated in the susceptible genotype at 2 dpi by Fg were highly enriched with genes encoding for protein kinase activity, particularly protein serine/threonine kinases (Fig. 7). Changes in nitrogen compound metabolic process, the other most important enriched category, were relatively modest (Additional file 2D).
The group of 1727 genes up-regulated only in Shaw at 4 dpi, was highly enriched with genes involved in regulation of gene expression, including ET-responsive, WRKY, Myb, bZIP, NAC-domain containing and other types of transcription factors (Additional file 2E). This group also includes the NF-X1-type zinc finger protein NFXL1, which has previously been shown to contribute to FHB susceptibility in wheat [15] (Fig. 12). These changes correlated with the regulation of several processes, including nitrogen compound and lipid metabolism, signal transduction and composition of membranes (Fig. 7).

Expression profiles of the five NFXL1 genes. a in the RNA-seq dataset; b cumulative expression of the four genes on chromosome 7 by RT-qPCR (gene specific assays could not be designed due to high sequence similarity among the four genes). Fg and H2O: treatments with Fg and water (control); 2d and 4d: 2 and 4 dpi; S: Shaw; HC: HC374; N: Nyubai; W: Wuhan 1
The group of 477 genes associated with susceptibility of Shaw at both 2 and 4 dpi was enriched in genes coding for protein kinase activity (Fig. 7), as observed at 2 dpi; however the up-regulation in Shaw of this larger number of kinase genes lasted through the two time points sampled. This group was also enriched in genes associated with oxalate oxidase activity and transport, especially MatE antiporters. More details are provided in Additional file 2F.
To further understand the dynamics of changes in regulation of gene expression in Shaw, the distribution of genes in key functional categories associated with expression regulation was examined using their gene description (Additional file 1A, cols C, BG-BI). This allowed the inclusion of genes without GO annotations. A shift can be observed in the types of kinases up-regulated uniquely in Shaw during the experiment (Table 7). A majority of the leucine rich repeat receptor kinases (LRR-RKs) were up-regulated in Shaw at 2 dpi, while most other types of kinases were up-regulated either at 4 dpi or at both time points. It is of note that only two mitogen-activated protein (MAP) kinases were up-regulated uniquely in Shaw among the 53 MAP kinases differentially expressed after Fg treatment. The majority of the transcription factors were uniquely up-regulated in Shaw at 4 dpi. Another group of genes involved in regulation via the proteasome, the F-box containing proteins, were also up-regulated in Shaw at 4 dpi or at both time points.
Gene association network analysis
The top 1% of the similarity matrix (Eq. 1 in Methods) was considered in a network consisting of 8946 wheat DEGs connected with 4,328,705 edges. The majority of network nodes were from the cluster C1, which exclusively occupied the top 53% of nodes (4766 genes). This cluster included a large number of genes that were strongly up-regulated by Fg infection, with the up-regulation in Shaw being the largest. The top 10% of nodes (895 genes, highlighted in Additional file 1A, col AB) in the networks were considered key hub genes. The lowest number of immediate neighbors (directly connected genes) among the key hub genes was 3030. The top node (TraesCS1D01G429900), having 6553 immediate neighbors, was a WRKY transcription factor (TF). There were 23 WRKY TF genes among the top 10% key hub genes and 109 in the entire network. An enrichment index (defined in the method section) was applied to illustrate the extent of enrichment of interesting groups of genes. For example in the case of WRKY TFs, there were 23 (2.57% = 23/895) in the key hub gene population, 109 (1.22% = 109/8946) in the entire network, 145 (0.50% = 145/28961) in the DEGs and 311 (0.28% = 311/110790) in the entire wheat genome; thus the enrichment index (E) for WRKY TFs was 1.78, 4.34, 9.15 in the DEGs, network and key hub gene populations, respectively. The WRKY TFs group was one of the two groups with the highest percentage of enrichment (Figs. 13a, Additional file 4). Glutathione S-transferases (GST) were also highly enriched, with 22 (E = 6.35) as key hub genes and 162 (E = 4.68) in the entire network (Fig. 13). Among the 8946 DEGs in the networks, 5340 were part of the Fg up-regulated group of genes (Fig. 6a, Additional file 4B) and 1019 were putatively associated with FHB susceptibility (Fig. 11).

Enrichment of gene groups in the association networks and in key hub gene populations. a proportion in the gene populations, b enrichment index. Details are in Additional file 4. The color legend in panel b is for both panels
The groups of genes annotated as fimbria adhesin EcpD, NFXL1 and erect panicle 2 protein were among highly enriched groups (Fig. 13b); fimbria adhesin EcpD and NFXL1 were up-regulated by Fg infection. There are only six NFXL1 genes in the entire wheat genome, of which five were differentially expressed, and four were identified as key hub genes (Figs. 12, 13b). The six genes annotated as erect panicle 2 protein formed two subgroups with different expression profiles and orthologs (Additional file 5).
Collectively, among the 2270 DEGs putatively associated with FHB susceptibility, over 66% were in the group of Fg up-regulated genes (Fig. 6a) and the majority (60%) was in the C1 cluster (Additional file 1A, cols AA, BG-BI). Among genes associated with FHB susceptibility, many (175) were also key hub genes, including 12 F-box proteins, 2 WRKY TFs, 2 JA ZIM, and 2 glutathione S-transferases (Additional file 4B).
DEGs encoding pathogenesis-related proteins
Of the 235 genes in the wheat genome known to be closely related to plant pathogenesis, based on the wheat IWGSC RefSeq v1.0 gene function annotation, description, gene ontology fields, and manual curation, 57 were DEGs in the analyzed dataset (Additional file 6). These included 29 pathogenesis-related protein 1 (PR1), 2 PR1–1, 9 PR4, 7 thaumatin-like, and other genes detailed in Additional file 6. PR1 genes are known to be associated with the SA pathway while PR4 and thaumatin genes are associated with JA and/or ET pathway. In the list of DEGs, 13 were putatively associated with FHB susceptibility and none with FHB resistance (Additional file 6, cols BD & BE). There were 33 PR genes that were also nodes in the gene association network. Two PR1 (TraesCS5B01G442900, TraesCS5D01G446800) were considered key hub genes and were associated with Fg challenge, but not with FHB susceptibility or resistance. All PR1, PR1–1 and PR-4 genes were up-regulated by Fg, but none of them were expressed higher in any resistant genotype than in the susceptible Shaw.
Genes related with hormonal effects in response to Fg challenge
Through an ortholog search in Arabidopsis thaliana, Brachypodium distachyon, Oryza sativa Japonica and Zea mays, and using the current and previous versions of wheat genome annotations (Survey genome version 2.2 and TGACv1 in Ensembl Plants), 542 wheat DEGs were identified as involved in phytohormone pathways (Additional file 7). Some annotations were assigned using orthologs, to be more specific. Nearly half of these hormone pathway genes (245) were involved in the network described above and 24 were key hub genes. There were significantly more DEGs involved in auxin, ET, and JA pathways compared with ABA, SA, cytokinin, and gibberellin pathways (Table 8). The majority of the DEGs associated with the JA and ET response pathways were up-regulated by Fg, with a higher level of expression in the susceptible Shaw (Table 9). The only hormone biosynthetic pathway with multiple DEGs was the JA pathway. Of the 357 DEGs up-regulated by Fg infection, 80 were associated with FHB susceptibility (Additional file 7, cols BO-BQ); only two were associated with FHB resistance: auxin transport protein TraesCS6B01G198200 in HC374 and ET-responsive transcription factor TraesCS4D01G298600 in Wuhan 1.
In the auxin signaling pathway, DEGs associated with regulation of IAA homeostasis included up-regulated efflux carrier family proteins and indole-3-acetic acid-amido synthetase GH3.3, and down-regulated influx transporters and efflux carrier components (Table 9). Those DEGs were respectively up- or down-regulated by Fg in all four genotypes, with a stronger differential expression in Shaw than in the resistant genotypes. The 17 SAUR-like auxin-responsive family proteins, all up-regulated by Fg, were heavily involved in the network with an average connectivity of 1738. Nine of the SAUR-like DEGs were associated with FHB susceptibility. In addition, 25 of the 41 auxin response factors and 13 of the 35 auxin-responsive proteins were down-regulated after Fg infection.
In the ET signaling pathway, DEGs annotated as positive regulators of the ET response pathway, ethylene-insensitive 3 and multiprotein-bridging factor MBF1 were up-regulated by Fg and expressed higher in the susceptible Shaw than any of the three resistant genotypes (Table 9, Additional file 7). MBF1C DEGs were also well connected in the network. One DEG (TraesCS2A01G396400, 1-aminocyclopropane-1-carboxylate synthase) was associated with ET biosynthesis; it was significantly up-regulated in all genotypes by Fg infection, and expressed at a higher level in Shaw.
Enzymes associated with JA biosynthesis, including allene oxide cyclase and synthase and 12-oxophytodienoate reductase, were up-regulated by Fg, with higher expression in FHB susceptible Shaw (Table 9, Additional file 7). Seven lipoxygenases were generally up-regulated by Fg in one or more genotypes. Three DEGs encoding LOX3/4 (TraesCS4A01G009400, TraesCS4D01G294100, and TraesCS2B01G333600) were highly involved in the association network. In the JA signaling pathway, the 38 DEGs encoding various JA ZIM domain proteins were all up-regulated by Fg and generally expressed at higher levels in Shaw than in the three FHB resistant genotypes. Five were associated with FHB susceptibility.
