
Abstract
Background
Aspergillus arachidicola is an aflatoxigenic fungal species, first isolated from the leaves of a wild peanut species native to Argentina. It has since been reported in maize, Brazil nut and human sputum samples. This aflatoxigenic species is capable of secreting both B and G aflatoxins, similar to A. parasiticus and A. nomius. It has other characteristics that may result in its misidentification as one of several other section Flavi species. This study offers a preliminary analysis of the A. arachidicola genome.
Results
In this study we sequenced the genome of the A. arachidicola type strain (CBS 117610) and found its genome size to be 38.9 Mb, and its number of predicted genes to be 12,091, which are values comparable to those in other sequenced Aspergilli. A comparison of 57 known Aspergillus secondary metabolite gene clusters, among closely-related aflatoxigenic species, revealed nearly half were predicted to exist in the type strain of A. arachidicola. Of its predicted genes, 691 were identified as unique to the species and 60% were assigned Gene Ontology terms using BLAST2GO. Phylogenomic inference shows CBS 117610 sharing a most recent common ancestor with A. parasiticus. Finally, BLAST query of A. flavus mating-type idiomorph sequences to this strain revealed the presence of a single mating-type (MAT1–1) idiomorph.
Conclusions
Based on A. arachidicola morphological, genetic and chemotype similarities with A. flavus and A. parasiticus, sequencing the genome of A. arachidicola will contribute to our understanding of the evolutionary relatedness among aflatoxigenic fungi.
Similar content being viewed by others


Background
As each new species that is added to Aspergillus section Flavi is characterized, it seems the numbers of species that are capable of producing B and G aflatoxins are increasing. In the last decade, seven novel B + G producing species have been characterized [1,2,3,4]. The potency of B aflatoxins is widely considered to be greater than that of G aflatoxins, but both secondary metabolites, especially aflatoxins B1 and G1, are regarded as carcinogenic and their purpose or function in nature has yet to be fully understood [5].
Aspergillus arachidicola (CBS 117610) was first sampled from an Arachis glabrata leaf in the Corrientes province of Argentina, and it was characterized and associated with Aspergillus section Flavi in 2008 [1]. It has characteristics similar to other section Flavi species [1], which may have resulted in repeated misidentifications because it was sampled in the same environment, exhibited similar morphological characteristics, and produced similar mycotoxins to other well-known and characterized species. Since its first association in South America with a single host (peanut plants), A. arachidicola has been reported in several hosts/environments such as a Brazil nut shell [6], a clinical sample from a respiratory tract biopsy [7], and a maize kernel [8]. All occurrences of A. arachidicola have been reported in similar regions of South America. A North American strain, previously reported as A. arachidicola isolated from a diseased alkali bee in 2008 [1], was eventually re-characterized as A. pseudonomius by Varga et al. in 2011 [2].
The phylogenetic association and predominating extrolite profile for A. arachidicola are similar to A. parasiticus, yet it has macro-morphological features (e.g. colony texture and color) that are more similar to A. flavus [1]. Interestingly, many of its reported characteristics, such as conidial ornamentation and seriation, were reported to be similar to both A. flavus and A. parasiticus [1]. Pildain and co-workers’ morphological observations for A. arachidicola correlate to a report of hybrid offspring resulting from experimental crosses between A. flavus and A. parasiticus parent strains, for which many features were considered “A. flavus-like, A. parasiticus-like or intermediate between the two species” [9]. Other newly characterized species have striking similarities to A. flavus and one or more B + G producing species [1,2,3,4, 10]. Perhaps A. arachidicola is one of several naturally-occurring hybrids to be sampled and characterized as a new species. In this study, we sequenced the genome of this aflatoxigenic fungus, performed genomic comparisons with closely-related aflatoxigenic species, and determined its mating-type to build our understanding of how aflatoxin production has evolved and the evolutionary relationships of aflatoxigenic fungi, especially when reports of phenomena such as inter-specific hybridization and horizontal gene transfer are taken into consideration.
Results
Genome information for A. arachidicola type strain CBS 117610
Our sequencing coverage for the A. arachidicola type strain was ~ 20×. This gave us 4 million reads with a median read length of 352 bp, 674 contigs greater than 1000 bp, and an N50 length of 127,297 bp. Raw sequence reads have been accessioned in the NCBI Sequence Read archive under the number SRR5569329. The A. arachidicola genome assembly is 38.9 Mb in size and includes 12,091 protein-encoding genes (Table 1). Additional sequencing quality statistics and predicted genomic information for this type strain are also shown in Table 1. The completeness of the assembly is relatively high as measured by a CEGMA percent completeness score of 95% and a BUSCO percent completeness score of 93% [11].
Genomic comparisons with other sequenced aflatoxigenic species
A comparison of genomes was undertaken involving the A. arachidicola type strain with those of closely-related aflatoxigenic species. The genome of A. arachidicola is most similar to the 39.82 Mb genome of the SU-1 type strain of A. parasiticus, which is larger than many other sequenced Aspergilli [12,13,14,15,16]. Since A. arachidicola is reported to share similarities with A. flavus and A. parasiticus, Table 2 includes various morphological, toxigenic and genomic comparisons for these three species, for which there are several observable similarities.
Sclerotium production in A. arachidicola type strain CBS 117610
So far, there is no reported evidence that A. arachidicola produces sclerotia. Not all strains of A. flavus are capable of producing sclerotia [17], while sclerotium production in A. parasiticus is said to be an occasional occurrence [18]. Olarte and co-workers [9] reported diminished sclerotium production among their observed A. flavus x A. parasiticus hybrid offspring. Several genes have been reported to promote development of sclerotia in various fungi, including Aspergilli. For example, Sclerotinia sclerotiorum ssp1 and ssp2 orthologs, identified in A. flavus and A. oryzae as sspA and sspB [19], were also found in CBS 117610. Another putative sclerotium production ortholog was found that corresponds to the S. sclerotiorum pac1 gene [20]. Also found in A. arachidicola was a putative sclR ortholog, originally described in A. oryzae [21], as well as fluP and aswA orthologs, originally described for A. flavus [22, 23]. None of these putative orthologs appeared transcriptionally broken in A. arachidicola. Even the putative velvet gene (veA) homolog in A. arachidicola, reported as a regulator of sclerotium development for A. parasiticus [24], exhibited 96.5 and 99.5% amino acid sequence identity when compared to A. flavus and A. parasiticus, respectively.
