
Abstract
Background
The progress of medicine, science, technology, education, and culture improves, year by year, quality of life and life expectancy of the populace. The modern human has a chance to further improve the quality and duration of his/her life and the lives of his/her loved ones by bringing their lifestyle in line with their sequenced individual genomes. With this in mind, one of genome-based developments at the junction of personalized medicine and bioinformatics will be considered in this work, where we used two Web services: (i) SNP_TATA_Comparator to search for alleles with a single nucleotide polymorphism (SNP) that alters the affinity of TATA-binding protein (TBP) for the TATA boxes of human gene promoters and (ii) PubMed to look for retrospective clinical reviews on changes in physiological indicators of reproductive potential in carriers of these alleles.
Results
A total of 126 SNP markers of female reproductive potential, capable of altering the affinity of TBP for gene promoters, were found using the two above-mentioned Web services. For example, 10 candidate SNP markers of thrombosis (e.g., rs563763767) can cause overproduction of coagulation inducers. In pregnant women, Hughes syndrome provokes thrombosis with a fatal outcome although this syndrome can be diagnosed and eliminated even at the earliest stages of its development. Thus, in women carrying any of the above SNPs, preventive treatment of this syndrome before a planned pregnancy can reduce the risk of death. Similarly, seven SNP markers predicted here (e.g., rs774688955) can elevate the risk of myocardial infarction. In line with Bowles’ lifespan theory, women carrying any of these SNPs may modify their lifestyle to improve their longevity if they can take under advisement that risks of myocardial infarction increase with age of the mother, total number of pregnancies, in multiple pregnancies, pregnancies under the age of 20, hypertension, preeclampsia, menstrual cycle irregularity, and in women smokers.
Conclusions
According to Bowles’ lifespan theory—which links reproductive potential, quality of life, and life expectancy—the above information was compiled for those who would like to reduce risks of diseases corresponding to alleles in own sequenced genomes. Candidate SNP markers can focus the clinical analysis of unannotated SNPs, after which they may become useful for people who would like to bring their lifestyle in line with their sequenced individual genomes.
Similar content being viewed by others

Background
Incessant progress in medical and biological sciences, advancement of technology, and education year in and year out improve quality of life and life expectancy of the population, creating comfortable conditions for active living. Nonetheless, there are numerous factors that adversely affect human health. They can include, for example, different kinds of environmental pollution, an increase in population density, which leads to the rapid spread of infections and parasitoses, and an increase in psychological stress. This situation not only reduces the quality of life and longevity of the individual but also has a deferred, long-term effect on the next generation, by acting as a mutagen [1]. The accumulating mutational load often worsens health and reduces the subsequent generation’s survival and adaptation to their habitat that ultimately reduces the chances of sustainable population reproduction.
The effects of the above factors limit individual reproductive potential: a concept used in population ecology to assess the evolutionary success of an individual [2] or a population [3]. In the 1970s, Eric Pianka defined reproductive potential as the most important conditional indicator reflecting a population’s ability to reproduce, survive, and develop under optimal ecological conditions [2,3,4,5]. In the context of human society, in the term “reproductive potential,” researchers can also include the mental state and physical state that allow a person to produce healthy offspring when social and physical maturity is achieved. Consequently, reproductive potential depends not only on physiological readiness for reproduction (primarily the reproductive system), but also on the general physical condition (with the exception of existing diseases that are incompatible with the implementation of reproduction) and on socio-economic status. With this in mind, everything is focused on individual ability for reproduction until the next generation becomes reproductive. In particular, not only the phenotype plays a role here, but so does the genotype, where most abilities of a given individual are encoded, both normal and mutational as well as epigenetic ones. It should also be noted that reproductive potential varies throughout the life cycle and does so in different ways for men and women. Ideally, the evaluation of reproductive potential would include not only the direct material and energy costs of reproduction but also the price of the risk associated with future reproductive attempts [5].
Predictive-preventive personalized medicine may help to improve individual reproductive success. Its methods include prediction (based on the analysis of the genome) of the probability of a specific disease, analysis of individual indicators, biomarkers (such as single nucleotide polymorphisms, SNPs [6, 7]), and the development of preventive and therapeutic measures for changing the physiological parameters of the reproductive potential in patients [8]. In particular, the analysis of SNP biomarkers allows a physician not only to make a prognosis for a patient regarding possible diseases that can reduce reproductive potential but also to adjust the prescribed treatment, taking into account individual characteristics and reactions to medicines.
In addition, according to Bowles’ lifespan theory [9], which links reproductive potential, quality of life, and life expectancy of an individual, it is possible timely to prevent diseases, which correspond to the alleles of the decoded genotype.
Within the framework of the biggest modern scientific project “1000 Genomes”, 10545 individual genomes have already been sequenced [10]. The “reference human genome” is publicly available via the Ensembl database [11] using the Web service UCSC Genome Browser [12]. A total of 100,877,027 SNPs have been experimentally identified and stored in the dbSNP database [6]. Database dbWGFP [13] containing 8.58 billion possible human whole-genome SNPs has already been created for accumulation of predictions, experimental data, clinical observations, and any other information relevant for biomedical analysis of individual genomes. For such an analysis, the most valuable biomedical SNP markers—within the framework of personalized medicine—are those that can differ between the individual human genomes of patients having some pathology and the reference human genome [14]. To find such markers, cohorts of patients with a given disease and healthy volunteers (as a control) are compared in a clinical study (e.g., [15]).
As far as human health is concerned, the clinical search for biomedical SNP markers is the only acceptable method. Nevertheless, it is so laborious and expensive that its application to all 8.58 billion potentially possible SNPs [13] and all known human pathologies is rather unlikely. Moreover, both Haldane’s dilemma [16] and Kimura’s theory of neutral evolution [17] independently predict that the absolute majority of SNPs in humans are neutral and do not affect health in any way; thus, it is unclear why it is necessary to verify them clinically. With this in mind, the mainstream clinical search for SNP markers of a given disease is currently limited by the simplest idea about heuristic handmade selection of candidate SNPs for clinical testing among unannotated SNPs on the basis of their location near the human genes that are already clinically associated with this disease (e.g., [18, 19]). Accordingly, computer-based preliminary analysis of unannotated SNPs can eliminate the absolute majority of neutral SNPs to make the clinical cohort-based search for biomedical SNP markers faster, cheaper, and more targeted [20]. There are many public Web services [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38] that facilitate the computer-based search for candidate SNP markers using various similarity measures based on whole-genome data in health [39], after treatment [40], and during a disease [41] or infection [42] to eliminate unannotated SNPs that bear the least resemblance to known biomedical SNP markers (i.e. to eliminate the most probable neutral SNPs). The Central Limit Theorem predicts that the accuracy of this similarity-based elimination of unannotated neutral SNPs increases with the increase in the size and diversity of whole-genome data under study [43].
Now, the best accuracy of this mainstream search corresponds to SNPs in protein-coding regions of genes [44], i.e., SNPs that damage proteins [45] whose defects are uncorrectable by treatment or lifestyle changes. On the contrary, the worst accuracy of this kind of search is seen for regulatory SNPs [11], which alter concentrations of proteins without any damage to the proteins, and such problems are correctable by medication and lifestyle. The best balance between the predictability and biomedical usefulness corresponds to the regulatory SNPs between nucleotide positions -70 and –20 upstream of a transcription start site (TSS) [46, 47] where TATA-binding protein (TBP) binds to the promoter at the very beginning of transcription initiation. This TBP–promoter complex is obligatory for any TSSes because the TBP knockout model animals (TBP−/−) are always inviable since their development cannot proceed past the blastula stage because their maternal supply of TBP is exhausted [48, 49]. Moreover, the TBP–promoter affinity linearly correlates with the transcription magnitude of the human gene containing this promoter [50]. This notion has been repeatedly confirmed experimentally (for review, see [51]). The canonical form of the TBP-binding site (TATA box, synonyms: Hogness box and Goldberg-Hogness box [52]) is the best-studied regulatory element among human gene promoters [47].