Many of the DEGs associated with ABA were up-regulated by Fg and expressed higher in Shaw. These included genes associated with regulation of ABA biosynthesis (ABA3), homeostasis (ABA 8′-hydrolase) and signaling (RING/U-box protein) (Table 9, Additional file 7). Of the five genes encoding ABA-responsive binding factor that were uniformly up-regulated by Fg and expressed progressively higher at 4 dpi in Shaw, four were also associated with FHB susceptibility.
Most of the DEGs associated with the SA pathway were up-regulated by Fg infection and showed a stronger response in Shaw (Table 9, Additional file 7). This included NPR1, salicylate o-methyltransferase, two NRR repressor homolog 1 genes, a receptor-like protein kinase, a putative protein kinase and accelerated cell death 11 (ACD11), which were all associated with FHB susceptibility. Three phytoalexin-deficient (PAD) 4–1 protein genes orthologous to Arabidopsis PAD4 (AT3G52430), and two MAPK genes (MPK3) were also up-regulated by Fg and expressed higher in Shaw than in the resistant genotypes.
Chromosome distribution of wheat DEG groups
The patterns of gene frequency distribution on wheat chromosomes among the DEGs and those putatively associated with FHB susceptibility were similar to the global gene distributions (Additional file 8). In contrast, among the various groups putatively associated with FHB resistance, the DEGs were more clustered. For example, between Nyubai and HC374, there were more common genes on chromosomes 6 (A, B, D), including two groups of homolog genes. The 107 genes specific to Wuhan1 were mostly from chromosomes 2, 4 and 5 (A, B, D), including 4 pairs of homolog genes on chromosomes 4; while the 50 genes specific to Nyubai were mostly from chromosomes 2 and 3 and included very few homologs. Although there were only 21 genes specific to HC374, 10 of them were located on chromosomes 6, including 9 acid beta-fructofuranosidase genes.
Transcriptomics of the pathogen – F. graminearum
Expression profiles of pathogen genes were quite uniform. The highest number of DEGs appeared in the susceptible Shaw at 4 dpi (Fig. 2b, Additional file 9), which was consistent with the abundance of pathogen mRNA reads, pathogen biomass estimated by GAPDH expression, and DON concentration (Fig. 1). There were very few pathogen genes that were preferentially expressed in any of the FHB resistant hosts, but over half (4443) of the DEGs were significantly (p < 0.05) preferentially expressed in the susceptible Shaw. Over 8805 pathogen genes showed differential expression between the host plants, especially between a resistant wheat genotype and the susceptible Shaw. There was a significant number of genes, over 50% at 2 dpi and 96% at 4 dpi, that were more highly expressed in the susceptible Shaw than in any of the resistant host plants, but no single gene was expressed significantly higher in a resistant plant than in Shaw (Table 10). A significant number of genes (4350) expressed higher in Wuhan 1 than in Nyubai at 4 dpi, consistent with the proportion of Fg reads in the two genotypes (Fig. 1a, Additional file 9C). Among the 8811 differentially expressed pathogen genes, 8802 were in one major cluster that was significantly correlated (p < 0.05) with the abundance of pathogen mRNA read (R = 0.95), pathogen biomass estimated by Fg-GAPDH (R = 0.87), and DON concentration (R = 0.92) (Additional file 9D).
Over half of the DEGs were consistent at both time points and 43% were consistent at 4 dpi across all three FHB resistant genotypes (FRS222222 = 4498, FRS020202 = 3774). There were only two genes having the DEFE pattern of FRS202020, expressed significantly higher at 2 dpi in Shaw than in all three resistant genotypes; one is a pectate lyase (FGRAMPH1_01T16515), and the other is a hydrolase (FGRAMPH1_01T12653). There were five pectate lyases among the 8811 pathogen DEGs, one (FGRAMPH1_01T11755) had very similar DEFE pattern (FRS002020) and was in the same small C2 cluster as FGRAMPH1_01T16515 (Fig. 14b). The other three pectate lyase genes were in the C1 cluster (Fig. 14a). There are totally seven pectate lyase genes in the entire Fg genome.

Expression profiles of five Fg pectate lyase genes. a in expression cluster C1; b in expression cluster C2. Fg: treatment with Fg; 2d and 4d: 2 and 4 dpi; S: Shaw; HC: HC374; N: Nyubai; W: Wuhan 1
The DEFE patterns FRS020202 and FT2000 indicate that differential expression between the susceptible Shaw and all three resistant genotypes at 4 dpi and the difference between the two time points were significant only in Shaw. Joining these patterns FRS020202∩FT2000 resulted in 2431 DEGs. The GO terms described in Additional file 2G for these DEGs were enriched with genes involved in regulation of transcription (GO:0003700, p < 5E-19; GO:0006355, p < 3E-9), aromatic compound metabolism (GO:0006725, p < 4E-07), and regulation of nitrogen compound metabolism (GO:0051171, p < 6E-07). An enrichment in acetyltransferase activity (GO:0016407, p < 0.002), mainly N-acetyltransferase activity (GO:0008080, p < 0.003) was also noted.
Discussion
In our dataset, the higher number of DEGs of both host and pathogen in the susceptible Shaw, especially at 4 dpi, was closely associated with higher Fg biomass and extent of infection. A similar phenomenon was noticed in another study on different genotypes (Wang L, Li Q, Liu Z, Surendra A, Pan Y, Li Y, et al: Integrated transcriptome and hormone profiling highlight the role of multiple phytohormone pathways in wheat resistance against fusarium head blight, submitted). Our analyses showed that distinct groups of genes were activated at different stages in the individual genotypes in response to Fg infection. This is evident in this study among both groups of genes associated with FHB resistance and susceptibility.
Genes associated with Fg challenge
About 40% of the DEGs in this dataset were up-regulated after Fg treatment, and the large majority were up-regulated in all four genotypes investigated, with the up-regulation in Shaw being the largest. Gene ontology analysis showed up-regulation of a number of specific protein kinases and transcription factors, suggesting reprogramming of gene regulatory pathways. Up-regulation of such molecular functions was observed in other wheat-Fg transcriptomic studies and with treatment of Fg mycotoxin DON [16,17,18]. We also observed up-regulation of key processes associated with biotic stress such as regulation of cell death, response to unfolded protein and regulation of immune response. The response to unfolded proteins is activated when the endoplasmic reticulum becomes overwhelmed by the cell’s biosynthetic demands and accumulates unfolded or misfolded proteins; it can lead to the initiation of cell death when the situation remains out of control [19]. Cell death also plays an important role in the immune response in plants [20]. Our analysis suggests that the intensity of the wheat response to Fg infection, associated with a massive reprogramming of gene transcription, overwhelmed the endoplasmic reticulum and may have triggered apoptosis, especially in the susceptible genotype. In addition, many up-regulated plant defense regulatory genes were also correlated with the regulation of cell death. Although cell death is a successful strategy to stop a biotrophic fungus, it may contribute to the success of a hemi-biotrophic fungus such as Fg.
Nitronate monooxygenases (EC1.13.12.16, NMO) are essential to mitigate nitro-oxidative cellular damage and contribute to the maintenance of redox balance in stress conditions [21]. In the fungus Magnaporthe oryzae, expression of this enzyme is associated with self-detoxification as well as suppression of the rice immune response. NMOs are known to oxidize alkyl nitronates to aldehydes and nitrite in plants and fungi, and to detoxify the metabolic inhibitor propionate-3-nitronate, toxic to all organisms that use succinate dehydrogenase [22, 23]. NMO is also found to be associated with multiple herbicide resistance in Lolium multiflorum [24]. The up-regulation of the wheat NMOs correlating with the level of Fg infection, in this study, suggests a potential role in the immune response of wheat.
Significant increases in aromatic amino acid metabolism, tryptophan biosynthesis in particular, and nitrogen compound metabolic process were also noted. Tryptophan is a precursor for many phenylpropanoids and for lignin; these contribute to strengthening of the primary cell wall, and are thought to participate in blocking of fungal progression in plant tissues (reviewed in [25]). Many studies have noted an up-regulation of tryptophan and phenylpropanoid biosynthesis pathways after Fg infection [17, 26,27,28].
About 21% of the DEGs were down-regulated by Fg treatment, and the majority was down-regulated to a larger extent in Shaw, especially at 4 dpi. There was an important effect on microtubule-based processes and on primary metabolic processes, including nitrogen compound metabolism, glucan biosynthesis, chlorophyll biosynthesis and photosynthesis. Processes associated with DNA repair and replication, gene silencing by RNA, translation and protein folding were also negatively affected. Down-regulation of primary metabolism and photosynthesis has been reported in different plant-pathogen interactions and has been suggested to be a mechanism to alleviate the energy costs associated with the up-regulation of other metabolic pathways for defense [5, 6]. Down-regulation of ribosomal proteins in an FHB-susceptible genotype by Fg and DON has been reported by Foroud et al. [26]. DON is known to inhibit protein synthesis. A negative effect of Fg infection on microtubule-based processes, DNA repair and replication, and protein folding has not, to our knowledge, been reported before. Microtubules-based processes contribute to many cellular activities, including intracellular transport, secretion, cell structure, and chromosome separation. It has been shown that disruption of microtubule networks suppresses cell wall-mediated defense in Arabidopsis thaliana [29]. Heat shock proteins are key components of protein folding; many of them have been reported to have a positive role in regulation of plant immunity [30]. The down-regulation of genes associated with protein folding in our experiments correlated with the up-regulation of the response to unfolded proteins.