Secondary metabolite gene clusters in A. arachidicola type strain CBS 117610
Since A. arachidicola forms part of a group of species known for aflatoxin production, a primary aim with this genome sequence is to investigate secondary metabolite clusters, specifically those involved in the production of mycotoxigenic compounds. The number of secondary metabolite (SM) clusters within A. arachidicola is inferred to be 56 based on analysis using the Secondary Metabolite Unique Regions Finder (SMURF) and 72 based on analysis using the Antibiotics-Secondary Metabolite Analysis Shell (antiSMASH) (Table 3), while closely-related A. parasiticus contains 61 and 89 SM clusters according to SMURF and antiSMASH, respectively. The discrepancies observed for SM counts relates to the antiSMASH algorithm, which is designed to predict more than 40 types of gene clusters (e.g., Type 1–3 PKS, NRPS, terpenes, etc.) [25], thus it is considered to provide a more comprehensive list of cluster predictions than SMURF. In contrast to antiSMASH, SMURF conducts cluster predictions for five general SM cluster categories [26].
One very important SM cluster involves the pathway to synthesize the carcinogenic compound known as aflatoxin. The A. arachidicola type strain is reported to produce both B and G aflatoxins [1], a genotype confirmed by the absence of the deletion in the norB/cypA region. Based on our observations, its aflatoxin gene cluster is similar in size (68 kb) and contains the same 25 genes known to comprise the aflatoxin pathway, in the same orientation, as other B + G producing species, such as A. bombycis, A. nomius and A. parasiticus (Additional file 1: Figure S1) [16]. Another important toxic secondary metabolite is cyclopiazonic acid (CPA), which has been associated with aflatoxin producing species [27], although it was first reported to be produced by Penicillium cyclopium [28]. Previous characterization of the CPA biosynthesis cluster for several A. flavus strains revealed three genes that are responsible for its production [29]. Although there are no reports of A. arachidicola producing CPA, BLAST queries of the nucleotide sequences, and subsequent comparison to the protein sequences, for A. flavus (AF36) maoA, dmaT and pks-nrps genes yielded sequence identities (93, 94 and 92%, respectively) within the A. arachidicola genome. Its CPA cluster spans 15,918 bp, adjacent to its aflatoxin gene cluster, separated by a genomic distance of 7766 bp. Both the aflatoxin and CPA genomic regions can be found on contig_10 of the A. arachidicola genome under GenBank accession number NEXV01000673. The three CPA genes in A. arachidicola are also oriented the same as those in the AF36 strain (Additional file 1: Figure S1). A candidate A. flavus biocontrol strain known as K49 has a substitution mutation in its pks-nrps gene at amino acid 703 that changes a serine (TCA) to a stop codon (TGA) and truncates 3202 amino acids [30]. Closer examination of these genes in A. arachidicola, compared to those from the functional CPA cluster in the AF36 biocontrol strain, as well as the non-functional cluster of K49, revealed a deletion mutation within the 1376 bp dmaT gene. This single deletion, at nucleotide 474, introduces a frameshift that alters the translation of downstream protein sequence. This shift in the translational reading frame results in generation of a stop codon at nucleotide position 568. Therefore, this shortened dmaT protein correlates with loss-of-function (Additional file 1: Figure S1). BLAST query of the CPA nucleotide and amino acid sequences to A. parasiticus yielded no evidence of putative homologs, which supports the lack of CPA production in A. parasiticus.
No known secondary metabolite gene clusters were predicted that relate to the production of other toxic secondary metabolites reportedly produced by A. arachidicola [1], such as aspergillic acid, chrysogine, oryzaechlorin and parasiticolide. First discovered in Penicillium chrysogenum, the six genes that are reported to comprise the chrysogine cluster have been very recently accessioned and published for several species, including A. nomius [31]. Only five chrysogine genes were reported to exist in Wollenberg’s examined A. nomius strain, and these were BLAST queried to the A. arachidicola genome. We found all of the putative homologs present on the same contig, and upon assembly they were found to be oriented similarly to those in A. nomius. The only exception was the direction of transcription for the chry6 homolog in A. arachidicola, which is the reverse of the chry6 gene in A. nomius. The homolog for the chry4 gene, reportedly absent in A. nomius [31], also could not be located within the A. arachidicola type strain genome. The characterization of the putative genes comprising the aspergillic acid cluster of A. flavus has yet to be published (Jeff Cary, personal communication), but the putative genes have been deposited in GenBank (accession numbers XM_002373770-XM_002373777). BLAST query of the A. flavus gene, and also the respective translated protein, sequences to A. arachidicola revealed that all the putative homologs are present on the same contig and are located within a 17,851 bp stretch of nucleotide sequence.
Interestingly, clusters were predicted for several compounds that are most often associated with the genus Penicillium, such as citrinin, patulin and penicillin (Table 4), yet these three mycotoxins have also been reported for some Aspergilli [32,33,34]. A quick BLAST query of patulin cluster genes (based on nucleotide and amino acid sequences from A. clavatus) revealed some putative homologs in A. arachidicola, but not organized into a cluster. For this reason we cannot support the prediction of the patulin cluster in A. arachidicola.