In our previous studies, we developed public Web service SNP_TATA_Comparator (http://beehive.bionet.nsc.ru/cgi-bin/mgs/tatascan/start.pl) [53] and applied it to predict candidate SNP markers within TATA boxes of human genes associated with obesity [54], autoimmune diseases [55], chronopathology [56], aggressiveness [57, 58], Alzheimer’s disease [59], and efficacy of anticancer chemotherapy [60] (for review, see [20]). In the present work, we applied our Web service [53] in the same way to human reproductive potential as the most common concept of population ecology dealing with the evolutionary success of either individuals [2] or populations [3].
Results
Tables 1, 2, 3, 4, 5, 6 and 7 present the results obtained by our Web service [53] for the 126 known and candidate reproductive-potential-related SNP markers in the TBP-binding sites of human gene promoters (see Methods: Supplementary Method, Additional file 1).
First, we analyzed all SNPs mapped within [−70; −20] regions upstream of transcription start sites for the human genes containing the known biomedical SNP markers that alter TBP’s binding to promoters of these genes (Tables 1, 2, 3, 4, 5 and 6). Let us first describe in more detail only one human gene in order to briefly review all the others.
Known and candidate reproductivity-related SNP markers of cancers
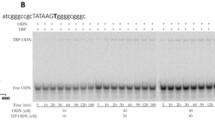
The human ESR2 gene (estrogen receptor β) contains a known SNP marker (Fig. 1a: rs35036378) of an ESR2-deficient primary pT1 breast tumor, which is needed in tamoxifen-based prophylaxis of cancer [61] as shown in Table 1. The prediction of our Web service [53] is consistent with this independent clinical observation (Fig. 1b: text box “Results”, line “Decision” contains the label “deficiency: significant”).

The result produced by SNP_TATA_Comparator [53] for reproductive potential-related SNP markers in the human ESR2 gene. Legend: a Unannotated SNPs (analyzed in this study) in the region [-70; -20] (where all proven TBP-binding sites (boxed) are located; double-headed arrow, ↔) of the human ESR2 gene promoter retrieved from dbSNP, rel. 147 [6] using the UCSC Genome Browser [12]. Dash-and-double-dot arrows: known and candidate SNP markers of reproductive potential are predicted by a significant change in the affinity of TBP for the human ESR2 gene promoter. b and c The results from our Web service SNP_TATA_Comparator [53] for the two SNP markers of reproductive potential: known marker rs35036378 [61] and candidate marker rs766797386 near the known TBP-binding site (boxed) of the human ESR2 gene promoter. Solid, dotted, and dashed arrows indicate queries in the reference human genome [10] by means of the BioPerl library [265]. Dash-and-dot arrows: estimates of significance of the alteration of gene product abundance in patients carrying the minor allele (mut) relative to the norm (ancestral allele, wt) expressed as a Z-score using package R [266]. Circles indicate the ancestral (wt) and minor (mut) alleles of the SNP marker labeled by its dbSNP ID [6]
Next, near this known biomedical SNP marker rs35036378, we found the unannotated SNP rs766797386, which can also decrease expression of the human ESR2 gene (Fig. 1c) and thus cause an ESR2-deficient primary pT1 tumor requiring prophylaxis by tamoxifen against breast cancer [61]. This result allowed us to suggest rs766797386 as a candidate SNP marker of a higher risk of breast cancer reducing reproductive potential.
Finally, using our secondary keyword search for these two SNP markers (hereinafter: see Methods: Additional file 2: Figure S1. dotted-line box, Additional file 2), we learned (hereinafter: see Table S1, Additional file 3) that cadmium (Cd) elevates the risk of a primary tumor’s becoming malignant [62], whereas mothers undergoing tamoxifen-based treatment should not breastfeed [63].
The human HSD17B1, PGR, and GSTM3 genes encode hydroxysteroid (17-β) dehydrogenase 1, progesterone receptor, and glutathione S-transferase μ3, respectively. Their promoters have the known SNP markers rs201739205, rs10895068, and rs1332018, which elevate risks of breast [64] and endometrial [65] cancers; a brain tumor in a fetus, newborn, or a child [66], respectively; as well as renal cancer and Alzheimer’s disease [67] (Table 1). Near these known biomedical SNP markers, there are four unannotated SNPs rs201739205, rs748743528, rs200209906, and rs750789679, which can similarly alter expression levels of the same genes according to the predictions of our Web service [53] (Table 1). Hence, we proposed them as the candidate SNP markers of the same diseases.
Besides, within the same promoters, we found four other unannotated SNPs rs755636251, rs544843047, rs748231432, and rs763859166, which can cause the opposite alterations in the expression of the corresponding genes (Table 1). Using our primary keyword search (hereinafter: see Methods, Additional file 2: Figure S1. two dashed-line boxes, Additional file 2), we found that both HSD17B1 overexpression and deficiency can elevate the risk of breast cancer [68], whereas GSTM3 deficiency can reduce these risks in people who never drink alcohol [69] (Table 1). In addition, Searles Nielsen and colleagues [66] suggested that another mechanism of GSTM3 overexpression can reduce the risk of a brain tumor in some children, as can rs748231432 and rs763859166 according to our results shown in Table 1.
Finally, using our secondary keyword search, we found eight retrospective clinical reviews [70,71,72,73,74,75,76]. The most interesting among them, in our opinion, is a report on a nontrivial balance between reproductive potential and the risk of cancers of reproductive organs [70]. It is interesting that only one SNP marker (rs605059; protein-coding region, HSD17B1) of a positive correlation between the lifespan and number of children in women is known so far [71]. It is also noteworthy that one of current theories is that aging is a stepwise reduction in reproductive potential of individuals where one of these steps is under the control of the luteinizing hormone, whose suppression by smoking can reduce the risk of Alzheimer’s disease [9].
The human IL1B, CYP2A6, CYP2B6, and DHFR genes encode interleukin 1β, xenobiotic monooxygenase, 1,4-cineole 2-exo-monooxygenase, and dihydrofolate reductase, respectively. Their promoters contain the known SNP markers (rs1143627 [77,78,79,80,81,82,83,84,85], rs28399433 [86, 87]) of nonreproductive organ cancer, as well as SNP markers (rs34223104 [88] and rs10168 [89]) of bioactivation and resistance to anticancer drugs, as shown in Table 2. Near these known SNP markers, we detected three unannotated SNPs, rs761592914, rs563558831, and rs750793297, which can alter expression levels of the same genes in the same manner (Table 2) and may be candidate SNP markers in this regard.
In addition, in the same gene regions, we found four other unannotated SNPs rs549858786, rs766799008, rs764508464, and rs754122321 that can have the opposite effect on the expression of the corresponding genes (Table 2). Using our primary keyword search, we found four articles [90,91,92,93] similar to those that were in the case of the known SNPs, where we learned about the correlations between the intensity of physiological and clinical manifestations under study [85,86,87,88,89] (Table 2). Finally, our secondary keyword search yielded 12 reviews [93,94,95,96,97,98,99,100,101,102,103,104,105], among which, the most relevant for us was the notion that Helicobacter pylori infection can cause not only cancer of non-reproductive organs, but can directly reduce human reproductive potential in both men and women [101].
Looking through Tables 1, 2, and Additional file 3: Table S1, one can see that a person increases his/her lifespan and reproductive potential when this person reduces the encounters with cancer risk factors.
Known and candidate reproductivity-related SNP markers of metabolism
Human LEP, GCG, GH1, and INS genes encode hormones leptin, glucagon, somatotropin, and insulin, respectively. There are four known biomedical SNP markers: rs201381696 (obesity [54, 106]), rs183433761 (resistance to obesity during a high-fat diet [54]), rs11568827 (short stature [107]), and rs5505 (type 1 diabetes after neonatal diabetes mellitus [108]) as presented in Table 3.
Near these known SNP markers, 10 candidate SNP markers rs200487063, rs34104384, rs757035851, rs796237787, rs768454929, rs761695685, rs774326004, rs777003420, rs563207167, and rs11557611 were first predicted by our Web service [53] and, then, were characterized by our primary keyword search (Table 3). The most interesting among these predictions [109,110,111,112,113,114,115,116], in our opinion, is the candidate SNP marker rs563207167 of neonatal macrosomia whose known clinical marker is hyperinsulinemia [115], which can be caused by the minor allele of this SNP according to our calculations (Table 3).