Overall, the response of wheat to Fg infection included a large reprogramming of cellular processes which were mostly shared between susceptible and resistant genotypes, although with a larger magnitude in the susceptible genotype. Wheat invested a lot of energy, both in susceptible and resistant genotypes, towards extensive transcriptional reprogramming. That effort, particularly in the susceptible genotype, appeared to be insufficient to mount an effective plant defense against FHB.
Differential gene expression associated with Fg treatment and susceptibility to FHB
In the susceptible Shaw, 543 wheat genes were uniquely up-regulated at 2 dpi and 2204 at 4 dpi; among these genes, 477 were detected at both 2 and 4 dpi. More than 30% of these genes were part of the gene association network, supporting a coordinated reprogramming of gene transcription. The earlier and higher differential expression of many genes in susceptible, FHB-challenged plants may be attributable to earlier and higher levels of Fg infection, triggering activation of plant defense and other responses. Although “plant susceptibility genes” have been documented such as PMR6 required for susceptibility to powdery mildew in Arabidopsis [31], the larger extent of genes with differential expression in susceptible, infected plants do not fall in this class.
A subgroup of LRR-RKs was up-regulated early (2 dpi) in Shaw and not in the resistant genotypes, followed by up-regulation of many other types of kinases and of many types of transcription factors at 4 dpi. Additional kinases and transcription factors were up-regulated only in Shaw at both time points (Table 7). LRR-RKs play important roles in plant immunity, by recognizing pathogen-associated molecular patterns (PAMPs) associated either with the pathogens or the attacked host and processing the extracellular signals through signaling cascades and defense interaction networks [31,32,33,34]. Specific LRR-RKs have been associated with resistance to specific pathogens in many crops, including in wheat (e.g. powdery mildew, [35]). However, the blocking or diverting of LRR-RK signaling by a pathogen has also been reported [31, 36]. Given the large transcription reprogramming happening in the susceptible genotype, one may ask if Fg can hijack wheat signaling. WRKY, Myb and ET-responsive transcription factors have been shown to be part of signaling cascades activated by LRR-RKs [9, 37, 38]. Very few LRR-RKs and associated networks have been characterized in wheat. So it is yet too early to confirm whether the LRR-RKs, transcription factors and kinases up-regulated only in Shaw in early infection are part of defense or other types of responses.
The ubiquitin/proteasome system has been shown to play an important role in regulating the plant immune response, including by degradation of receptors activating the immune response [39, 40]. In Shaw, we observed the up-regulation of many F-box proteins, used by the ubiquitin/proteasome system to recognize proteins targeted for degradation. Identification of the targets for those F-box proteins will be required to determine if they contribute to the FHB susceptibility in Shaw.
Differential gene expression associated with Fg treatment and resistance against FHB
DEGs common to the three resistant genotypes
The 12 DEGs associated with FHB resistance that were up-regulated in all three FHB-resistant genotypes examined in this study were enriched in functions associated with early defense response in plants. Four kinase proteins, two LRR-RKs and two with lectin domains, were strongly up-regulated in the FHB-resistant genotypes. Kinases of this type are referred to as pattern recognition receptors (PRRs). Upon recognition of specific PAMPs by PRRs, the PAMP-triggered immunity response is activated, leading to activation of MAP kinases, ROS production and transcriptional reprogramming including up-regulation of MAPK, WRKY, MYB and ET-responsive transcription factors [41,42,43]. A large number of predicted PRRs are present in wheat; however very few have been characterized. One or more of the kinases identified here could contribute to the perception of Fg in the FHB-resistant genotypes.
The three glutathione S-transferases (GSTs) were part of a group of 261 GSTs, which were differentially expressed in the analyzed dataset, and only one of the three was part of an association network. Modulation of the redox state of glutathione by GSTs regulates early signaling events in biotic stresses such as fungal infections, including activation of the essential regulator of systemic acquired resistance NPR1 [44]. In the npr1–3 mutant of Arabidopsis thaliana, the expression of GST (At2g47730) and SOBIR1 (At2g31880) is severely impaired as compared to the wild type when subjected to SA perturbation [45]. GSTs are known for their roles in maintaining the physiological redox state of the cell, protection of the cell against oxidative damages and detoxification of xenobiotics [46, 47]. Although up-regulation of GSTs has been observed in FHB-resistant as well as FHB-susceptible wheat material following Fg infection, increased levels of specific GST family members has been observed in wheat and barley in association with FHB resistance [26, 27, 48, 49].
Two components of the phosphatidylinositol signaling pathway were expressed at higher level in the three FHB-resistant genotypes, a phosphatidylinositol 4-phosphate 5-kinase (PIP5K, EC:2.7.1.68) and a hyccin-like gene. Hyccin is part of a complex that localizes phosphatidylinositol 4-kinase to the plasma membrane and stabilizes its activity [50]. In Arabidopsis cells, phosphatidylinositol 4-kinase and PIP5K, involved in two consecutive steps in phosphatidylinositol phosphorylation, are both activated within minutes by SA treatment [51]. Phosphatidylinositol 4-kinase has been shown to be part of a complex that negatively regulates SA-dependent gene expression and defense response [52]. The higher expression of hyccin and PIP5K in FHB-resistant material could be interpreted as a tone-down of the SA-dependent defense response in the FHB resistant genotypes. This is consistent with our analysis of DEGs associated with hormone pathways (see below).
Purple acid phosphatases (PAPs, EC 3.1.3.2), which was up-regulated in the three FHB resistant genotypes but not in Shaw, have important roles in intracellular and extracellular phosphorus scavenging and recycling, including P-remobilization during tissue senescence, however their physiological roles are poorly understood [53]. Recently, the purple acid phosphatase PAP5 from A. thaliana has been shown to be required for maintenance of basal resistance and to work upstream of SA accumulation [54]. Further work will be required to understand their contribution to defense response in wheat.
The MatE transmembrane transporter is part of the large family of multidrug and toxic compound extrusion (MATE) proteins that are known to transport secondary metabolites and xenotic compounds. Although only a few of those transporters have a characterized function, some have been identified as key players in stress and pathogen response. For example, the rice MATE1 and MATE2 and Arabidopsis ADS1 are negative regulators of disease resistance [55, 56] while Arabidopsis EDS5 is associated with disease tolerance via a SA pathway [57]. The MatE gene TraesCS2B01G296000 identified in this study as associated with resistance is not homologous to any of the characterized MATE proteins.
Other DEGs associated with resistance
We identified a higher number of DEGs with similar expression profiles between HC374 and Nyubai than in the other two comparisons, both in correlation analysis and in the DEFE analysis. This suggests that the progeny line HC374, resulting from a cross between Nyubai and Wuhan 1, is genetically closer to Nyubai than Wuhan 1 in terms of resistance to FHB. This is consistent with the fact that HC374 and Nyubai share two QTLs on chromosomes 3BS and 5A, while HC374 and Wuhan 1 share one weaker QTL on chromosome 2DL [4].
Among the 17 DEGs strongly up-regulated by Fg in both Nyubai and HC374, there were four LRR containing proteins (including TraesCSU01G181100), two with transmembrane domains and possible receptor activity as PRR, and two located either in the cytoplasm or nucleus that could play a role in elicitor-triggered immunity (ETI). Notably, the receptor protein kinase TraesCS6A01G417900 is orthologous to the Arabidopsis chitin elicitor receptor kinase CERK1. This kinase has been shown to be required for the non-host defense response of Arabidopsis to Fusarium oxysporum [58]. The barley ortholog HvCERK1 has been shown to be up-regulated by Fg and to contribute to FHB resistance [59]. Transient silencing of HvCERK1 is also associated with lower expression of a MAPK and two WRKY genes as well as lower production of phenylpropanoid and flavonoid metabolites. The up-regulation of phenylalanine ammonia lyase, a key enzyme in the biosynthesis of phenylpropanoids, in this group of genes common between Nyubai and HC374 is consistent with the possibility that TraesCS6A01G417900 has CERK1-like activity in wheat and may contribute to resistance to FHB in the two genotypes. Also in common between Nyubai and HC374 were genes associated with secondary metabolism, including an acetylglucosaminyltransferase, a key enzyme for the biosynthesis of terpenes, sesquiterpene synthase, and other genes associated with stress response and detoxification (OMA1, peroxidase, MatE transporter). Increased expression of biosynthesis genes for phenylpropanoid and terpenoid compounds has been observed in wheat carrying the Fhb1 gene for FHB resistance, located on the chromosome 3BS [60]. This gene is also carried by both Nyubai and HC374.
The group of 50 DEGs unique to Nyubai contains an additional 15 kinases and receptor-like kinases including many that were up-regulated at much higher levels at 2 and 4 dpi in Nyubai in response to Fg infection. The increased expression of multiple kinases has been associated with FHB resistance in other wheat material [27, 48, 49]. Up-regulation of numerous kinases suggests an effort (or an attempt) to modulate gene expression. However, higher up-regulation of known gene sets associated with defense and detoxification was not observed in the time frame analyzed. In contrast, the 107 DEGs unique to Wuhan 1 contained a large number of defense related genes that were transiently up-regulated at a higher level at 2 dpi in that genotype compared to the other genotypes. These observations were supported by network analysis and suggest the use of different defense strategies by these resistant parents.