Another predicted cluster in A. arachidicola relates to the compound ustiloxin B, which is both a mycotoxin and a phytotoxin that was first discovered in the rice pathogen, Ustilaginoidea virens [35]. This particular secondary metabolite has been reportedly produced by A. flavus [36], but in our antiSMASH analysis the ustiloxin B cluster was not predicted to exist in A. flavus NRRL 3357. However, SMURF has identified the ustiloxin B cluster in NRRL 3357 as cluster #31 (Jeff Cary, personal communication). Whether or not these SM clusters are remnants from a shared ancestor between distant fungi, or if they were inherited through horizontal gene transfer, is unknown but worthy of further studies.
Secondary metabolite cluster comparisons with other sequenced aflatoxigenic species
There are a number of known secondary metabolite gene clusters that have been reported for aflatoxigenic Aspergillus species. “Known” clusters refers to those that antiSMASH outputs as having homology in the GenBank file. A comparison of 57 known clusters, between A. arachidicola and its most closely-related species, A. parasiticus, revealed 20 of them as predicted to exist in both species (Table 4). Clusters predicted by antiSMASH to exist in only A. arachidicola and A. parasiticus were those associated with the production of huperzine A and ustiloxin B. Huperzine A is a compound that is being studied for its effectiveness against cognitive decline in elderly patients [37]. There were 10 known clusters predicted to be in A. parasiticus that were not predicted for A. arachidicola, and seven clusters predicted to exist in A. arachidicola were not predicted to exist in A. parasiticus. Of those seven predicted not to exist in A. parasiticus, five were also found in A. flavus. The only other cluster predicted to exist in A. arachidicola and one of the other examined species is affiliated with the production of Yanuthone D, a compound with antibiotic and antifungal properties [38], which was also predicted to exist in A. nomius. The cluster associated with the production of notoamide and stephacidin compounds was predicted to exist only in A. arachidicola. The notoamides and stephacidins are prenylated indole alkaloids that are of interest to biomedical researchers for many potentially beneficial properties that may include anti-tumor, insecticidal and antibacterial properties [39]. One of the predicted clusters with 100% identity to those in the other examined species were those for the production of aflavarin, a metabolite with anti-insectan properties [40] that has recently been associated with sclerotium production in A. flavus [23, 41].
Gene ontology for A. arachidicola type strain CBS 117610
For the 691 genes determined to be unique to A. arachidicola, among four closely related species (A. parasiticus, A. flavus L, A. flavus S and A. nomius), the Fisher’s Exact test showed that 19 of the 32 most significantly-enriched Gene Ontology (GO) terms were associated with molecular function, 12 were associated with biological process, and only one associated with cellular components. However, the 91 sequences associated with the biological process of oxidation reduction were observed having the highest enrichment levels (adjusted p-value = 9.19E-4) of all the GO terms/categories (Table 5). The second highest enrichment (second-lowest p-value) was shared by sequences related to the molecular functions of oxidoreductase activity (n = 23) and heme binding (n = 25), both having an adjusted p-value of 2.58E-2.
Comparisons of sequenced Aspergillus genomes
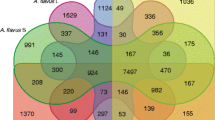
Of its 12,091 predicted genes, orthology analysis (Fig. 1) revealed that A. arachidicola shares 8106 genes with four other species examined from section Flavi. The lowest number of unique genes shared among more than one species with A. arachidicola was 26, and included A. nomius and A. flavus L. The 691 genes inferred as unique to A. arachidicola are the lowest number for this group, since A. flavus S (AF70), A. nomius (NRRL 13137), A. parasiticus (SU-1) and A. flavus L (NRRL 3357) harbor more unique genes (790, 1036, 1238 and 1812, respectively). The highest quantity of shared unique genes between A. arachidicola and any of the other species examined is with A. parasiticus (n = 332), which means that both species share the same 332 genes that are not observed in the other examined species. The lowest quantity of shared genes is with A. flavus L-type (n = 70). Coincidentally, A. arachidicola shares 100 genes (the second highest quantity) with the other B + G producing strain examined, A. nomius. The number of genes shared among all three B + G species examined, when compared to those shared by all five species, is less than 2.5% (n = 201).

Venn diagram including several species from section Flavi. A Venn diagram quantifies unique and orthologous genes for multiple species in section Flavi. This diagram of overlapping shapes includes species names, gene counts, and color-shading: CBS 117610 (A. arachidicola) is shaded blue, SU-1 (A. parasiticus) is shaded red, AF70 (A. flavus S) is shaded brown, NRRL 3357 (A. flavus L) is shaded yellow, and NRRL 13137 (A. nomius) is shaded green
Phylogenomic comparisons allowed us to infer a species tree for A. arachidicola with other Aspergillus species, and the outgroup taxa Penicillium chrysogenum. Our findings indicate that this species shares a most recent common ancestor with A. parasiticus (Fig. 2), which supports the findings of Pildain et al. [1]. This common ancestor of A. arachidicola and A. parasiticus diverged from the most recent common ancestor of the B-producing morphotypes of A. flavus, both of which share a common ancestor. Prior to the speciation event that gives rise to A. flavus, A. parasiticus and A. arachidicola is an event that that split their most recent common ancestor from the predecessor of A. nomius and A. bombycis.