Finally, our secondary keyword search produced 31 original articles [105, 117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146], e.g., showing that a maternal high-fat diet elevates the risk of hypertrophy in offspring via fetal hyperinsulinemia programmed epigenetically [141]. It is also relevant that bupropion used as an antidepressant against smoking in pregnancy can cause hyperinsulinemia in newborn children [142].
Human genes NOS2, STAR, APOA1, CETP, SOD1, TPI1, and GJA5 code for inducible nitric oxide synthase 2, steroidogenic acute regulatory protein, apolipoprotein A1, cholesteryl ester transfer protein, Cu/Zn superoxide dismutase, triosephosphate isomerase, and connexin 40, respectively. Their promoters contain eight known biomedical SNP markers shown in Table 4.
Around these known biomedical SNP markers, we found six unannotated SNPs rs544850971, rs17231520, rs569033466, rs757176551, rs781835924, and rs587745372, which can alter expression levels of the human genes containing them according to in silico predictions of our Web service [53] (Table 4). Next, we carried out our primary keyword search where [147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165] the most interesting finding (in our opinion) is the clinical association between a SOD1 deficiency and asthenospermia [151], as one can see in Table 4. Finally, we performed our secondary keyword search, which yielded 21 literary sources [155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175]. For instance, bisphenol A pollution in men can increase the risk of congenital heart morphogenesis disorders in their offspring as Lobmo and colleagues [174] have reported.
As readers can see in Tables 3, 4, and Additional file 3: Table S1, deviations from normal metabolism in parents (e.g., starvation, stress, dietary changes, and polluted environment) can epigenetically program pathologies of the development in their offspring (e.g., [141]). Therefore, a person can increase his/her reproductive potential and lifespan by keeping one’s metabolism normal.
Known and candidate reproductivity-related SNP markers related to blood
Human genes HBB, HBD, HBG2, ACKR1, MBL2, MMP12, and F2 encode subunits β, δ, and γ2 (fetal) of hemoglobin as well as glycoprotein D, mannan-binding lectin, macrophage elastase, and serine protease, respectively. Table 5 shows 10 known SNP markers (rs397509430, rs33980857, rs34598529, rs33931746, rs33981098, rs34500389, and rs35518301) of both malaria resistance and thalassemia [176] as well as rs2814778 (both malaria resistance and low white-blood-cell count [177, 178]), rs72661131 (variable immunodeficiency [179], preeclampsia [180], and stroke [181]), and rs2276109 (lower risks of psoriasis [182], systemic sclerosis [183], and asthma [184]).
Using our Web service [53], we found seven candidate SNP markers rs63750953, rs281864525, rs117785782, rs34166473, rs745580140, rs562962093, and rs572527200, which can alter expression of the human genes containing them, as is the case for the above SNP markers, which can affect the human reproductive potential [185, 186] (Table 5). In addition, using our primary keyword search, we identified three more candidate SNP markers: rs567653539 (reduced risks of recurrent vulvovaginal infections [187]), rs572527200 (high risk of ovarian hyper stimulation syndrome [188]), rs564528021, and rs752364393 (high risk of pre-eclampsia [189]). Finally, we performed our secondary keyword search, which yielded 22 reviews [162, 190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210], the most important of which (in our opinion) mentions pre-eclampsia as a leading cause of maternal and fetal mortality and morbidity worldwide [162], as readers can see in Additional file 3: Table S1.
Human genes THBD, PROC, F8, F3, F7, F9, and F11 code for thrombomodulin, and blood coagulation factors XIV, 8, 3, 7, 9, and 11, respectively (Table 6). There are three known SNP markers rs13306848 (thrombosis [211]), rs563763767 (myocardial infarction and thrombosis [212]), and F7:-33a→c (moderate bleeding [213]) located within the promoters of these genes, which are listed in Table 6.
Within 90-bp proximal regions of these promoters, we selected 30 candidate SNP markers of tumor invasion [214], thrombosis, inflammation and pregnancy loss [215,216,217], ovarian cancer [218], hemorrhage [219], angioneurotic edema [220], hemophilia B [221], and myocardial fibrosis [222] (Table 6). We predicted them using our Web service [53] and a primary keyword search, as described above in detail. Finally, our secondary keyword search produced 29 reviews [101, 223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249,250]. The most interesting among them, in our opinion, is the fact that Homo sapiens is the longest-lived species among great apes (Hominidae) in the postreproductive period. Most often, this period in the life of a human is accompanied by various types of dementia and atherosclerosis, whereas cardiomyopathy and myocardial fibrosis predominate in great apes [248].
Looking through Tables 5, 6, and Additional file 3: Table S1, readers can see that by reducing the risk of blood diseases, a person can increase his/her lifespan and reproductive potential.
Candidate SNP markers of reproductivity-related genes
In addition, using a standard keyword search in the PubMed database, we found articles on human reproductive potential. On this basis, we selected a set of 22 human genes—AR, CAT, CLCA4, CYP1B1, CYP17A1, DAZ1, DAZ2, DAZ3, DAZ4, DEFB126, DNMT1, GNRH1, LHCGR, MTHFR, NR5A1, PARP1, PYGO2, SRD5A2, SRY, TACR3, TET1, and TSSK2—whose promoters do not contain known biomedical SNP markers. This gene set represents a wide variety of known reproductivity-related physiological markers, such as enzymes, transcription factors, hormones, and their receptors. Table 7 presents the results obtained using our Web service [53].
None of the SNPs can statistically significantly alter TBP’s affinity for the promoters of human genes CAT, CLCA4, CYP1B1, DAZ1, DAZ2, DAZ3, DAZ4, DEFB126, GNRH1, LHCGR, PARP1, PYGO2, SRD5A2, SRY, TACR3, TET1, and TSSK2 being analyzed (data not shown). Within promoters of five remaining genes (AR, MTHFR, DNMT1, CYP17A1, and NR5A1), in the same way, we found 24 candidate SNP markers (Table 7). Our primary keyword search associated them with androgenetic alopecia and androgen-induced premature senescence in adult men [251], preeclampsia [252], adverse pregnancy outcomes [253], epigenetic disorders of fetal/newborn brain development [254, 255], activation of protooncogenes in cancer [256], hyperandrogenism in polycystic ovary syndrome [257], fertility impairments [258], adrenal tumors and endometriosis [259] (Table 7).
As a cross-validation test, we unexpectedly found the ratio 5:19 of the candidate SNP markers in the reproductivity-related genes (Table 7) decreasing versus increasing TBP-promoter affinity. In contrast, the well-known whole-genome ratio 2:1 of SNPs reducing versus SNPs increasing affinity of the transcription factors for human gene promoters has been identified by two independent teams [260, 261]. According to binomial distribution, this difference between the candidate SNP markers in the reproductivity-related genes (Table 7) and all SNPs of the human genome is statistically significant (α < 0.000005). This statistical significance reflects the stronger pressure of natural selection against underexpression of the reproductivity-related genes. This unexpected finding indicates higher robustness of this specific sort of human genes on a whole-genome scale and is consistent with the commonly accepted meaning of the term “reproductive potential” as a mainstream concept in population ecology, which defines this term as a measure of evolutionary success of either human individuals [2] or populations [3]. This match between our predictions (Table 7) and one of the mainstream biomedical concepts [2, 3] support the plausibility of the candidate SNP markers predicted here.