Only a small number of genes unique to the hybrid HC374 were up-regulated relative Nyubai and Wuhan 1; the close genetic relationship with its parental genotypes explains this at least in part. Surprisingly, 9 of the 12 differentially expressed acid beta-fructofuranosidases, or acid invertases, in our dataset had a much higher up-regulation in HC374 at 4 dpi. Cell wall invertases have been shown to be essential for proper sugar partitioning and signaling to induce defense response during pathogen attack [61].
Eight DEGs with similar strong up-regulation of expression in Wuhan 1 and Nyubai, but not in HC374 were enriched in genes with secondary metabolic transfer activity (transferases, cytochrome P450, blue copper proteins). This could be an attempt by both genotypes either to detoxify xenobiotics or to produce/modify secondary metabolites. The observation that most of the DEGs were expressed at much lower level in HC374 suggests that they are not essential to FHB resistance. However, up-regulation of glycosyltransferases and P450s has been observed in FHB-resistant barley and wheat, including Chevron, CM82036, Dream, Nobeokabouzu and Sumai 3 [27, 48, 62, 63].
Genes associated with hormonal pathways and pathogenesis-related proteins in response to FHB challenge
There are contradictory reports in the literature related to hormonal signaling pathways and their contribution to FHB resistance in wheat. Makandar et al. [64, 65] showed that SA-induced expression of PR1 was associated with FHB resistance. In contrast, many studies have observed an increase in JA level and pathway activity in FHB resistant genotypes [16, 25, 62]. There have been suggestions that the regulation of pathogenesis-related genes by plant hormones is genotype-dependent in wheat [66], and evidence that the timing of activation of the SA and JA pathways is critical to determine resistance or susceptibility to Fg in Arabidopsis [67]. Observations in wheat supported this finding [68]. In addition, ET signaling is shown to facilitate Fg infection in Arabidopsis and wheat [69].
In the genotypes characterized here, the majority of the DEGs associated with ET and JA pathways showed a stronger up-regulation by Fg in the susceptible Shaw. This included the positive regulators of ET response ethylene insensitive 3 and MBF1C. MBF1C is a transcriptional coactivator that modulates ET-response signal transduction. Overexpression of MBF1C gene in Arabidopsis thaliana has been shown to confer enhanced tolerance to bacterial infection, heat and osmotic stresses [70]. Genes for at least three steps in the biosynthesis of JA, allene oxide synthase and cyclase, and 12-oxophytodienoate reductase, were more strongly up-regulated in Shaw than in the 3 resistant genotypes. Many lipoxygenases, involved in the biosynthesis of JA and other oxylipins, were also up-regulated by Fg. Nalam et al. [71] have shown that inactivation of the lipoxygenase TaLpx-1, essential to oxylipin synthesis, resulted in enhanced resistance against Fg in wheat; they also showed that inactivation of the ortholog gene LOX1 in Arabidopsis lead to a stronger SA pathway activity and attenuation of JA signaling during Fg infection.
There was also up-regulation of many JA ZIM domain protein genes, including 5 associated with FHB susceptibility; these are repressor of JA signaling. However, conjugation of JA to isoleucine by amido synthetases, including indole-3-acetic acid-amido synthetase GH3, triggers the degradation of the JA ZIM domain proteins and induces signaling [72]. Given that many indole-3-acetic acid-amido synthetase GH3.3 were up-regulated in the genotypes characterized, it is possible that JA-isoleucine conjugation occurred. Up-regulation of PR4 and thaumatin genes by Fg is suggestive of JA signaling activity.
There was a much smaller number of DEGs up-regulated by Fg associated with the SA pathway. These included DEGs annotated as NPR1, NRR repressor homolog 1, salicylate o-methyltransferase, accelerated cell death 11 (ACD11), phytoalexin-deficient 4 (PAD 4) and MPK3. In Arabidopsis, these genes contribute to the regulation of SA levels and signaling pathways. Salicylate o-methyltransferase regulates SA homeostasis. NPR1 is a positive transcriptional co-regulator of SA signaling and a negative one for JA, and it is needed for PR1 expression [73, 74]; NRR repressor 1 negatively regulates NPR1-mediated transcriptional activation [75]. ACD11 is a ceramide-1-phosphate transfer protein that modulates SA-dependent programmed cell death [76]. PAD 4 promotes SA accumulation and regulates the crosstalk between SA and JA/ET pathways [77]. MPK3 inhibit SA accumulation and repress some defense response pathways [78]. Although the function of the SA-associated DEGs suggested a complex response, the up-regulation of a large number of genes from the PR1 family was consistent with SA signaling up-regulation; however this activity was associated more with susceptibility than resistance to FHB, as suggested by the 5 PR1 genes up-regulated only in Shaw during the time period examined.
Auxin signaling is a key component of growth and development in healthy plants. However contradictory reports exist in the literature about its role in disease. In Arabidopsis, auxin signaling has been reported both to promote disease susceptibility to bacterial infection and contribute to resistance to necrotrophic fungi [79, 80]. In rice, SAUR-like genes have been shown to negatively regulate auxin synthesis and transport [81]; suppression of auxin activity promotes basal immunity [82]. The role of auxin in the response to Fg infection in wheat is poorly characterized. Biselli et al. [17] observed that genes associated with auxin metabolism and signaling were induced by FHB in both the susceptible and resistant wheat genotypes that they characterized, but at a higher level in the susceptible one. We observed that many genes from two families associated with negative regulation of auxin activity, the SAUR-like and GH3.3 genes were strongly up-regulated by Fg infection, especially in Shaw. GH3.3 can conjugate auxin (in addition to JA) to amino acids, contributing to auxin homeostasis by inactivation [82]. Auxin efflux carrier proteins and influx transporters were respectively up- and down-regulated by Fg infection; these are involved in regulation of auxin homeostasis and their differential expression profiles suggest an attempt to move or keep auxin out of cells. In addition, many of the auxin-response factors and auxin-responsive protein genes were down-regulated by Fg infection, consistent with a negative regulation of auxin activity. Qi et al. [83] showed that large amounts of auxin were detected at 4 dpi with Fg in a susceptible wheat genotype, possibly produced by the fungus. Fg may use auxin as a susceptibility factor and the up-regulation of SAUR-like, GH3.3 and efflux carrier protein genes may be an attempt by wheat to deal with the excess of auxin.
The expression profiles of genes associated with the ABA signaling pathway suggested activity towards reduction of ABA signaling, especially in the susceptible Shaw. The up-regulation by Fg of the two RING/U-box superfamily protein genes, orthologous to Arabidopsis AIP2 – a negative regulator of ABA signaling, and the five cytochrome P450 family ABA 8′-hydroxylase genes, homologous to Arabidopsis CYP707A family – key players in the regulation of ABA levels via catabolism [84], supports that interpretation. Surprisingly, the three molybdenum cofactor sulfurase genes (ABA3), known as key positive regulators of ABA biosynthesis, were also significantly up-regulated by Fg. Overexpression of ABA3 has been reported to be associated with enhanced abiotic stress tolerance in Arabidopsis, maize and rice [85, 86]. Our dataset suggests an association of ABA activity with FHB susceptibility. It has been shown that exogenous treatment of wheat heads with ABA accelerates apparition of disease symptoms in a susceptible wheat cultivar [83].
Overall, our analysis of hormonal pathways indicates that increased gene expression activity in the JA, ET, SA, auxin and ABA pathways were associated with FHB susceptibility in the genotypes characterized. The number of DEGs associated with FHB susceptibility, especially from the auxin, ET and JA pathways, as well as the expression profile of many pathogenesis-related genes up-regulated by Fg treatment supports this association. The large number of up-regulated genes in hormone pathways and pathogenesis-related gene families illustrates the large effort made by wheat to mount a defense response that is either not sufficient or not productive in the genotypes characterized.
Transcriptomics of the pathogen
Of the five pectate lyase genes highly expressed in Shaw, the two expressed at higher levels at 2 dpi may contribute to initial penetration of the plant host cell wall rather than propagation of infection within the host. Plant pathogenic fungi secreted cell-wall degrading enzymes such as pectate lyases are important in degradation of plant pectin, a polymer of galacturonic acid found in the middle lamella of the cell wall. Pectinases are the first enzymes to be secreted by fungal pathogens when they attack plant cell walls [87, 88]. Inactivation of a pectate lyase in Fusarium solani reduced the ability of the fungus to infect plant tissues [89].
Significant enrichment of pathogen acetyltransferase expression after infection was evident in this study, especially in the susceptible host Shaw at 4 dpi. It is well known that plant immune response during pathogen infection requires extensive transcriptional reprogramming involving histone acetylation. Pathogens interfere with this process by using effector proteins encoding acetyltransferases that can directly acetylate host proteins to alter immunity [90, 91].
We found very few Fg genes up-regulated at a higher level in resistant genotypes as compared with the susceptible one at 2 and 4 dpi. This is in contrast with the findings of Hofstad et al. [92], who found 112 genes expressed at a higher level in their FHB resistant genotype at 4 dpi. Fungal strain or host genotype differences may explain this difference.