Phylogenomic tree of sequenced Aspergillus species. This tree includes inferred patterns of ancestry by phylogenomic comparisons of multiple Aspergillus species (A. fumigatus, A. nidulans, A. bombycis, A. nomius, A. arachidicola, A. parasiticus, A. flavus L and A. flavus S) with Penicillium chrysogenum as the outgroup taxa
A. arachidicola CBS 117610 contains a MAT1–1 idiomorph
The A. arachidicola type strain contains a single MAT1–1 gene (Fig. 3), which means this strain is heterothallic (self-infertile). Previous research reported a possible heterothallic existence for most of the species in section Flavi, with each species containing a single mating-type idiomorph [42]. The ability of this species to outcross has not yet been reported. Other heterothallic Aspergillus species, such as A. flavus and A. parasiticus, have a mating-type gene flanked by two conserved genes in close proximity: one for DNA lyase (APN1) and one for cytoskeleton assembly control (SLA2). The flanking of the MAT locus by these genes was recently reported to represent an ancestral configuration in fungi [43]. Although these two genes are consistently found to flank the MAT locus in fungi, the genomic distances separating them may vary [15, 16, 42]. For the MAT1–1 gene in A. arachidicola, the genomic distance to the APN1 gene was determined to be 1831 bp. Both the MAT1–1 and APN1 genes are located on contig_148, which in GenBank has accession number NEXV01000431. The genomic distance separating SLA2 from the MAT idiomorph could not be determined because the SLA2 gene was located on a separate contig (contig_382; GenBank accession NEXV01000197) with no overlap. Whether this is because in A. arachidicola it is much farther between these genes, or because of a data quality issue, is unknown. The chromosomal location of the mating-type locus in A. flavus and A. parasiticus is reported to be Chromosome VI [42]. Although it has been reported that the mating-type locus in heterothallic fungi will reside in similar chromosomal locations [44], this has not yet been confirmed for A. arachidicola. Comparison of the A. arachidicola MAT1–1 gene’s amino acid sequence to other MAT1–1 gene sequences from closely-related species (A. flavus, A. parasiticus, A. nomius, A. alliaceus and A. fumigatus) revealed 45.5% overall identity, and its identity to both A. flavus and A. parasiticus was 96% (data not shown). There were only two amino acid substitutions that distinguished A. arachidicola MAT1–1 sequence from A. flavus and A. parasiticus, and they were both highly conserved. Of the four substitutions that distinguished A. arachidicola and A. parasiticus from A. flavus, one was highly conserved, one was semi-conserved, and two were non-synonymous. The five substitutions that separated A. arachidicola and A. flavus from A. parasiticus included two highly conserved and three non-synonymous. Across all species examined, 30% of the 371 aligned amino acids showed synonymous substitutions, while 24.5% of amino acid substitutions were non-synonymous.

The mating-type locus of A. arachidicola CBS 117610. The schematic diagram for its mating-type locus shows the orientation and physical distance shared among the MAT1–1 gene and the adjacent upstream and downstream genes. The black vertical line in MAT1–1 represents the approximate location of a 52 bp intron
Discussion
Genomic comparisons with other sequenced aflatoxigenic species
Species within section Flavi have genomes that are reported to be larger than other aspergilli, which may be the result of gene acquisition [13], or a large percentage of repetitive DNA, as has been suggested for A. oryzae [45]. The A. arachidicola genome is predicted to contain 1.65% repetitive DNA content (Table 2), which is larger than the other examined species, but does not explain all of the differences in genome sizes. For example, A. arachidicola has more repetitive DNA than A. parasiticus, but A. parasiticus has a 1 Mb larger genome. The reason for A. parasiticus and A. arachidicola having noticeably larger genomes is unclear, but could relate to either of the aforementioned reasons. Further research is needed to support or refute these possibilities. With regard to the aflatoxin gene cluster, A. flavus has a large deletion in the norB/cypA region that reportedly prevents synthesis of G aflatoxins [46]. Perhaps the smaller Aspergillus genomes are the result of numerous genomic deletions, which do not exist in G-producing species such as A. parasiticus. Sequencing the genomes of more B + G aflatoxin producing species may offer more insights into this.
Sclerotium production in A. arachidicola type strain CBS 117610
We examined A. arachidicola homologs for several genes that have been reported to regulate sclerotium production in Aspergilli or other fungi, and found that none of them should be non-functional in CBS 117610. Either the true gene associated with sclerotium production has yet to be described, or there are several genes necessary to stimulate sclerotium production in these fungi. Another possibility is that we have yet to determine the environmental conditions necessary to stimulate sclerotium production in this fungus.
Secondary metabolite gene clusters in A. arachidicola type strain CBS 117610
We found an intact AF gene cluster that is oriented, and of similar length/composition, to those of other B + G aflatoxin species. We also found that the clustered genes responsible for the production of CPA are present in this fungus, although a single deletion in one of those genes (dmaT) renders the pathway non-functional. If a random recombination event could replace this non-functional dmaT gene in CBS 117610 with a functional dmaT, then A. arachidicola could become an AF and CPA producing species.
Using antiSMASH, we were unable to predict the presence of clustered genes for several mycotoxins reportedly produced by A. arachidicola, such as those responsible for the production of chrysogine. Although chrysogine production was reported for some of the sampled A. arachidicola strains, Pildain et al. [1] did not specify if CBS 117610 produced this compound. Therefore, any inferences on the genes necessary for chrysogine production in A. arachidicola would involve sequencing its relative genomic region in strains or closely-related species that are known to produce it. No complete clusters could be detected via BLAST query and alignment analysis. Since most mycotoxins are the products of gene clusters, it will not be possible to determine the identity of the gene clusters associated with these secondary metabolites until they are experimentally established. Software limitations could also group smaller gene clusters with larger, adjacent clusters. For example, during our antiSMASH analysis the (smaller) CPA cluster was merged with the much larger and adjacent aflatoxin cluster. The antiSMASH output reports the best BLAST hit of any known cluster for each predicted cluster, so the merging of these two clusters initially obscured the presence of the CPA cluster. Other known SM clusters, predicted for other B + G producing species were also predicted to exist in A. arachidicola, which were not reported for this species in its first characterization. A complete metabolic profile for A. arachidicola will be necessary to determine the presence of any of the compounds reportedly synthesized via these predicted SM clusters.
Gene ontology for A. arachidicola type strain CBS 117610
In our GO analysis, we found the highest levels of enrichment for the biological process of oxidation reduction. Oxidation-reduction reactions (i.e. redox) are reported to correlate with several important facets of fungal biology such as cell differentiation, virulence and growth [47]. Molecular functions for oxidoreductase activity and heme binding were also found to be highly enriched in our GO analysis. Oxidoreductase is an enzyme that is linked with oxidation reduction reactions [48], and heme binding involves fungal acquisition of iron from the host in order to facilitate its survival and growth [49].