Verification procedures for the selected candidate SNP markers predicted here
Different public Web services [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38, 53] have their advantages and disadvantages in eliminating unannotated neutral SNPs. To optimize such knowledge, a comparison between the results of these Web services and experimental data as an independent commonly accepted uniform platform seems to be a necessary step for prediction of candidate SNP markers in silico [15, 20, 59]. Keeping this in mind, we selected some of the 126 candidate SNP markers predicted here—rs563763767, rs33981098, rs35518301, rs1143627, rs72661131, rs1800202, and rs7277748—and measured equilibrium dissociation constant KD of TBP–DNA complexes using an electrophoretic mobility shift assay (EMSA) in vitro (see Methods). The results are shown in Fig. 2, for example, panels A and B present electropherograms and their graphical representation in the case of ancestral and minor alleles, respectively, of the candidate SNP marker rs33981098 within the human HBB gene promoter. Here, readers can see that this SNP reduces the TBP–DNA affinity in half: from 44 nM in the norm (wt) to 90 nM in pathology (mut); this finding supports our prediction, namely, the twofold decrease in the estimate of TBP–DNA affinity from 5 to 9 nM (Table 5). Overall, panel C shows the coordinate plane of the predicted (axis X) and the measured (axis Y) ratio of KD;MUT/KD;WT values of minor versus ancestral alleles of each SNP being verified. As one can see in this figure, there is a significant correlation between our predictions in silico and our measurements in vitro in four statistical tests, namely: linear correlation (r), Spearman’s rank correlation (R), Kendall’s rank correlation (τ), and Goodman–Kruskal generalized correlation (γ) test, which confirm one another’s results. Therefore, the correlations between our predictions and experimental data are robust in terms of the variation of statistical criteria that supports the candidate reproductive-potential-related SNP markers predicted here.

Experimental verification of the selected candidate SNP markers by an electrophoretic mobility shift assay (EMSA) in vitro. Legend: a and b Examples of electropherograms in the case of ancestral (panel A: norm, wild-type, wt) and minor (panel b: minor) alleles of the candidate SNP marker rs33981098 within the human HBB gene promoter and the corresponding diagrams of experimental values. c The significant correlations between the ratio of KD values of the equilibrium dissociation constant of the TBP–ODN complex, which were either measured in vitro (Y-axis) or in silico predicted (X-axis). Solid and dashed lines or curves denote the linear regression and boundaries of its 95% confidence interval, calculated using software Statistica (StatsoftTM, USA). Circles denote the ancestral and minor alleles of the candidate SNP markers rs563763767, rs33981098, rs35518301, rs1143627, rs72661131, rs1800202, and rs7277748 being verified; r, R, τ, γ, and α are linear correlation, Spearman’s rank correlation, Kendall’s rank correlation, Goodman–Kruskal generalized correlation, and their significance, respectively.
Besides the conventional EMSA, we used two modern high-performance methods. Figure 3 shows the results of high-resolution spectrometry on SX.20 (Applied Photophysics, UK), where a stopped-flow fluorescence assay in vitro in real-time mode was applied to the selected candidate SNP marker rs1800202 (see Methods). As readers can see in Table 4, we predicted in silico that the KD value of TBP’s binding affinity for this gene’s wild-type promoter (ancestral alleles), 1 nM, can be weakened by the minor allele of this SNP to 4 nM, in agreement with the experimental data: 1 versus 6 nM, respectively (Table 4). This is one more argument in favor of the significance of the candidate reproductive-potential-related SNP markers predicted here.

The kinetics of binding to and bending of the ODN corresponding to the selected SNP marker rs1800202. Legend: a The ancestral allele, ODN 5′-ctcTATATAAgtggg-3′. b The minor allele, ODN 5′-ctcTATAgAAgtggg-3′. ODN concentration was 0.1 μM. TBP concentration was between 0.1 and 1.0 μM as indicated near the corresponding curve of the time series. KD values, a 1 nM and b 6 nM, were obtained as the output of the Dynafit software (Biokin, USA) when we used the corresponding time-series data as input for this software
Finally, we conducted transfection of the human cell line hTERT-BJ1 (human fibroblasts) in culture, using the pGL 4.10 vector carrying a reporter LUC gene whose transcription is initiated by either ancestral or minor alleles of the selected candidate SNP marker rs28399433 of the human CYP2A6 promoter (Table 2). The results are depicted in Fig. 4. As shown in Table 2, the low affinity of TBP for the minor allele of this SNP relative to the norm (ancestral allele) is consistent with the ex vivo underexpression of a reporter LUC gene carrying the minor allele of this SNP within the pGL 4.10 vector. This ex vivo observation independently confirms our prediction that this SNP can reduce the affinity of TBP for the promoter of the human CYP2A6 gene (Table 2).

Cell culture verification of the selected candidate SNP marker rs28399433 in cell line hTERT-BJ1 (human fibroblasts) transfected with the pGL 4.10 vector carrying a reporter LUC gene. Legend: Dark gray bar, the original vector pGL 4.10 (Promega, USA) without any insertions, which served as an independent control; open bars, ancestral allele (wild type, WT); light gray bar, minor allele (rs28399433). The height of the bars and their error bars correspond to the mean estimates and boundaries of their 95% confidence intervals calculated from five independent experiments. All differences are statistically significant at the confidence level of α < 0.05
Thus, three independent experiments indicate that the candidate reproductive-potential-related SNP markers predicted here using our Web-service [53] seem to have statistically significant effects and are not neutral.
Discussion
In this work, we limited our research to SNPs altering TBP’s affinity for human gene promoters (according to predictions made by our Web service [53]) and thereby altering the expression of these genes; this is because the TBP-binding site is the best-studied transcription-regulatory element [47]. Using our Web service [53], we analyzed over 1000 SNPs between nucleotide positions -70 and -20 upstream of more than 50 protein-coding regions documented in the Ensembl database [11] and found only 126 candidate reproductive-potential-related SNP markers (Tables 1,2, 3, 4, 5, 6 and 7). This 8-fold reduction in the number of possible SNPs can make the clinical cohort-based search for such biomedical SNP markers faster, cheaper, and more targeted, indeed.
For clinical verification of the candidate SNP markers predicted here, we heuristically set up their prioritization based on Fisher’s Z-tests as rank ρ-values from the “best” (A) to the “worst” (E) in alphabetical order (Tables 1, 2, 3, 4, 5, 6 and 7). With this in mind, our findings do not mean that all the eliminated SNPs (data not shown) cannot be considered candidate reproductive-potential-related SNP markers. This is because they may alter transcription factor-binding sites without disrupting the TBP-binding site (e.g., rs11568827, rs796237787, and rs16887226). To perform this sort of analysis for any of them, there are many public Web services [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38] whose research capabilities may be enhanced when they are used in combination with our Web service [53].
It is also worth mentioning that 126 candidate SNP markers predicted here are whole-genome landmarks indicative of either elevated or reduced reproductive potential relative to the norm and can be expected to be present in patients as minor alleles of these SNPs [20]. For example, 10 candidate SNP markers of thrombosis (rs563763767, rs781855957, rs13306848, rs568801899, rs779755900, rs749456955, rs746842194, rs754815577, rs768753666, rs774688955) cause overproduction of coagulation inducers (Table 6). In pregnant women, Hughes syndrome provokes thrombosis with a fatal outcome, although this syndrome can be diagnosed and cured even at the earliest stages of its development [230,231,232] (Additional file 3: Table S1). Thus, in women carrying any of the above SNPs, preventive treatment of this syndrome [230,231,232] before a planned pregnancy can reduce the risk of death. Table 6 shows that seven SNPs (rs563763767, rs779755900, rs749456955, rs746842194, rs754815577, rs768753666, rs774688955) among the 10 mentioned above elevate the risk of myocardial infarction. Hence, a woman with some of these SNPs can improve her longevity by bringing her lifestyle in line with the knowledge that the risk of myocardial infarction elevates with total number of pregnancies, the age of the mother, as well as in pregnancy under the age of 20, in multiple pregnancies, in menstrual cycle irregularity, hypertension, preeclampsia, and in women smokers [233,234,235,236] (Additional file 3: Table S1).
Finally, during our keyword search in the PubMed database, we encountered a large variety of research articles, clinical cases, laboratory data, retrospective reviews, and empirical findings—on human reproductive potential in various life situations—from sociologists, geneticists, legal scholars, clinicians, bioinformaticians, pharmacists, psychologists, pedagogues, physiologists, economists, and other relevant experts such as specialists on management, insurance, environmental protection, health care, and law enforcement (Tables 1, 2, 3, 4, 5, 6 and 7, and Additional file 3: Table S1). This observation means that this vital knowledge is very much in demand for the general population, but it is too scattered for practice use. As one can see in Tables 1, 2, 3, 4, 5, 6 and 7 and Additional file 3: Table S1, 126 candidate reproductive-potential-related SNP markers predicted here may serve as valid whole-genome landmarks near which the above authors can organize their main research on how the evolutionary success of an individual [2] or a population [3] could be enhanced. Consequently, the results of these studies can be directly addressed to people who would like to change their lifestyle in view of the possible risks of diseases. This approach becomes possible within the framework of predictive-preventive personalized medicine based on the sequenced individual genomes.