Conclusions
This study explored global transcriptomic profiles of four different wheat genotypes treated with Fg. In both the resistant and susceptible genotypes, transcriptional reprogramming was evident upon FHB challenge. In the susceptible genotype Shaw, a sub-group of LRR-RKs was uniquely up-regulated by Fg, followed by up-regulation of many transcription factors and numerous genes associated with defense response. However, this activity was not sufficient to prevent the spread of Fg infection. Differential gene expression patterns associated with the resistant genotypes, including patterns common to two or three of them and patterns unique to individual resistant genotypes, indicated multi-facetted defense responses that were distinct for each resistant genotype.
Methods
Plant material and RNA extraction and sequencing
Four wheat (Triticum aestivum) genotypes (i.e. cultivars or lines) were used in this study: Wuhan 1 (Type II FHB resistant), Nyubai (Type I FHB resistant), HC374 (a FHB resistant double haploid line derived from crossing Wuhan 1 with Nyubai), and Shaw (a FHB susceptible cultivar). Seeds from the three resistant genotypes were kindly provided by Dr. George Fedak (Ottawa Research and Development Centre, Agriculture and Agri-Food Canada). Somers et al. [4] identified 4 QTLs for FHB resistance located on chromosomes 2DL (carried by Wuhan 1 and HC374), 3BS (Nyubai and HC374), 4B (Wuhan 1) and 5A (Nyubai and HC374).
Wheat plants were grown in controlled-environment cabinets with 16 h light at 20 °C and 8 h dark at 16 °C until mid-anthesis then transferred to growth chambers at anthesis. Heads at mid-anthesis were point inoculated with either water (control), or Fg (strain DAOM233423). Plant growth, inoculation and infection conditions were described in detail previously [93]. Inoculated spikelet samples were collected in triplicate (from 5 heads per replicate) at 2 and 4 days post inoculation (dpi). In total, 48 samples were collected; total RNA, containing both the plant and pathogen RNA present in the samples, was extracted using the TriReagent (Molecular Research Center Inc.) followed by a treatment with DNase I (RNase-Free DNase set, Qiagen) onto columns (RNeasy Mini kit, Quiagen) according to instructions provided by the manufacturer. RNA was processed for deep paired-end RNA sequencing using Illumina HiSeq 2500 by the National Research Council Canada sequencing service.
RNA-seq data processing
The International Wheat Genome Sequencing Consortium (IWGSC) RefSeq v1.0 complete reference genome and corresponding annotation v1.0 [94] were used as reference for the analysis of wheat RNA-seq data. Following recommendations of IWGSC, the chromosome-partitioned version of the Chinese Spring version 1.0 genome was used and the gff3 file was reformatted accordingly. IWGSC provided both high and low confidence gene models for options; in this study, only the high confidence gene models were used. Fusarium graminearum reference genome (Fusarium graminearum str. PH-1) was obtained from EnsemblFungi [95]. Wheat and Fg genomes were combined into a host-pathogen pan-genome, and annotation data from both species were combined into a pan-annotation (Liu Z, Li Y, Pan Y, Wang L, Ouellet T, Fobert P: Strategy in wheat-Fusarium dual genome RNA-seq data processing, in preparation). This pan-genome contains 124,935 gene models, 14,145 from Fg and 110,790 from wheat. The RNA-seq reads were preprocessed by trimming adaptor sequences, filtering low-quality reads (Phred Score < =20 [96]) and eliminating short reads (length < = 20 bps) using FASTX Toolkit [97, 98]. After filtering, barcode and adaptor removal, an average of 24.5 million paired reads per sample were retained for subsequent read mapping through the RNA-seq data processing procedures. The cleaned RNA-seq reads were mapped against the pan-genome using STAR v2.5.3a [99] to generate gene-level counts. DESeq2 [100] was used for data normalization and subsequent DEG analysis for each pairwise comparison. Normalized read counts along with log2 fold change and p-values were used for downstream data analysis.
Data reduction and feature pattern identification
Absolute value of log2 fold change > = 1 and p-values <=0.01 were applied to filter all datasets. For differential analysis, we retained only genes with a minimum of 100 reads in at least one of the samples compared. We further applied the Differential Expression Feature Extraction method (DEFE, Pan Y, Li Y, Liu Z, Surendra A, Wang L, Foroud NA, Goyal RK, Ouellet T, Fobert PR: Differential expression feature extraction and its application in wheat RNA-seq data analysis, forthcoming) to identify differentially expressed genes. Gene expression data presented in the result section are log2 transformed using a pseudo-count of +1.
Gene ontology enrichment analysis and orthology
Gene ontology (GO) annotations were compiled by combining three versions of Triticum aestivum: (1) IWGSC RefSeq v1.0 annotation from [94], (2) Ensembl Plants TGACv1 (from Ensembl Plants release 34) and (3) IWGSC survey genome 2.2 (from Ensembl Plants release 28 [101]). The mapping of gene IDs between the three versions was based on the tables obtained from URGI (between IWGSC RefSeq v1.0 and Ensembl Plants TGACv1) and Ensembl Plants (between Ensembl Plants TGACv1 and IWGSC survey genome 2.2). The resulting GO annotations are available in Additional file 10. Gene ontology enrichment analysis was conducted using Gene Ontology Analyzer [102].
Orthologs in Arabidopsis thaliana (Araport11, from TAIR: [103]) and Brachypodium distachyon (Bdistachyon_314, from JGI: [104]) were obtained for Triticum aestivum genes. One-to-one reciprocal best hit (RBH) was performed between each of the Triticum aestivum sub-genome A, B and D, respectively, on one side and the Araport11 or Bdistachyon_314 on the other.
Association network and pathway analysis
Gene association network analysis of the differentially expressed gene dataset was conducted using the WGCNA R package [105]. A Pearson correlation coefficient matrix C was first computed, and then the positive adjacency matrix and negative adjacency matrix were computed, respectively: A+ = (C+)e, A− = (| C−| )e, where C+ and C− are the positive and negative correlation coefficient matrices and e is the power for soft-thresholding. Based on these adjacency matrices, a TOM similarity matrix was generated for each adjacency matrix, and the two TOM matrices were merged into a similarity matrix using:
Gene association network analysis was performed using the similarity matrix (S in Eq. 1) as edge weights. The top 1% weight in S was considered as valid edges in the network.
Hierarchical clustering was employed based on the similarity matrix (1-S as distance measure) to cluster genes. Dynamic tree cutting was used to generate gene clusters/modules using the dynamicTreeCut R package [105]. We digitalized the experimental traits (Fg treatment, time post inoculation) as described in [106]. The five FHB-related traits (Fg treatment, Fg_time, %Fg, Fg_GAPDH, and DON, Fig. 6, Additional file 1C) were used for Pearson correlation analysis between each of them on one side and expression profile of each gene or centroid (eigen gene resulted from WGCNA) of each cluster on the other side. Results of these correlation analyses are presented in Additional file 1A (Cols BD-BE) and Additional file 1D.
An enrichment index was defined to describe the extent of enrichment of certain group of genes among a subset of the wheat genome, such as the DEGs dataset or a set of network nodes. The global abundance, g is defined by the proportion of this group of genes in the wheat genome, which is the global frequency of this group of genes, f, divided by the size of the wheat genome, G: g = f/G. Similarly, the abundance (d) of this group of genes in the subset of the genome (D) is defined by its proportion in the subset of the genome, which is the frequency (fd) of this group of genes in the subset fd: d = fd/D. An enrichment index (E) is defined as E = d/g.
RT-qPCR validation
The cDNA synthesis of all RNA samples was carried out with the RETROscript® reverse transcription kit (Fisher Scientific, Waltham, MA, USA), using 2 μg of each RNA sample into a 20-μl reaction volume with oligo(dT)18 primer, and all manipulations followed the manufacturer’s protocol. SensiFast SYBR No-Rox kit (Bioline) and the MJ Research PTC200 thermal Cycler with Chromo 4 detector were utilized to performed RT-qPCRs, with 10 min at 95 °C, 35 cycles of 30 s at 95 °C, 30 s at melting temperature, and 1 min at 72 °C, melting curve from 55 °C to 95 °C, read every 1 °C, hold 5 s. Two technical replicates were done for each of the three biological replicates. Genes selected for RT-qPCR analysis respected two of three criteria: 1) being expressed at a sufficiently high level to be detectable by RT-qPCR analysis; 2) having sufficient long region(s) of unique sequence, allowing the design of genome-specific primers; or 3) having a closely related expression profile to their homologous genes from wheat sub-genomes A and B. Primers used are described in Additional file 11. Fungal biomass was estimated using expressed levels of GAPDH (FGSG_06257). The 2-ΔΔCt method [107] was used to calculate FC values, and the relative expression levels were normalized against three wheat reference genes, IAAOx (TraesCS2A01G246300), AOx (TraesCS2A01G327600), and hn-RNP-Q (TraesCS2A01G390200), as calculated by Vandesompele et al. [108], and rescaled using the lowest value as 1 among compared samples.