Comparisons of sequenced Aspergillus genomes
Our orthology analysis predicted that A. arachidicola CBS 117610 contains the least amount of unique genes among the species examined, and that there are a greater number of shared unique genes between A. arachidicola and A. parasiticus. The more aflatoxigenic species are examined and included in orthology analysis, the likelihood increases of discerning genes that could improve species delimitation via diagnostic PCR. What has yet to be determined is whether or not “unique” genes are wholly unique. For example, if the shuffling of genetic material (i.e. recombination) results in genes that are a composite from two or more different genomes, then orthology analysis might consider them unique when they are merely lacking identity with other examined species. More thorough comparisons of the unique genes in sequenced genomes of closely related species will either support or refute this. Alternatively, these unique genes could relate to certain gene clusters that are found in A. arachidicola but not in other closely related species. Our phylogenomic inferences support previous reports that A. arachidicola and A. parasiticus share a most recent common ancestor [1]. This may correlate with their sharing the greatest number of unique genes. Phylogenomics involving even more aflatoxigenic species will better refine our understanding of the evolution of toxic secondary metabolite clusters, and offer insights regarding the potential impacts of recombination on these clusters within mycotoxigenic Aspergillus species.
A. arachidicola CBS 117610 contains a MAT1–1 idiomorph
We found that this strain has the MAT1–1 mating-type, which could account for a lack of sclerotium production in the A. arachidicola type strain. It remains to be seen whether the other sampled A. arachidicola isolates are of the same mating type. According to Horn et al. [50], there may be roles affiliated with each mating type, and that strains capable of producing conidia and sclerotia are hermaphroditic. If conidia provide the donor (i.e. paternal) genetic material, and sclerotia provide the receptor (i.e. maternal) genetic material, then perhaps CBS 117610 is a true “male” strain since it fails to produce sclerotia. The mating types of other sampled A. arachidicola isolates, specifically MAT1–2 strains, would need to be characterized, and additional comparisons made, to support or refute this. Further, if no A. arachidicola strains are found that produce sclerotia, perhaps inter-specific mating experiments involving A. arachidicola conidia and sclerotia from closely related species (A. flavus or A. parasiticus) might reveal its reproductive potential. Experimental crosses involving A. flavus and A. parasiticus proved that inter-specific hybridization is possible [9].
Conclusions
Aspergillus arachidicola is one of several recently-characterized fungi that seems to share morphological, genomic and chemotype similarities to several other historical Aspergillus species. It contains several secondary metabolite gene clusters (functional as well as non-functional) that warrant further study. Obviously, there is more research required to support or refute the potential for A. arachidicola to be a naturally-occurring hybrid species. For example, there would need to be more A. arachidicola isolates examined. Other than the type strain, only six were reported by Pildain et al. [1]. However, there have been few additional reports of A. arachidicola isolates sampled [6,7,8]. Hybrid organisms can suffer fitness disadvantages for adaptability to certain niches, based on inherited traits from either parent [51]. Inter- and intra-specific mating experiments would need to be undertaken to determine the fecundity of A. arachidicola, since it has been reported that hybrid offspring often suffer infertility [9, 51]. And thorough comparisons (e.g. SM production) would need to be made between the inter-specific hybrids reported by Olarte et al. [9] and multiple A. arachidicola strains. Given the increasing numbers of reports that highlight sexual potential (both intra- and inter-specific) of fungi, as well as evidence of horizontal gene transfer, it is important not to discount the potential impacts these phenomena may have on speciation and the evolutionary relatedness of fungal organisms.
Methods
Genome sequencing and annotation of A. arachidicola CBS 117610
The CBS 117610 genome was sequenced using a Personal Genome Machine (PGM) from Life Technologies (Grand Island, New York). A loopful of CBS 117610 spores were inoculated in Yeast Extract Sucrose (YES) liquid medium (Sigma-Aldrich, Saint Quentin-Fallavier, France), and kept in agitation in an orbital incubator at 170 rpm, at 27 °C, for five days. DNA extraction was performed by grinding a portion of mycelium in a 5 ml mortar on ice, followed by its addition to 5.5 ml lysis buffer (20 mM Tris-HCl, 250 mM NaCl, 25 mM EDTA 0.5 M pH = 8, 1% SDS). The content was transferred to a 15 ml tube and 12.5 μl of Proteinase K (20 mg/ml) (EMD Millipore, Billerica, MA) were added, then the samples were incubated for 30 min up to 1 h at 37 °C, followed by an additional 10 min at 65 °C. Thereafter, one volume of phenol/chloroform (7:3, v:v) was added, and samples were vigorously shaken and centrifuged at 3080 x g for one hour. Supernatant was recovered into a new tube, and 6 μl RNAse A (100 mg/ml) (Serva electrophoresis GmbH, Heidelberg, Germany) were added prior to incubation for 2–3 h at 37 °C. Next, one volume of chloroform was added and centrifuged at 3080 x g for 10 min. Supernatant was recovered into a new tube, and one volume of isopropanol was added. At this point, samples were softly shaken for 2 h in a horizontal shaker and kept overnight at 4 °C. The next day, samples were centrifuged at 13,000 x g for 30 min. The supernatant was eliminated and the pellet carefully washed with 300 μl of 70% ethanol, then centrifuged at 13,000 x g for 15 min, followed by a gentle aspiration of the supernatant. Finally, the pellet was re-suspended with 30 μl of pure water. DNA samples were quantified using a NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE, USA). Library preparation and sequencing were conducted according to previously reported protocols [15, 16]. A total of 4.1 million reads were used for genome assembly with SPAdes (version 3.9.0) [52]. BUSCO (version 1.22) [11] was used to train Augustus with ortholog information from the “fungi” lineage. Annotation software pipelines such as MAKER [53], GeneMark [54], Augustus [55], as well as the Swiss-Prot database for detecting protein homology, were used as previously described [15, 16]. A transcriptome assembly from A. parasiticus, made using Trinity, was also used as EST evidence within MAKER. We then used NCBI’s Genome Annotation Generator [https://github.com/genomeannotation/GAG] for annotation format conversion, and deposited the annotation under BioProject project number PRJNA385482.