Conclusions
In keeping with Bowles’ lifespan theory [9], a large body of useful literature can be packaged into readable portions relevant to candidate reproductive-potential-related SNP markers for people who would like to reduce the risks of diseases corresponding to known alleles in own sequenced genome. After clinical validation, these candidate SNP markers may become useful for physicians (to improve treatment of patients) and for the general population (lifestyle choices improving longevity).
Methods
DNA sequences
We analyzed SNPs retrieved from the dbSNP database, v.147 [6] between nucleotide positions -70 and -20 upstream of the protein-coding regions documented by the Ensembl database [11] using the public Web service “UCSC Genome Browser” [12] as shown in Fig. 1a.
Synthetic double-helical deoxyoligonucleotides (ODNs)
The ODNs identical to ancestral and minor alleles of the selected SNPs— rs563763767, rs33981098, rs35518301, rs1143627, rs72661131, rs1800202, and rs7277748—were synthesized and purified (BIOSYN, Novosibirsk, Russia).
Preparation and purification of recombinant full-length human TBP
Recombinant human TBP (full-length native amino acid sequence) was expressed in Escherichia coli BL21 (DE3) cells transformed with the pAR3038-TBP plasmid (a generous gift from Prof. B. Pugh, Pennsylvania State University) as described elsewhere [262] with two modifications: the IPTG concentration was 1.0 instead of 0.1 mM, and the induction time was 3 instead of 1.5 h (for more details, see [263]).
EMSA
The above ODNs were labeled with 32P on both strands by means of T4 polynucleotide kinase (SibEnzyme, Novosibirsk) with subsequent annealing by heating to 95°C (at equimolar concentrations) and slow cooling (no less than 3 h) to room temperature. Equilibrium dissociation constants (KD) for each TBP–ODN complex were measured using a conventional protocol [263] including titration of a fixed amount of the above-mentioned recombinant TBP, 0.3 nM, with the increasing concentrations of each ODN to reach an equilibrium, whose timing was determined independently for each ODN in advance. The binding experiments were conducted at 25°C in a buffer consisting of 20 mM HEPES-KOH pH 7.6, 5 mM MgCl2, 70 mM KCl, 1 mM EDTA, 100 μg/ml BSA, 0.01% of NP-40, and 5% of glycerol. The ТВР–ODN complexes were separated from the unbound ODN using an EMSA, and their abundance levels were measured. The results of these measurements were input into conventional software OriginPro 8, whose output was a KD value expressed in nanomoles per liter, nM.
Stopped-flow fluorescence measurements
The ODNs identical to both ancestral and minor alleles of the selected SNP rs1800202, (i.e., 5′-ctcTATATAAgtggg-3′ and 5′-ctcTATAgAAgtggg-3′, respectively) were labeled at their 5′-termini with fluorescent dyes TAMRA and FAM (BIOSYN, Novosibirsk, Russia). Combining a fixed concentration (0.1 μM) of ODNs with various concentrations (0.1, 0.2, 0.4, 0.6, 0.8, or 1.0 μM) of the above TBP, we analyzed six time-series of the fluorescence expressed in conventional units using high-resolution spectrometer SX.20 (Applied Photophysics, UK). The results of these measurements served as input into the Dynafit software (Biokin, USA), whose output was the above KD values (for more details, see [264]).
Cell culture, transfection, and reporter assays
Cell line hTERT-BJ1 (human fibroblasts) was cultivated in a complete medium consisting of Dulbecco’s modified Eagle’s medium/Nutrient mixture F-12 Ham, supplemented with 10% (v/v) of fetal bovine serum (Sigma), penicillin (100 U/mL), and streptomycin (100 μg/mL; BioloT). The culture was maintained at 37°C in a humidified atmosphere containing 5% of CO2 until the desired degree of confluence. The proximal core promoter (177 bp long) containing either the ancestral allele or minor allele of the selected candidate SNP marker rs28399433 (5′-tcaggcagTATAAAggcaaac-3′ or 5′- tcaggcagTAgAAAggcaaac-3′, respectively) was cloned into the pGL 4.10 vector (Promega, USA) and cotransfected with pRL-TK using Screen Fect A (InCella) as described elsewhere [265]. Next, the cells were cultured in 6-well plates for 24 h. Luciferase activity was determined using the Dual-Luciferase Reporter Assay Kit (Promega, USA) All the experiments were conducted five times independently at 80–85% confluence.
DNA sequence analysis in silico
We analyzed DNA sequences between nucleotide positions -70 and -20 upstream of the protein-coding regions in the human genes retrieved from the human reference genome using the standard BioPerl library [266] via our Web service [53] in the case of ancestral alleles of SNPs under study, as described in Fig. 1b. In the case of minor alleles of these SNPs, we created sequences by hand using the above DNA sequences according to the description of these alleles from database dbSNP [6] as described in Fig. 1c. Next, clicking on the “Calculate” button (Fig. 1b, and c), we computed the maximal –ln(KD) value and its standard deviation ± ε of the affinity of TBP for the [–70; -20] region (where all the known sites are located) for both ancestral and minor alleles of the human gene promoter being analyzed. On this basis, using a package R [267], our Web service [54] made its statistical decision whether the analyzed SNP can alter the expression of the human gene under study as described in Additional file 1 [268,269,270,271,272,273,274]. Earlier, we tested these estimates using independent data from more than a hundred our own experiments [275,276,277,278,279,280,281,282,283,284,285] and the experiments of other authors (for review, see [51]). Finally, as soon as we predicted either SNP-caused significant overexpression or SNP-driven significant underexpression of the human genes being analyzed (as clinically relevant physiological markers), we conducted a manual two-step keyword search in NCBI databases [286] as described in detail in Additional file 2 [287].
Abbreviations
- ACKR1 :
-
atypical chemokine receptor 1
- APOA1 :
-
apolipoprotein A1
- AR :
-
androgen receptor
- CAT :
-
catalase
- CETP :
-
cholesteryl ester transfer protein
- CLCA4 :
-
chloride channel accessory 4
- CYP17A1 :
-
cytochrome p450 family 17 subfamily A member 1
- CYP1B1 :
-
cytochrome P450 family 1 subfamily B member 1
- CYP2A6 :
-
cytochrome P450 family 2 subfamily A member 6
- CYP2B6 :
-
cytochrome P450 family 2 subfamily B Member 6
- DAZ1 (2, 3, 4) :
-
deleted in azoospermia 1 (2, 3, 4, respectively)
- DEFB126 :
-
defensin β 126
- DHFR :
-
dihydrofolate reductase
- DNMT1 :
-
DNA methyltransferase 1
- EMSA:
-
electrophoretic mobility shift assay
- ESR2 :
-
estrogen receptor 2
- F2 (3, 7, 8, 9, 11) :
-
coagulation factor II (III, VII, VIII, IX, XI, respectively)
- GCG :
-
glucagon
- GH1 :
-
growth hormone 1
- GJA5 :
-
gap junction protein α5
- GNRH1 :
-
gonadotropin releasing hormone 1
- GSTM3 :
-
glutathione S-transferase μ3
- HBB :
-
hemoglobin subunit β
- HBD :
-
hemoglobin subunit δ
- HBG2 :
-
hemoglobin subunit γ2
- HSD17B1 :
-
hydroxysteroid 17-β dehydrogenase 1
- IL1B :
-
interleukin 1 β
- INS :
-
insulin
- Kd :
-
equilibrium dissociation constant
- LEP :
-
leptin
- LHCGR :
-
luteinizing hormone (choriogonadotropin receptor)
- Ln:
-
natural logarithm
- MBL2 :
-
mannose binding lectin 2
- MMP12 :
-
matrix metallopeptidase 12
- MTHFR :
-
methylenetetrahydrofolate reductase
- Mut:
-
minor allele of SNPs. Genes
- NOS2 :
-
nitric oxide synthase 2
- NR5A1 :
-
nuclear receptor subfamily 5 group A member 1
- PARP1 :
-
poly(ADP-ribose) polymerase 1
- PGR :
-
progesterone receptor
- PROC :
-
protein C (inactivator of coagulation factors Va and VIIIa)
- PYGO2 :
-
pygopus family PHD finger 2
- SNP:
-
single nucleotide polymorphism
- SOD1 :
-
superoxide dismutase 1
- SRD5A2 :
-
steroid 5 α-reductase 2
- SRY :
-
sex determining region Y
- STAR :
-
steroidogenic acute regulatory protein
- TACR3 :
-
tachykinin receptor 3
- TBP:
-
TATA-binding protein
- TET1 :
-
Tet methylcytosine dioxygenase 1
- TF:
-
transcription factor
- THBD :
-
thrombomodulin
- TPI1 :
-
triosephosphate isomerase 1
- TSS:
-
transcription start site
- TSSK2 :
-
testis specific serine kinase 2
- WT:
-
wild type (norm)
References
Axelsson J, Bonde JP, Giwercman YL, Rylander L, Giwercman A. Gene-environment interaction and male reproductive function. Asian J Androl. 2010;12:298–307.