Abbreviations
- ABA:
-
Abscisic acid
- AOx:
-
Amine oxidase
- AP2/ERF:
-
APETALA2/ethylene-responsive transcription factor
- bHLH:
-
basic helix-loop-helix domain containing transcription factor
- bZIP:
-
basic leucine zipper domain containing transcription factor
- Ct :
-
Threshold cycle
- DAMP/PAMP:
-
Damage/pathogen associated molecular pattern
- DEFE:
-
Differential expression feature extraction
- DEG:
-
Differentially expressed gene
- DON:
-
Deoxynivalenol
- dpi:
-
days post inoculation
- ET:
-
Ethylene
- ETI:
-
Elicitor-triggered immunity
- FC:
-
Fold change
- Fg :
-
Fusarium graminearum
- FHB:
-
Fusarium head blight
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- GO:
-
Gene ontology
- hn-RNP-Q:
-
heterogeneous nuclear ribonucleoprotein Q
- IAAOx:
-
Indole-3 acetaldehyde oxidas
- IWGSC:
-
International Wheat Genome Sequencing Consortium
- JA:
-
Jasmonic acid
- LOX:
-
Lipoxygenase
- LRR-RK:
-
Leucine-rich repeat receptor kinase
- MADS box:
-
MADS box domain containing transcription factor
- MAP:
-
Mitogen-activated protein
- MAPK:
-
Mitogen-activated protein kinase
- MYB:
-
Myeloblastosis domain containing transcription factor
- NAC:
-
NAC domain containing transcription factor
- NMO:
-
Nitronate monooxygenases
- NPR:
-
Nonexpresser of PR (pathogenesis-related) genes
- NRR:
-
Negative regulator of resistance
- OMA:
-
Metalloendopeptidase
- PAD:
-
Phytoalexin-deficient
- PIP5K:
-
Phosphatidylinositol 4-phosphate 5-kinase
- PR:
-
Pathogenesis-related
- PRR:
-
Pattern recognition receptor
- QTL:
-
Quantitative trait locus
- ROS:
-
Reactive oxygen species
- SA:
-
Salicylic acid
- SAUR:
-
Small auxin-up RNA
- TF:
-
Transcription factor
- TOM:
-
Topology overlap matrix
- WGCNA:
-
Weighted Gene Correlation Network Analysis
- WRKY:
-
WRKY domain containing transcription factor
- ZIM:
-
Zinc-finger inflorescence meristem
References
Bai G, Shaner G. Management and resistance in wheat and barley to fusarium head blight. Annu Rev Phytopathol. 2004;42:135–61.
Mesterházy Á, Bartók T, Mirocha CG, Komoróczy R. Nature of wheat resistance to fusarium head blight and the role of deoxynivalenol for breeding. Plant Breed. 1999;118:97–110.
Buerstmayr H, Ban T, Anderson JA. QTL mapping and marker-assisted selection for fusarium head blight resistance in wheat: a review. Plant Breed. 2009;128:1–26.
Somers DJ, Fedak G, Savard M. Molecular mapping of novel genes controlling fusarium head blight resistance and deoxynivalenol accumulation in spring wheat. Genome. 2003;46:555–64.
Bolton MD. Primary metabolism and plant defense – fuel for the fire. Mol Plant-Microbe Interact. 2009;22:487–97.
Kangasjärvi S, Neukermans J, Li S, Aro EM, Noctor G. Photosynthesis, photorespiration, and light signalling in defence responses. J Exp Bot. 2012;63:1619–36.
Rojas CM, Senthil-Kumar M, Tzin V, Mysore KS. Regulation of primary plant metabolism during plant-pathogen interactions and its contribution to plant defense. Front Plant Sci. 2014;5:17.
Pieterse CM, Van der Does D, Zamioudis C, Leon-Reyes A, Van Wees SC. Hormonal modulation of plant immunity. Annu Rev Cell Dev Biol. 2012;28:489–521.
Tsuda K, Somssich IE. Transcriptional networks in plant immunity. New Phytol. 2015;206:932–47.
Glazebrook J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol. 2005;43:205–27.
Kazan K, Gardiner DM. Transcriptomics of cereal-Fusarium graminearum interactions: what we have learned so far. Mol Plant Pathol. 2018;19:764–78.
Moore JW, Loake GJ, Spoel SH. Transcription dynamics in plant immunity. Plant Cell. 2011;23:2809–20.
Weirauch MT, Hughes TR. A catalogue of eukaryotic transcription factor types, their evolutionary origin, and species distribution. Subcell Biochem. 2011;52:25–73.
Erayman M, Turktas M, Akdogan G, Gurkok T, Inal B, Ishakoglu E, et al. Transcriptome analysis of wheat inoculated with fusarium graminearum. Front Plant Sci. 2015;6:867.
Ouellet T, Balcerzak M, Leung W, Hattori J, Martin T, Gulden S, et al. DON induces genes that increase wheat susceptibility to fusarium head blight. Milwaukee: Proceedings of the National Fusarium Head Blight Forum; 2013. p. 76. https://scabusa.org/pdfs/forum13_proc_complete.pdf
Xiao J, Jin X, Jia X, Wang H, Cao A, Zhao W, et al. Transcriptome-based discovery of pathways and genes related to resistance against fusarium head blight in wheat landrace Wangshuibai. BMC Genomics. 2013;14:197.
Biselli C, Bagnaresi P, Facioli P, Hu X, Balcerzak M, MAttera MG, et al. Comparative transcriptome profiles of near-isogenic hexaploid wheat lines differing for effective alleles at the 2DL FHB resistance QTL. Front Plant Sci. 2018;9:37.
Desmond OJ, Manners JM, Stephens AE, Maclean DJ, Schenk PM, Gardiner DM, et al. The Fusarium mycotoxin deoxynivalenol elicits hydrogen peroxide production, programmed cell death and defence responses in wheat. Mol Plant Pathol. 2008;9:435–45.
Ruberti C, Kim S-J, Stefano G, Brandizzi F. Unfolded protein response in plants: one master, many questions. Curr Opin Plant Biol. 2015;27:59–66.
Coll NS, Epple P, Dang JL. Programmed cell death in the plant immune system. Cell Death Differ. 2011;18:1247–56.
Marroquin-Guzman M, Hartline D, Wright JD, Elowsky C, Bourret TJ, Wilson RA. The Magnaporthe oryzae nitrooxidative stress response suppresses rice innate immunity during blast disease. Nature Microbiol. 2017;2:17054.
Francis K, Nishino SF, Spain JC, Gadda G. A novel activity for fungal nitronate monooxygenase: detoxification of the metabolic inhibitor propionate-3-nitronate. Arch Biochem Biophys. 2012;521:84–9.
Francis K, Smitherman C, Nishino SF, Spain JC, Gadda G. The biochemistry of the metabolic poison propionate 3-nitronate and its conjugate acid, 3-nitropropionate. IUBMB Life. 2013;65:759–68.
Mahmood K, Mathiassen SK, Kristensen M, Kudsk P. Multiple herbicide resistance in Lolium multiflorum and identification of conserved regulatory elements of herbicide resistance genes. Front Plant Sci. 2016;7:1160.
Walter S, Nicholson P, Doohan FM. Action and reaction of host and pathogen during fusarium head blight disease. New Physiol. 2010;185:54–66.
Foroud N, Ouellet T, Laroche A, Oosterveen B, Jordan MC, Ellis BE, Eudes F. Differential transcriptome analyses of three wheat genotypes reveal different host response pathways associated with fusarium head blight and trichothecene resistance. Plant Pathol. 2012;61:296–314.
Kugler KG, Siegwart G, Nussbaumer T, Ametz C, Spannagl M, Steiner B, et al. Quantitative trait loci-dependent analysis of a gene co-expression network associated with Fusarium head blight resistance in bred wheat (Triticum aestivum L.). BMC Genomics. 2013;14:728.
Schweiger W, Steiner B, Ametz C, Siegwart G, Wiesenberger G, Berthiller F, et al. Transcriptomic characterization of two major Fusarium resistance quantitative trait loci (QTLs), Fhb1 and Qfhs.ifa-5A, identifies novel candidate genes. Mol Plant Pathol. 2013;14:772–85.
Lee AH-Y, Hurley B, Felsensteiner C, Yea C, Ckurshumova W, Bartetzko V, et al. A bacterial acetyltransferase destroys plant microtubule networks and blocks secretion. PLoS Pathog. 2012;8:e1002523.
Park C-J, Seo Y-S. Heat shock proteins: a review of the molecular chaperone for plant immunity. Plant Pathol J. 2015;31:323–33.
Eckardt NA. Plant disease susceptibility genes? Plant Cell. 2002;14:1983–6.
Tang D, Wang G, Zhou J-M. Receptor kinases in plant-pathogen interactions: more than pattern recognition. Plant Cell. 2017;29:618–37.
Smakowska-Luzan E, Mott GA, Parys K, Stegmann M, Howton TC, Layeghifard M, et al. An extracellular network of Arabidopsis leucine-rich repeat receptor kinases. Nature. 2018;553:342–6.
Lannoo N, Van Damme EJM. Lectin domains at the frontiers of plant defense. Front Plant Sci. 2014;5:397.
Rajaraman J, Douchkov D, Hensel G, Stefanato FL, Gordon A, Ereful N, et al. An LRR/malectin receptor-like kinase mediates resistance to non-adapted and adapted powdery mildew fungi in barley and wheat. Front Plant Sci. 2016;7:1836.
Dou D, Zhou JM. Phytopathogen effectors subverting host immunity: different foes, similar battleground. Cell Host Microbe. 2012;12:484–95.