Genomic comparisons among Aspergillus species
The antiSMASH [27] and SMURF [28] programs were used to predict SM clusters in A. arachidicola. Phylogenomic analysis was performed by detecting orthologous proteins within other fungi using Proteinortho (version 5.13) [56], aligning them using MUSCLE (version 3.8.31) [57], and concatenating them into a 2 Mb amino acid alignment using GBLOCKS (version 0.91) [58]. We inferred our phylogenetic tree using RAxML-HPC (version 8.1.17) [59] using data and conditions as previously described [15, 16]. Gene Ontology term enrichment was performed using goseq (version 1.28.0) [60] using the “hypergeometric” option. Repetitive elements were identified using RepeatMasker (version 4.0.7) [61] with the RepBase [62] library and species set to fungus.
The sclerotium-related genes involved BLAST queries of nucleotide and amino acid sequences to those of A. arachidicola. All SM gene cluster comparisons involved BLAST queries to the A. arachidicola genome, then they were aligned to its contig sequences for distance mapping. Similarly, the mating-type (MAT) locus comparisons were performed by BLAST query of A. flavus MAT, APN and SLA genes to the A. bombycis genome. Distance mapping between the examined genes/clusters were performed using Sequencher software (Gene Codes Corporation, Ann Arbor, MI).
Abbreviations
- antiSMASH:
-
Antibiotics & Secondary Metabolite Analysis Shell
- APN1:
-
DNA lyase gene
- BLAST:
-
Basic local alignment search tool
- bp:
-
Base pairs
- BUSCO:
-
Benchmarking Universal Single-Copy Orthologs
- CBS:
-
Centraalbureau voor Schimmelcultures
- CEGMA:
-
Core Eukaryotic Genes Mapping Approach
- CPA:
-
Cyclopiazonic acid
- GO:
-
Gene Ontology
- JCVI:
-
J. Craig Venter Institute
- JGI:
-
Joint Genome Institute
- MAT:
-
Mating-type
- NCBI:
-
National Center for Biotechnology Information
- NRPS:
-
Non-ribosomal peptide synthase
- NRRL:
-
Northern Regional Research Laboratory
- OMST:
-
O-methylsterigmatocystin
- PCR:
-
Polymerase Chain Reaction
- PGM:
-
Personal genome machine
- PKS:
-
Polyketide synthase
- RAxML:
-
Randomized axelerated maximum likelihood
- SLA2:
-
Cytoskeleton assembly control gene
- SM:
-
Secondary Metabolite
- SMURF:
-
Secondary metabolite unique regions finder
References
Pildain MB, Frisvad JC, Vaamonde G, Cabral D, Varga J, Samson RA. Two novel aflatoxin-producing Aspergillus species from Argentinean peanuts. Int J Syst Evol Microbiol. 2008;58:725–35.
Varga J, Frisvad JC, Samson RA. Two new aflatoxin producing species, and an overview of Aspergillus section Flavi. Stud Mycol. 2011;69:57–80.
Gonçalves SS, Stchigel AM, Cano JF, Godoy-Martinez PC, Colombo AL, Guarro J. Aspergillus novoparasiticus: a new clinical species of the section Flavi. Med Mycol. 2012;50:152–60.
Soares C, Rodrigues P, Peterson SW, Lima N, Venâncio A. Three new species of Aspergillus section Flavi isolated from almonds and maize in Portugal. Mycologia. 2012;104:682–97.
Santini A, Ritieni A. Aflatoxins: risk, exposure and remediation. In: Razzaghi-Abyaneh M, editor. Rijeka: Aflatoxins - recent advances and future prospects: InTech; 2013. https://doi.org/10.5772/52866.
Gonçalves JS, Ferracin LM, Carneiro Vieira ML, Iamanaka BT, Taniwaki MH, Pelegrinelli Fungaro MH. Molecular analysis of Aspergillus section Flavi isolated from Brazil nuts. World J Microbiol Biotechnol. 2012;28:1817–25.
Negri CE, Gonçalves SS, Xafranski H, Bergamasco MD, Aquinob VR, Castroc PTO, Colomboa AL. Cryptic and rare Aspergillus species in Brazil: prevalence in clinical samples and in vitro susceptibility to triazoles. J Clin Microbiol. 2014;52:3633–40.
Viaro HP, da Silva JJ, de Souza Ferranti L, Bordini JG, Massi FP, Pelegrinelli Fungaro MH. The first report of A. novoparasiticus, A. arachidicola and A. pseudocaelatus in Brazilian corn kernels. Int J Food Microbiol. 2017;243:46–51.
Olarte RA, Worthington CJ, Horn BW, Moore GG, Singh R, Monacell JT, Dorner JW, Stone EA, Xie D-Y, Carbone I. Enhanced diversity and aflatoxigenicity in interspecific hybrids of Aspergillus flavus and Aspergillus parasiticus. Mol Ecol. 2015;24:1889–909.
Frisvad JC, Skouboe P, Samson RA. Taxonomic comparison of three different groups of aflatoxin producers and a new efficient producer of aflatoxin B1, sterigmatocystin and 3-O-methylsterigmatocystin, Aspergillus rambellii sp. nov. Syst Appl Microbiol. 2005;28:442–53.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2.
Linz JE, Wee J, Roze LV. Aspergillus parasiticus SU-1 genome sequence, predicted chromosome structure, and comparative gene expression under aflatoxin-inducing conditions: evidence that differential expression contributes to species phenotype. Eukaryot Cell. 2014;13:1113–23.