Pianka ER. Natural selection of optimal reproductive tactics. Amer Zool. 1976;16:775–84.
Chapman RN. Animal ecology with special reference to insects. NY: McGraw-Hill Book Co; 1931.
Chapman RN. The quantitative analysis of environmental factors. Ecology. 1928;9:111–22.
Pianka ER. On r and К selection. Amer Natur. 1970;104:592–7.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11.
Deplancke B, Alpern D, Gardeux V. The genetics of transcription factor DNA binding variation. Cell. 2016;166:538–54.
Ortega VE, Meyers DA. Pharmacogenetics: implications of race and ethnicity on defining genetic profiles for personalized medicine. J Allergy Clin Immunol. 2014;133:16–26.
Bowles JT. The evolution of aging: a new approach to an old problem of biology. Med Hypotheses. 1998;51:179–221.
Telenti A, Pierce LC, Biggs WH, di Iulio J, Wong EH, Fabani MM, et al. Deep sequencing of 10,000 human genomes. Proc Natl Acad Sci U S A. 2016;113:11901–6.
Zerbino DR, Wilder SP, Johnson N, Juettemann T, Flicek PR. The Ensembl regulatory build. Genome Biol. 2015;16:56.
Haeussler M, Raney BJ, Hinrichs AS, Clawson H, Zweig AS, Karolchik D, et al. Navigating protected genomics data with UCSC Genome Browser in a Box. Bioinformatics. 2015;31:764–6.
Wu J, Wu M, Li L, Liu Z, Zeng W, Jiang R. dbWGFP: a database and web server of human whole-genome single nucleotide variants and their functional predictions. Database (Oxford). 2016;2016:baw024.
Trovato GM. Sustainable medical research by effective and comprehensive medical skills: overcoming the frontiers by predictive, preventive and personalized medicine. EPMA J. 2014;5:14.
Yoo SS, Jin C, Jung DK, Choi YY, Choi JE, Lee WK, et al. Putative functional variants of XRCC1 identified by RegulomeDB were not associated with lung cancer risk in a Korean population. Cancer Genet. 2015;208:19–24.
Haldane JBS. The Cost of Natural Selection. J Genet. 1957;55:511–24.
Kimura M. Evolutionary rate at the molecular level. Nature. 1968;217:624–6.
Varzari A, Bruch K, Deyneko IV, Chan A, Epplen JT, Hoffjan S. Analysis of polymorphisms in RIG-I-like receptor genes in German multiple sclerosis patients. J Neuroimmunol. 2014;277:140–4.
Varzari A, Deyneko IV, Tudor E, Turcan S. Polymorphisms of glutathione S-transferase and methylenetetrahydrofolate reductase genes in Moldavian patients with ulcerative colitis: Genotype-phenotype correlation. Meta Gene. 2016;7:76–82.
Ponomarenko M, Rasskazov D, Chadaeva I, Sharypova E, Ponomarenko P, Arkova O, et al. SNP_TATA_Comparator: genomewide landmarks for preventive personalized medicine. Front Biosci (Schol Ed). 2017;9:276–306.
Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O’Donnell CJ, de Bakker PI. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–9.
Barenboim M, Manke T. ChroMoS: an integrated web tool for SNP classification, prioritization and functional interpretation. Bioinformatics. 2013;29:2197–8.
Riva A. Large-scale computational identification of regulatory SNPs with rSNP-MAPPER. BMC Genomics. 2012;13:S7.
Macintyre G, Bailey J, Haviv I, Kowalczyk A. is-rSNP: a novel technique for in silico regulatory SNP detection. Bioinformatics. 2010;26:i524–30.
Ponomarenko JV, Furman DP, Frolov AS, Podkolodny NL, Orlova GV, Ponomarenko MP, et al. ACTIVITY: a database on DNA/RNA sites activity adapted to apply sequence-activity relationships from one system to another. Nucleic Acids Res. 2001;29:284–7.
Chen CC, Xiao S, Xie D, Cao X, Song CX, Wang T, et al. Understanding variation in transcription factor binding by modeling transcription factor genome-epigenome interactions. PLoS Comput Biol. 2013;9:e1003367.
Deyneko IV, Bredohl B, Wesely D, Kalybaeva YM, Kel AE, Blocker H, et al. FeatureScan: revealing property-dependent similarity of nucleotide sequences. Nucleic Acids Res. 2006;34:W591–5.
Chen CY, Chang IS, Hsiung CA, Wasserman WW. On the identification of potential regulatory variants within genome wide association candidate SNP sets. BMC Med Genomics. 2014;7:34.
Rasskazov DA, Antontseva EV, Bryzgalov LO, Matveeva MY, Kashina EV, Ponomarenko PM, et al. rSNP_Guide-based evaluation of SNPs in promoters of the human APC and MLH1 genes associated with colon cancer. Russ J Genet App Res. 2014;4:245–53.
Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–7.
Andersen MC, Engstrom PG, Lithwick S, Arenillas D, Eriksson P, Lenhard B, et al. In silico detection of sequence variations modifying transcriptional regulation. PLoS Comput Biol. 2008;4:e5.
Ponomarenko JV, Orlova GV, Frolov AS, Gelfand MS, Ponomarenko MP. SELEX_DB: a database on in vitro selected oligomers adapted for recognizing natural sites and for analyzing both SNPs and site-directed mutagenesis data. Nucleic Acids Res. 2002;30:195–9.
Saccone SF, Bolze R, Thomas P, Quan J, Mehta G, Deelman E, et al. SPOT: a web-based tool for using biological databases to prioritize SNPs after a genome-wide association study. Nucleic Acids Res. 2010;38:W201–9.
Podkolodnyy NL, Afonnikov DA, YuYu V, Bryzgalov LO, Ivanisenko VA, Demenkov PS, et al. Program complex SNP-MED for analysis of single-nucleotide polymorphism (SNP) effects on the function of genes associated with socially significant diseases. Russ J Genet App Res. 2014;4:159–67.
Fu Y, Liu Z, Lou S, Bedford J, Mu XJ, Yip KY, et al. FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 2014;15:480.
Deyneko IV, Kalybaeva YM, Kel AE, Blocker H. Human-chimpanzee promoter comparisons: property-conserved evolution? Genomics. 2010;96:129–33.
Coetzee SG, Rhie SK, Berman BP, Coetzee GA, Noushmehr H. FunciSNP: an R/bioconductor tool integrating functional non-coding data sets with genetic association studies to identify candidate regulatory SNPs. Nucleic Acids Res. 2012;40:e139.
Bryzgalov LO, Antontseva EV, Matveeva MY, Shilov AG, Kashina EV, Mordvinov VA, et al. Detection of regulatory SNPs in human genome using ChIP-seq ENCODE data. PLoS One. 2013;8:e78833.
Ni Y, Hall AW, Battenhouse A, Iyer VR. Simultaneous SNP identification and assessment of allele-specific bias from ChIP-seq data. BMC Genet. 2012;13:46.
Hein M, Graver S. Tumor cell response to bevacizumab single agent therapy in vitro. Cancer Cell Int. 2013;13:94.