Zhang Y, Wang Y, Fang B, Gannon P, Ding P, Li X, Zhang Y. Arabidopsis snc2-1D activates receptor-like protein-mediated immunity transduced through WRKY70. Plant Cell. 2010;22:3153–63.
Yuan N, Yuan S, Li Z, Zhou M, Wu P, Hu Q, et al. STRES INDUCED FACTOR 2, a leucine-rich repeat kinase regulates basal plant pathogen defense. Plant Phys. 2018;176:3062–80.
Devoto A, Miskett PR, Shirasu K. Role of ubiquitination in the regulation of plant defence against pathogens. Curr Opin Plant Biol. 2003;6:307–11.
Goff KE, Ramonell KM. The role and regulation of receptor-like kinases in plant defense. Gene Regul Systems Bio. 2007;1:167–75.
Libault M, Wan J, Czechowski T. Identification of 118 Arabidopsis transcription factor and 30 ubiquitin-ligase genes responding to chitin, a plant-defense elicitor. Molec Plant Microbe Interact. 2007;20:900–11.
Wan J, Zhang X-C, Neece D, Ramonell KM, Clough S, Kim SY, et al. A LysM receptor-like kinase plays a critical role in chitin signaling and fungal resistance in Arabidopsis. Plant Cell. 2008;20:471–81.
Shimizu T, Nakano T, Takamizawa D, Desaki Y, Ishii-Minami N, Nishizawa Y, et al. Two LysM receptor molecules, CEBiP and OsCERK1, cooperatively regulate chitin elicitor signaling in rice. Plant J. 2010;64:204–14.
Mou Z, Fan WH, Dong XN. Inducers of plant systemic acquired resistance regulate NPR1 function through redox changes. Cell. 2003;113:935–44.
Pan Y, Pylatuik JD, Ouyang J, Famili AF, Fobert PR. Discovery of functional genes for systemic acquired resistance in Arabidopsis thaliana through integrated data mining. J Bioinforma Comput Biol. 2004;2:639–55.
Gullner G, Kômives T. Defense reactions of infected plants: roles of glutathione and glutathione S-transferase enzymes. Acta Phytopathologica et Entomologica Hungarica. 2006;41:3–10.
Dubreuil-Maurizi C, Poinssot B. Role of glutathione in plant signaling under biotic stress. Plant Signal Behav. 2012;7:210–2.
Huang Y, Li L, Smith KP, Muehlbauer GJ. Differential transcriptomic responses to Fusarium graminearum infection in two barley quantitative trait loci associated with fusarium head blight resistance. BMC Genomics. 2016;17:387.
Dhokane D, Karre S, Kushalappa AC, McCartney C. Integrated metabolon-transcriptomics reveals fusarium head blight candidate resistance genes in wheat QTL-Fhb2. PLoS One. 2016;11:a0155851.
Baskin JM, Wu X, Christiano R, Oh MS, Schauder CM, Gazzerro E, et al. The leukodystrophy protein FAM126A (hyccin) regulates PtdIns(4)P synthesis at the plasma membrane. Nat Cell Biol. 2016;18:132–8.
Krinke O, Ruelland E, Valentová O, Vergnolle C, Renou J-P, Taconnat L, et al. Phosphatidylinositol 4-kinase activation is an early response to salicylic acid in Arabidopsis suspension cells. Plant Physiol. 2007;144:1347–59.
Antignani V, Klocko AL, Bak G, Chandrasekaran SD, Dunivin T, Nielsen E. Recruitment of PLANT U-Box13 and the PI4Kβ1/β2 phosphatidylinositol 4-kinases by the small GTPase RabA4B plays important roles during salicylic acid-mediated plant defense signaling in Arabidopsis. Plant Cell. 2015;27:243–61.
Tian J, Liao H. The role of intracellular and secreted purple acid phosphatases in plant phosphorus scavenging and recycling. Ann Plant Rev. 2015;48:265–88.
Ravichandran S, Stone SL, Benkel B, Prithiviraj B. Purple acid phosphatase5 is required for maintaining basal resistance against Pseudomonas syringae in Arabidopsis. BMC Plant Biol. 2013;13:107.
Tiwari M, Sharma D, Singh M, Tripathi RD, Trivedi PK. Expression of OsMATE1 and OsMATE2 alters development, stress responses and pathogen susceptibility in Arabidopsis. Sci Rep. 2014;4:3974.
Sun X, Gilroy EM, Chini A, Nurmberg PL, Hein I, Lacomme C, et al. ADS1 encodes a MATE-transporter that negatively regulates plant disease resistance. New Phytol. 2011;192:471–82.
Nawrath C, Heck S, Parinthawong N, Metraux JP. EDS5, an essential component of salicylic acid-dependent signaling for disease resistance in Arabidopsis, is a member of the MATE transporter family. Plant Cell. 2002;14:275–86.
Huaping H, Xiaohui J, Lunying W, Junsheng H. Chitin elicitor receptor kinase 1 (CERK1) is required for the non-host defense response of Arabidopsis to Fusarium oxysporum f. Sp cubense. Eur J Plant Pathol. 2017;147:571–8.
Karre S, Kumar A, Dhokane D, Kusahlappa AC. Metabolo-transcriptome profiling of barley reveals induction of chitin elicitor receptor kinase gene (HvCERK1) conferring resistance against Fusarium graminearum. Plant Mol Biol. 2017;93:247–67.
Gunnaiah R, Kushalappa AC, Duggavathi R, Fox S, Somers DJ. Integrated metabolo-proteomic approach to decipher the mechanisms by which wheat QTL (Fhb1) contributes to resistance against Fusarium graminearum. PLoS One. 2012;7:e40695.
Tauzin AS, Giardina T. Sucrose and invertases, a part of the plant defense response to the biotic stresses. Front Plant Sci. 2014;5:293.
Gottwald S, Samans B, Lück S, Friedt W. Jasmonate and ethylene dependent defence gene expression and suppression of fungal virulence factors: two essential mechanisms of fusarium head blight resistance in wheat? BMC Genomics. 2012;13:369.
Kosaka A, Tomohiro B, Manickavelu A. Genome-wide transcriptional profiling of wheat infected with Fusarium graminearum. Genomics Data. 2015;5:260–2.
Makandar R, Essig JS, Schapaugh MA, Trick HN, Shah J. Genetically engineered resistance to fusarium head blight in wheat by expression of Arabidopsis NPR1. Mol Pant-Microbe Interact. 2006;19:123–9.
Makandar R, Nalam VJ, Lee H, Trick HN, Dong Y, Shah J. Salicylic acid regulates basal resistance to fusarium head blight in wheat. Mol Plant-Microbe Interact. 2012;25:431–9.
Pordel R. The role of plant hormones in fusarium head blight of wheat: Thesis, University of Lethbridge; 2017. p. 138. https://www.uleth.ca/dspace/handle/10133/5013
Makandar R, Nalam V, Chaturvedi R, Jeannotte R, Sparks AA, Shah J. Involvement of salicylate and jasmonate signaling pathways in Arabidopsis interaction with Fusarium graminearum. Mol Plant-Microbe Interact. 2010;23:861–70.
Ding L, Xu H, Yi H, Yang L, Kong Z, Zhang L, et al. Resistance to hemi-biotrophic F graminearum infection is associated with coordinated and ordered expression of diverse defense signaling pathways. PLoS ONE. 2011;6:e19008.
Chen X, Steed A, Travella S, Keller B, Nicholson P. Fusarium graminearum exploits ethylene signaling to colonize dicotyledonous and monocotyledonous plants. New Phytol. 2009;182:975–83.
Suzuki N, Rizhsky L, Liang H, Shuman J, Shulaev V, Mittler R. Enhanced tolerance to environmental stress in transgenic plants expressing the transcriptional coactivator multiprotein bridging factor 1c. Plant Physiol. 2005;139:1313–22.
Nalam VJ, Alam S, Keereetaweep J, Venables B, Burdan D, Lee H, et al. Facilitation of Fusarium graminearum infection by 9-lipoxygenases in Arabidopsis and wheat. Mol Plant-Microbe Interact. 2015;28:1142–52.
Goossens J, Fernández-Calvo P, Schweizer F, Goossens A. Jasmonates: signal transduction components and their roles in environmental stress response. Plant Mol Biol. 2016;91:673–89.
Kinkerma M, Fan W, Dong X. Nuclear lecalization of NPR1 is required for activation of PR gene expression. Plant Cell. 2000;12:2339–50.
Spoel SH, Koornneef A, Claessens SM, Korzelius JP, Van Pelt JA, et al. NPR1 modulates cross-talk between salicylate- and jasmonate-dependent defense pathways through a novel function in the cytosol. Plant Cell. 2003;15:760–70.
Chen M, Canlas PE, Ronald PC. Strong suppression of systemic acquired resistance in Arabidopsis by NRR is dependent on its ability to interact with NPR1 and its putative repression domain. Mol Plant. 2008;1:552–9.
Simanshu DK, Zhai X, Munch D, Hofius D, Markham JE, Bielawski J, Bielawska A, Malinina L, Molotkovsky JG, Mundy JW, Patel DJ. Arabidopsis accelerated-cell-death 11, ACD11, is a ceramide-1-phosphate transfer protein and intermediary regulator of phytoceramide levels. Cell Rep. 2015;6:388–99.
Zheng Z, Qamar SA, Chen Z, Mengiste T. Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 2006;48:592–605.