Machida M, Asai K, Sano M, Tanaka T, Kumagai T, Terai G, Kusumoto K-I, Arima T, Akita O, Kashiwagi Y, Abe K, Gomi K, Horiuchi H, Kitamoto K, Kobayashi T, Takeuchi M, Denning DW, Galagan JE, Nierman WC, Yu J, Archer DB, Bennett JW, Bhatnagar D, Cleveland TE, Fedorova ND, Gotoh O, Horikawa H, Hosoyama A, Ichinomiya M, Igarashi R, et al. Genome sequencing and analysis of Aspergillus oryzae. Nature. 2005;438:1157–61.
Payne GA, Nierman WC, Wortman JR, Pritchard BL, Brown D, Dean RA, Bhatnagar D, Cleveland TE, Machida M, Yu J. Whole genome comparison of Aspergillus flavus and A. oryzae. Med Mycol. 2006;44:9–11.
Moore GG, Mack BM, Beltz SB. Genomic sequence of the aflatoxigenic filamentous fungus Aspergillus nomius. BMC Genomics. 2015;16:551.
Moore GG, Mack BM, Beltz SB, Gilbert MK. Draft genome sequence of an aflatoxigenic Aspergillus species, A. bombycis. Genome Biol Evol. 2016;8:3297–300.
Hua ST, Mcalpin CE, Chang P-K, Sarreal SL. Characterization of toxigenic and atoxigenic Aspergillus flavus isolates from pistachio. Mycotoxin Res. 2012;28:67–75.
Klich MA. Identification of common aspergillus species. Centraalbureau voor Shimmelcultures: Utrecht; 2002.
Li M, Rollins JA. The development-specific protein (Ssp1) from Sclerotinia sclerotiorum is encoded by a novel gene expressed exclusively in sclerotium tissues. Mycologia. 2009;101:34–43.
Rollins JA. The Sclerotinia sclerotiorum pac1 gene is required for sclerotial development and virulence. Mol Plant-Microbe Interact. 2003;16:785–95.
Jin FJ, Takahashi T, Matsushima K-I, Hara S, Shinohara Y, Maruyama J, Kitamoto K, Koyama Y. SclR, a basic helix-loop-helix transcription factor, regulates hyphal morphology and promotes sclerotial formation in Aspergillus oryzae. Eukaryot Cell. 2011;10:945–55.
Chang P-K, Scharfenstein LL, Ehrlich KC, Diana Di Mavungu J. The Aspergillus flavus fluP-associated metabolite promotes sclerotial production. Fungal Biol. 2016;120:1258–68.
Chang P-K, Scharfenstein LL, Li RW, Arroyo-Manzanares N, De Saeger S, Di Mavungu JD. Aspergillus flavus aswA, a gene homolog of Aspergillus nidulans oefC, regulates sclerotial development and biosynthesis of sclerotium-associated secondary metabolites. Fungal Genet Biol. 2017;104:29–37.
Calvo AM, Bok J, Brooks W, Keller NP. veA is required for toxin and sclerotial production in Aspergillus parasiticus. Appl Environ Microbiol. 2004;70:4733–9.
Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Michael A, Fischbach MA, Weber T, Takano E, Breitling R. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39:W339–46.
Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH, Fedorova ND. SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol. 2010;47:736–41.
Chang P-K, Ehrlich KC. Cyclopiazonic acid biosynthesis by Aspergillus flavus. Toxin Rev. 2011;30:79–89.
Holzapfel CW. The isolation and structure of cyclopiazonic acid, a toxic metabolite of Penicillium cyclopium Westling. Tetrahedron. 1968;24:2101–19.
Chang P-K, Horn BW, Dorner JW. Clustered genes involved in cyclopiazonic acid production are next to the aflatoxin biosynthesis gene cluster in Aspergillus flavus. Fungal Genet Biol. 2009;46:176–82.
Chang PK, Abbas HK, Weaver MA, Ehrlich KC, Scharfenstein LL, Cotty PJ. Identification of genetic defects in the atoxigenic biocontrol strain Aspergillus flavus K49 reveals the presence of a competitive recombinant group in field populations. Int J Food Microbiol. 2012;154:192–6.
Wollenberg RD, Saei W, Westphal KR, Klitgaard CS, Nielsen KL, Lysøe E, Gardiner DM, Wimmer R, Sondergaard TE, Sørensen JL. Chrysogine biosynthesis is mediated by a two-module nonribosomal peptide synthetase. J Nat Prod. 2017;80:2131–5.
Shah AJ, Tilburn J, Adlard MW, Arst Jr HN. pH regulation of penicillin production in Aspergillus nidulans. FEMS Microbiol Lett. 1991;77:209–12.
Lopez-Diam TM, Flannigan B. Production of patulin and cytochalasin E by Aspergillus clavatus during malting of barley and wheat. Int J Food Microbiol. 1997;35:129–36.
Zargar S. Inhibitory effect of various aqueous medicinal plant extracts on citrinin production a fungal biomass by Penicillium notatum and Aspergillus niger. IAIM. 2014;1:1–8.
Wang X, Wang J, Lai D, Wang W, Dai J, Zhou L, Liu Y. Ustiloxin G, a new cyclopeptide mycotoxin from rice false smut balls. Toxins. 2017;9:54.
Umemura M, Nagano N, Koike H, Kawano J, Ishii T, Miyamura Y, Kikuchi M, Tamano K, Yu J, Shin-ya K, Machida M. Characterization of the biosynthetic gene cluster for the ribosomally synthesized cyclic peptide ustiloxin B in Aspergillus flavus. Fungal Genet Biol. 2014;68:23–30.
Ha GT, Wong RK, Zhang Y. Huperzine a as potential treatment of Alzheimer's disease: an assessment on chemistry, pharmacology, and clinical studies. Chem Biodivers. 2011;8:1189–204.
Holm DK, Peterson LM, Klitgaard A, Knudsen PB, Jarczynska ZD, Nielsen KF, Gotfredsen CH, Larsen TO, Mortensen UH. Molecular and chemical characterization of the biosynthesis of the 6-MSA-derived meroterpenoid Yanuthone D in Aspergillus niger. Chem Biol. 2014;21:519–29.