Hu J, Locasale JW, Bielas JH, O'Sullivan J, Sheahan K, Cantley LC, et al. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol. 2013;31:522–9.
Leschner S, Deyneko IV, Lienenklaus S, Wolf K, Bloecker H, Bumann D, et al. Identification of tumor-specific Salmonella Typhimurium promoters and their regulatory logic. Nucleic Acids Res. 2012;40:2984–94.
Ponomarenko MP, Ponomarenko JV, Frolov AS, Podkolodnaya OA, Vorobyev DG, Kolchanov NA, et al. Oligonucleotide frequency matrices addressed to recognizing functional DNA sites. Bioinformatics. 1999;15:631–43.
Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick's Online Mendelian Inheritance in Man (OMIM). Nucleic Acids Res. 2009;37:D793–6.
Mitsuyasu H, Izuhara K, Mao XQ, Gao PS, Arinobu Y, Enomoto T, et al. Ile50Val variant of IL4R-alpha upregulates IgE synthesis and associates with atopic asthma. Nat Genet. 1998;19:119–20.
Savinkova LK, Ponomarenko MP, Ponomarenko PM, Drachkova IA, Lysova MV, Arshinova TV, et al. TATA box polymorphisms in human gene promoters and associated hereditary pathologies. Biochemistry (Mosc). 2009;74:117–29.
Ponomarenko M, Mironova V, Gunbin K, Savinkova L. Hogness Box. In: Maloy S, Hughes K, editors. Brenner's Encyclopedia of Genetics. vol. 3. 2nd ed. San Diego: Academic Press, Elsevier Inc. 2013. p. 491–4
Martianov I, Viville S, Davidson I. RNA polymerase II transcription in murine cells lacking the TATA binding protein. Science. 2002;298:1036–9.
Muller F, Lakatos L, Dantonel J, Strahle U, Tora L. TBP is not universally required for zygotic RNA polymerase II transcription in zebrafish. Curr Biol. 2001;11:282–7.
Mogno I, Vallania F, Mitra RD, Cohen BA. TATA is a modular component of synthetic promoters. Genome Res. 2010;20:1391–7.
Ponomarenko PM, Suslov VV, Savinkova LK, Ponomarenko MP, Kolchanov NA. A precise equation of equilibrium of four steps of TBP binding with the TATA box for prognosis of phenotypic manifestation of mutations. Biofizika. 2010;55:400–14.
Lifton R, Goldberg M, Karp R, Hogness D. The organization of the histone genes in Drosophila melanogaster: Functional and evolutionary implications. Cold Spring Harb Symp Quant Biol. 1978;42:1047–51.
Ponomarenko M, Rasskazov D, Arkova O, Ponomarenko P, Suslov V, Savinkova L, et al. How to use SNP_TATA_Comparator to find a significant change in gene expression caused by the regulatory SNP of this gene’s promoter via a change in affinity of the TATA-binding protein for this promoter. Biomed Res Int. 2015;2015:359835.
Arkova OV, Ponomarenko MP, Rasskazov DA, Drachkova IA, Arshinova TV, Ponomarenko PM, et al. Obesity-related known and candidate SNP markers can significantly change affinity of TATA-binding protein for human gene promoters. BMC Genomics. 2015;16:S5.
Ponomarenko MP, Arkova O, Rasskazov D, Ponomarenko P, Savinkova L, Kolchanov N. Candidate SNP markers of gender-biased autoimmune complications of monogenic diseases are predicted by a significant change in the affinity of TATA-binding protein for human gene promoters. Front Immunol. 2016;7:130.
Ponomarenko P, Rasskazov D, Suslov V, Sharypova E, Savinkova L, Podkolodnaya O, et al. Candidate SNP markers of chronopathologies are predicted by a significant change in the affinity of TATA-binding protein for human gene promoters. Biomed Res Int. 2016;2016:8642703.
Chadaeva IV, Ponomarenko MP, Rasskazov DA, Sharypova EB, Kashina EV, Matveeva MY, et al. Candidate SNP markers of aggressiveness-related complications and comorbidities of genetic diseases are predicted by a significant change in the affinity of TATA-binding protein for human gene promoters. BMC Genomics. 2016;17:995.
Chadaeva I, Rasskazov D, Sharypova E, Savinkova L, Ponomarenko P, Ponomarenko M. Candidate SNP markers of social dominance, which may affect the affinity of the TATA-binding protein for human gene promoters. Russ J Genet Appl Res. 2017;7:523–37.
Ponomarenko P, Chadaeva I, Rasskazov D, Sharypova E, Kashina E, Drachkova I, et al. Candidate SNP markers of familial and sporadic Alzheimer’s diseases are predicted by a significant change in the affinity of TATA-binding protein for human gene promoters. Front Aging Neurosci. 2017;9:231.
Turnaev II, Rasskazov DA, Arkova OV, Ponomarenko MP, Ponomarenko PM, Savinkova LK, et al. Hypothetical SNP markers that significantly affect the affinity of the TATA-binding protein to VEGFA, ERBB2, IGF1R, FLT1, KDR, and MET oncogene promoters as chemotherapy targets. Mol Biol (Mosk). 2016;50:161–73.
Philips S, Richter A, Oesterreich S, Rae JM, Flockhart DA, Perumal NB, et al. Functional characterization of a genetic polymorphism in the promoter of the ESR2 gene. Horm Cancer. 2012;3:37–43.
Strumylaite L, Bogusevicius A, Ryselis S, Pranys D, Poskiene L, Kregzdyte R, et al. Association between cadmium and breast cancer. Medicina (Kaunas). 2008;44:415–20.
Helewa M, Levesque P, Provencher D, Lea RH, Rosolowich V, Shapiro HM, et al. Breast cancer, pregnancy, and breastfeeding. J Obstet Gynaecol Can. 2002;24:164–80. quiz:181-4
Peltoketo H, Piao Y, Mannermaa A, Ponder BA, Isomaa V, Poutanen M, et al. A point mutation in the putative TATA box, detected in nondiseased individuals and patients with hereditary breast cancer, decreases promoter activity of the 17 β-hydroxysteroid dehydrogenase type 1 gene 2 (EDH17B2) in vitro. Genomics. 1994;23:250–2.
De Vivo I, Huggins GS, Hankinson SE, Lescault PJ, Boezen M, Colditz GA, et al. A functional polymorphism in the promoter of the progesterone receptor gene associated with endometrial cancer risk. Proc Natl Acad Sci USA. 2002;99:12263–8.
Searles Nielsen S, Mueller BA, Preston-Martin S, Farin FM, Holly EA, et al. Childhood brain tumors and maternal cured meat consumption in pregnancy: differential effect by glutathione S-transferases. Cancer Epidemiol Biomarkers Prev. 2011;20:2413–9.
Tan X, Wang Y, Han Y, Chang W, Su T, Hou J, et al. Genetic variation in the GSTM3 promoter confer risk and prognosis of renal cell carcinoma by reducing gene expression. Br J Cancer. 2013;109:3105–15.
He W, Gauri M, Li T, Wang R, Lin SX. Current knowledge of the multifunctional 17β-hydroxysteroid dehydrogenase type 1 (HSD17B1). Gene. 2016;588:54–61.
Mitrunen K, Hirvonen A. Molecular epidemiology of sporadic breast cancer. The role of polymorphic genes involved in oestrogen biosynthesis and metabolism. Mutat Res. 2003;544:9–41.
Jasienska G. Reproduction and lifespan: Trade-offs, overall energy budgets, intergenerational costs, and costs neglected by research. Am J Hum Biol. 2009;21:524–32.
Scarabino D, Scacchi R, Pinto A, Corbo RM. Genetic basis of the relationship between reproduction and longevity: a study on common variants of three genes in steroid hormone metabolism--CYP17, HSD17B1, and COMT. Rejuvenation Res. 2015;18:464–72.
Kalogera E, Dowdy SC, Bakkum-Gamez JN. Preserving fertility in young patients with endometrial cancer: current perspectives. Int J Womens Health. 2014;6:691–701.
Hambrecht A, Jandial R, Neman J. Emerging role of brain metastases in the prognosis of breast cancer patients. Breast Cancer (Dove Med Press). 2011;3:79–91.