Frei dit Frey N, Garcia AV, Bigeard J, Zaag R, Bueso E, Garmier M, et al. Functional analysis of Arabidopsis immune-related MAPKs uncovers a role for MPK3 as negative regulator of inducible defences. Genome Biol. 2014;15:R87.
Navarro L, Dunoyer P, Jay F, Arnold B, Dharmasiri N, Estelle M, et al. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science. 2006;312:436–9.
Llorente F, Muskett P, Sanchez-Vallet A, Lopez G, Ramos B, Sanchez-Rodriguez C, et al. Repression of the auxin response pathway increases Arabidopsis susceptibility to necrotrophic fungi. Mol Plant. 2008;1:496–509.
Kant S, Bi YM, Zhu T, Rothstein SJ. SAUR39, a small auxin-up RNA gene, acts as a negative regulator of auxin synthesis and transport in rice. Plant Physiol. 2009;151:691–701.
Ding X, Cao Y, Huang L, Zhao J, Xu C, Li X, Wang S. Activation of the indole-3-acetic acid-amido synthetase GH3-8 suppresses expansin expression and promotes salicylate- and jasmonate-independent basal immunity in rice. Plant Cell. 2008;20:228–40.
Qi PF, Balcerzak M, Rocheleau H, Leung W, Wei Y-M, Zheng Y-L, Ouellet T. Jasmonic acid and abscisic acid play important roles in host-pathogen interaction between Fusarium graminearum and wheat during the early stages of fusarium head blight. Physiol Mol Plant Pathol. 2016;93:39–48.
Saito S, Hirai N, Matsumoto C, Chigashi H, Ohta D, Sakata K, Mizutani M. Arabidopsis CYP707As encode (+)-abscisic acid 8′-hydrolase, a key enzyme in the oxidative catabolism of abscisic acid. Plant Physiol. 2004;134:1239–49.
Lu Y, Li Y, Zhang J, Xiao Y, Yue Y, Duan L, Zhang M, Li Z. Overexpression of Arabidopsis molybdenum cofactor sulfurase gene confers drought tolerance in maize (Zea mays L.). PLoS One. 2013;8:e52126.
Xiong L, Ishitani M, Lee H, Zhu JK. The Arabidopsis LOS5/ABA3 locus encodes a molybdenum cofactor sulfurase and modulates cold stress- and osmotic stress-responsive gene expression. Plant Cell. 2001;13:2063–83.
De Lorenzo G, Castoria R, Bellincampi D, Cervone F. Fungal invasion enzymes and their inhibition. In: Carrol GC, Tudzinski P, editors. Plant Relationships. The Mycota (A comprehensive treatise on fungi as experimental systems for basic and applied research), Vol 5. Berlin: Springer; 1997. p. 61–83.
Idnurm A, Howett BJ. Pathogenicity genes of phytopathogenic fungi. Mol Plant Pathol. 2001;2:241–55.
Crawford MS, Kolattukudy PE. Pectate lyase from Fusarium solani f. sp. pisi: purification, characterization, in vitro translation of the mRNA and involvement in pathogenicity. Arch Biochem Biophys. 1987;258:196–205.
Kong L, Qiu X, Kang J, Wang Y, Chen H, Huang J, et al. A phytophthora effector manipulates host histone acetylation and reprograms defense gene expression to promote infection. Curr Biol. 2017;27:981–91.
Walley JW, Shen Z, McReynolds MR, Schmelz EA, Briggs SP. Fungal-induced protein hyperacetylation in maize identified by acetylome profiling. Proc Natl Acad Sci. 2018;115:210–5.
Hofstad AN, Nussbaumer T, Akhunov E, Shin S, Kugler KG, Kisler C, et al. Examining the transcriptional response in wheat FHB1 near-isogenic lines to Fusarium graminearum infection and deoxynivalenol treatment. Plant Genome. 2016;9:32.
Gou L, Hattori J, Fedak G, Balcerzak M, Sharpe A, Visendi P, et al. Development and validation of Thinopyrum elongatum expressed molecular markers specific for the long arm of chromosome 7E. Crop Sci. 2016;56:354–64.
IWGSC RefSeq v1.0 Reference Genome and Annotation. https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Annotations/v1.0/. Accessed 30 May 2017.
Fusarium graminearum reference genome. http://fungi.ensembl.org/, Release 35. Accessed 30 May 2017.
Phred - Quality Base Calling, http://www.phrap.com/phred/. Accessed 14 Apr 2016.
http://hannonlab.cshl.edu/fastx_toolkit/. Accessed 1 June 2017.
De Wit P, Pespeni MH, Ladner JT, Barshis DJ, Seneca F, Jaris H, et al. The simple fool’s guide to population genomics via RNA-Seq: an introduction to high-throughput sequencing data analysis. Mol Ecol Resour. 2012;12:1058–67.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Ensembl Plant: http://plants.ensembl.org/. Accessed 1 Nov 2017.
Tchagang AB, Gawronski A, Bérubé H, Phan S, Famili F, Pan Y. GOAL: a software tool for assessing biological significance of genes groups. BMC Bioinformatics. 2010;11:229.
Arabidopsis thaliana genome, Araport11, from TAIR: https://www.arabidopsis.org/. Accessed 1 Nov 2017.
Brachypodium distachyon genome, Bdistachyon_314, from JGI: https://phytozome.jgi.doe.gov/. Accessed 1 Nov 2017.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
Pan Y, Ouellet T, Phan S, Tchagang A, Fauteux F, Tulpan D. Digitization of trait representation in microarray data analysis on wheat infected by Fusarium graminearum. In: Proceedings of 2015 IEEE conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), August 12-15, 2015 Niagara falls, Canada.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods. 2001;25:402–8.
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034.
Acknowledgements
The authors would like to thank the International Wheat Genome Sequencing Consortium (IWGSC) for pre-publication access to wheat reference genome IWGSC RefSeq v1.0, the DNA Technologies Unit at the National Research Council Canada, Saskatoon, SK for performing the RNA-seq sequencing, Winnie Leung for the design of the NFXL1 primers, and three anonymous reviewers for constructive comments. This work was resulted from collaborations under Canadian Wheat Alliance.
Funding
This project was supported by Canadian Agriculture and Agri-Food Growing Forward 2 program (project J-000412) and Genomics Research and Development Initiative (projects J-000008 and J-001580) awarded to TO, “National Wheat Improvement Program” awarded to CM by Western Grains Research Foundation and Agriculture and Agri-Food Canada, and National Research Council Canada’s “Canadian Wheat Improvement” project A1–011652 awarded to YP. The funding bodies had no role in design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets generated during the current study are available in the NCBI GEO repository (GSE113128). The analyzed data are available as Additional files to this article.
Author information
Authors and Affiliations
Contributions
YP & TO performed functional genomics analysis and drafted the manuscript; TO designed the experiment and HR generated the data, YP & ZL performed RNA-seq data preprocessing, DEG analysis, gene orthology analysis, and compiled gene ontology data; FF performed clustering and principal component analysis. YW and CM contributed to data analysis and interpretation. All authors contributed to writing and approved final version for submission.
Corresponding authors
Ethics declarations
Ethics approval
Ethics approval is not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
The 28,961 differentially expressed wheat genes. This file also includes (B) statistics of differential expression feature extraction (DEFE) patterns associated with these genes, (C) values of FHB-related traits, (D) Pearson correlation between centroids (eigenvalues) of the clusters and the FHB-related traits. (XLSX 19261 kb)
Additional file 2:
Results of GO enrichment analyses. (XLSX 1033 kb)
Additional file 3:
FHB resistant genes across all three resistant genotypes (A), common between two genotypes (B), and unique to individual genotypes (C). (XLSX 172 kb)
Additional file 4:
Enrichment indices of gene groups in various gene populations: DEGs, FHB-resistant, FHB-susceptible, up- and down-regulated by Fg, networks, key hub genes. (XLSX 2244 kb)
Additional file 5:
Annotation of the six genes currently annotated to encode erect panicle 2 protein need to be updated. Two groups of genes with annotation of “Erect panicle 2 protein” in the “Human-Readable-Description” provided with IWGSC RefSeq v1.0. Annotation of these six genes needs to be revisited. (PDF 228 kb)
Additional file 6:
The 57 Pathogenesis-Related genes. (XLSX 45 kb)
Additional file 7:
The 542 phytohormone pathway genes. (XLSX 419 kb)
Additional file 8:
The chromosomal distribution of various gene populations. (XLSX 31 kb)
Additional file 9:
The 8811 differentially expressed pathogen genes. This file also include (B) statistics of differential expression feature extraction (DEFE) pattern associated with each gene, (C) values of FHB-related traits, and (D) Pearson correlation between centroids of the clusters and the FHB-related traits. (XLSX 5432 kb)
Additional file 10:
Gene ontology annotations compiled from three versions of the wheat genome: IWGSC Survey genome 2.2, TGACv1 and IWGSC-RefSeqv1.0. (XLSX 18572 kb)
Additional file 11:
Primers used for RT-qPCR analyses in this study. (XLSX 13 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pan, Y., Liu, Z., Rocheleau, H. et al. Transcriptome dynamics associated with resistance and susceptibility against fusarium head blight in four wheat genotypes. BMC Genomics 19, 642 (2018). https://doi.org/10.1186/s12864-018-5012-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5012-3