Sunderhaus JD, Sherman DH, Williams RM. Studies on the biosynthesis of the stephacidin and notoamide natural products: a stereochemical and genetic conundrum. Israel J Chem. 2011;51:442–52.
TePaske MR, Gloer JB, Wicklow DT, Dowd PF. Aflavarin and β-Aflatrem: new anti-insectan metabolites from the sclerotia of Aspergillus flavus. J Nat Prod. 1992;55:1080–6.
Cary JW, Han Z, Yin Y, Lohmar JM, Shantappa S, Harris-Coward PY, Mack B, Ehrlich KC, Wei Q, Arroyo-Manzanares N, Uka V, Vanhaecke L, Bhatnagar D, Yu J, Nierman WC, Johns MA, Sorensen D, Shen H, De Saeger S, Diana Di Mavungu J, Calvo AM. Transcriptome analysis of Aspergillus flavus reveals veA-dependent regulation of secondary metabolite gene clusters, including the novel aflavarin cluster. Eukaryot Cell. 2015;14:983–97.
Ramirez-Prado JH, Moore GG, Horn BW, Carbone I. Characterization and population analysis of the mating-type genes in Aspergillus flavus and Aspergillus parasiticus. Fungal Genet Biol. 2008;45:1292–9.
Tsui CK-M, DiGuistini S, Wang Y, Feau N, Dhillon B, Bohlmann J, Hamelin RC. Unequal recombination and evolution of the mating-type (MAT) loci in the pathogenic fungus Grosmannia clavigera and relatives. G3-Genes Genom Genet. 2013;3:465–80.
Metzenberg RL, Glass NL. Mating type and mating strategies in Neurospora. BioEssays. 1990;12:53–9.
Chang P-K, Ehrlich KC. What does genetic diversity of Aspergillus flavus tell us about Aspergillus oryzae? Int J Food Microbiol. 2010;138:189–99.
Ehrlich KC, Chang P-K, Yu J, Cotty PJ. Aflatoxin biosynthesis cluster gene cypA is required for G aflatoxin formation. Appl Environ Microb. 2004;70:6518–24.
Breitenbach M, Weber M, Rinnerthaler M, Karl T, Breitenbach-Koller L. Oxidative stress in fungi: its function in signal transduction, interaction with plant hosts, and lignocellulose degradation. Biomol Ther. 2015;5:318–42.
Ichinose H, Wariishi H. Heterologous expression and mechanistic investigation of a fungal cytochrome P450 (CYP5150A2): involvement of alternative redox partners. Arch Biochem Biophys. 2012;518:8–15.
Caza M, Kronstad JW. Shared and distinct mechanisms of iron acquisition by bacterial and fungal pathogens of humans. Front Cell Infect Microbiol. 2013;3:80.
Horn BW, Gell RM, Singh R, Sorensen RB, Carbone I. Sexual reproduction in Aspergillus flavus sclerotia: acquisition of novel alleles from soil populations and uniparental mitochondrial inheritance. PLoS One. 2016; https://doi.org/10.1371/journal.pone.0146169.
Garbelotto M, Gonthier P, Nicolotti G. Ecological constraints limit the fitness of fungal hybrids in the Heterobasidion annosum species complex. Appl Environ Microbiol. 2007;73:6106–11.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin LV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.
Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, Holt C, Sánchez Alvarado A, Yandell M. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188–96.
Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005;33:6494–506.
Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19-Suppl 2:215–25.
Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho: detection of (co-) orthologs in large-scale analysis. BMC Bioinformatics. 2011;12:124.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–52.
Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90.
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14.
Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinform. 2009;25:4.10.1–4.10.14.
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7.
Acknowledgements
The authors would like to acknowledge the referees who took the time to review and help to improve this body of work.
Funding
This study was funded by the Agricultural Research Service, a division of the United States Department of Agriculture.
Availability of data and materials
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession NEXV00000000. The version described in this paper is version NEXV01000000. A direct link to the data can be found at https://www.ncbi.nlm.nih.gov/bioproject/?term=NEXV00000000.
Author information
Authors and Affiliations
Contributions
GM conceived of the study, prepared the genomic library, conducted sequence analysis and comparisons, and drafted the manuscript as well as its tables and figs. BM performed the genome annotations, all bioinformatics-related analyses, generated raw files for figures, accessioned genomic sequence to NCBI, and contributed to manuscript preparation. SB performed genomic sequencing, and contributed to manuscript preparation. OP cultured the CBS 117610 type strain, extracted DNA from the strain and shipped purified template to our lab in New Orleans for sequencing, and contributed to manuscript preparation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Comparison of aflatoxin and CPA gene clusters for A. arachidicola and several aflatoxin B + G species’ type strains. The schematic diagram (A) shows the orientation and relative sizes (bp) of genes in the aflatoxin gene cluster of A. arachidicola (CBS 117610; red), A. parasiticus (SU-1; green), A. nomius (NRRL 13137; blue) and A. bombycis (NRRL 26010; purple). Panel B shows the orientation and relative sizes (bp) of genes in the CPA gene cluster of A. arachidicola (CBS 117610; red), A. nomius (NRRL 13137; blue) and A. bombycis (NRRL 26010; purple). The A. parasiticus type strain did not contain a cluster of CPA genes. Panel C shows the orientation and distance (bp) separating the aflatoxin and CPA gene clusters in A. arachidicola (CBS 117610; red). The respective gene clusters and their distances, in A. nomius and A. bombycis, were not found to share the same contig; therefore, they could not be determined. (PDF 587 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Moore, G.G., Mack, B.M., Beltz, S.B. et al. Genome sequence of an aflatoxigenic pathogen of Argentinian peanut, Aspergillus arachidicola. BMC Genomics 19, 189 (2018). https://doi.org/10.1186/s12864-018-4576-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4576-2