Wright DD, Whitney J. Multiple hamartoma syndrome (Cowden's syndrome): case report and literature review. Gen Dent. 2006;54:417–9.
Molokwu JC, Prizment AE, Folsom AR. Reproductive characteristics and risk of kidney cancer: Iowa Women's Health Study. Maturitas. 2007;58:156–63.
Schnoller TJ, Jentzmik F, Al Ghazal A, Zengerling F, de Petriconi R, Hefty R, et al. Renal masses in pregnancy. Diagnostics and therapeutic management. Urologe A. 2011;50:1064–7.
El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402.
Wang Y, Kato N, Hoshida Y, Yoshida H, Taniguchi H, Goto T, et al. Interleukin-1β gene polymorphisms associated with hepatocellular carcinoma in hepatitis C virus infection. Hepatology. 2003;37:65–71.
Martinez-Carrillo DN, Garza-Gonzalez E, Betancourt-Linares R, Monico-Manzano T, Antunez-Rivera C, Roman-Roman A, et al. Association of IL1B -511C/-31T haplotype and Helicobacter pylori vacA genotypes with gastric ulcer and chronic gastritis. BMC Gastroenterol. 2010;10:126.
Wu KS, Zhou X, Zheng F, Xu XQ, Lin YH, Yang J. Influence of interleukin-1 β genetic polymorphism, smoking and alcohol drinking on the risk of non-small cell lung cancer. Clin Chim Acta. 2010;411:1441–6.
Borkowska P, Kucia K, Rzezniczek S, Paul-Samojedny M, Kowalczyk M, Owczarek A, et al. Interleukin-1β promoter (-31T/C and -511C/T) polymorphisms in major recurrent depression. J Mol Neurosci. 2011;44:12–6.
Strandberg L, Mellstrom D, Ljunggren O, Grundberg E, Karlsson MK, Holmberg AH, et al. IL6 and IL1B polymorphisms are associated with fat mass in older men: the MrOS Study Sweden. Obesity (Silver Spring). 2008;16:710–3.
Hayashi F, Watanabe M, Nanba T, Inoue N, Akamizu T, Iwatani Y. Association of the -31C/T functional polymorphism in the interleukin-1β gene with the intractability of Graves' disease and the proportion of T helper type 17 cells. Clin Exp Immunol. 2009;158:281–6.
Wang X, Jiang F, Liang Y, Xu L, Li H, Liu Y, et al. Interleukin-1β-31C/T and -511T/C polymorphisms were associated with preeclampsia in Chinese Han population. PLoS One. 2014;9:e106919.
Ramos BR, Mendes ND, Tanikawa AA, Amador MA, dos Santos NP, dos Santos SE, et al. Ancestry informative markers and selected single nucleotide polymorphisms in immunoregulatory genes on preterm labor and preterm premature rupture of membranes: a case control study. BMC Pregnancy Childbirth. 2016;16:30.
Pitarque M, von Richter O, Oke B, Berkkan H, Oscarson M, Ingelman-Sundberg M. Identification of a single nucleotide polymorphism in the TATA box of the CYP2A6 gene: impairment of its promoter activity. Biochem Biophys Res Commun. 2001;284:455–60.
Pianezza ML, Sellers EM, Tyndale RF. Nicotine metabolism defect reduces smoking. Nature. 1998;393:750.
Zukunft J, Lang T, Richter T, Hirsch-Ernst KI, Nussler AK, Klein K, et al. A natural CYP2B6 TATA box polymorphism (-82T--> C) leading to enhanced transcription and relocation of the transcriptional start site. Mol Pharmacol. 2005;67:1772–82.
Al-Shakfa F, Dulucq S, Brukner I, Milacic I, Ansari M, Beaulieu P, et al. DNA variants in region for noncoding interfering transcript of dihydrofolate reductase gene and outcome in childhood acute lymphoblastic leukemia. Clin Cancer Res. 2009;15:6931–8.
Xie C, Wen X, Ding P, Liu T, He Y, Niu Z, et al. Influence of CYP2A6*4 genotypes on maternal serum cotinine among Chinese nonsmoking pregnant women. Nicotine Tob Res. 2014;16:406–12.
Rao Y, Hoffmann E, Zia M, Bodin L, Zeman M, Sellers EM, et al. Duplications and defects in the CYP2A6 gene: identification, genotyping, and in vivo effects on smoking. Mol Pharmacol. 2000;58:747–55.
Shimizu M, Koyama T, Kishimoto I, Yamazaki H. Dataset for genotyping validation of cytochrome P450 2A6 whole-gene deletion (CYP2A6*4) by real-time polymerase chain reaction platforms. Data Brief. 2015;5:642–5.
Skubisz MM, Tong S. Of leaves and butterflies: how methotrexate came to be the savior of women. Obstet Gynecol. 2011;118:1169–73.
Kralíkova E, Bajerova J, Raslova N, Rames J, Himmerova V. Smoking and pregnancy: prevalence, knowledge, anthropometry, risk communication. Prague Med Rep. 2005;106:195–200.
Chow EJ, Stratton KL, Leisenring WM, Oeffinger KC, Sklar CA, Donaldson SS, et al. Pregnancy after chemotherapy in male and female survivors of childhood cancer treated between 1970 and 1999: a report from the Childhood Cancer Survivor Study cohort. Lancet Oncol. 2016;17:567–76.
Dooley MA, Nair R. Therapy insight: preserving fertility in cyclophosphamide-treated patients with rheumatic disease. Nat Clin Pract Rheumatol. 2008;4:250–7.
Leader A, Lishner M, Michaeli J, Revel A. Fertility considerations and preservation in haemato-oncology patients undergoing treatment. Br J Haematol. 2011;153:291–308.
Yiu ZZ, Warren RB, Mrowietz U, Griffiths CE. Safety of conventional systemic therapies for psoriasis on reproductive potential and outcomes. J Dermatolog Treat. 2015;26:329–34.
Schwartz AG, Ray RM, Cote ML, Abrams J, Sokol RJ, Hendrix SL, et al. Hormone use, reproductive history, and risk of lung cancer: The Women's Health Initiative Studies. J Thorac Oncol. 2015;10:1004–13.
Zhong GC, Liu Y, Chen N, Hao FB, Wang K, Cheng JH, et al. Reproductive factors, menopausal hormone therapies and primary liver cancer risk: a systematic review and dose-response meta-analysis of observational studies. Hum Reprod Update. 2016;23:126–38.
Moretti E, Figura N, Collodel G, Ponzetto A. Can Helicobacter pylori infection influence human reproduction? World J Gastroenterol. 2014;2:5567–74.
Laurberg P, Andersen SL. Graves'-Basedow disease in pregnancy. New trends in the management and guidance to reduce the risk of birth defects caused by antithyroid drugs. Nuklearmedizin. 2015;54:106–11.
Sichel DA, Cohen LS, Robertson LM, Ruttenberg A, Rosenbaum JF. Prophylactic estrogen in recurrent postpartum affective disorder. Biol Psychiatry. 1995;38:814–8.
Luke B. Adverse effects of female obesity and interaction with race on reproductive potential. Fertil Steril. 2017;107:868–77.
Leisegang K, Bouic PJ, Menkveld R, Henkel RR. Obesity is associated with increased seminal insulin and leptin alongside reduced fertility parameters in a controlled male cohort. Reprod Biol Endocrinol. 2014;12:34.
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–32.
Horan M, Millar DS, Hedderich J, Lewis G, Newsway V, Mo N, et al. Human growth hormone 1 (GH1) gene expression: complex haplotype-dependent influence of polymorphic variation in the proximal promoter and locus control region. Hum Mutat. 2003;21:408–23.
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–5.
Tumer N, Erdos B, Matheny M, Cudykier I, Scarpace PJ. Leptin antagonist reverses hypertension caused by leptin overexpression, but fails to normalize obesity-related hypertension. J Hypertens. 2007;25:2471–8.
Beltowski J. Central vs. peripheral leptin excess in the pathogenesis of obesity-associated hypertension. J Hypertens. 2008;26:827–8.
Takagi Y, Kinoshita K, Ozaki N, Seino Y, Murata Y, Oshida Y, et al. Mice deficient in proglucagon-derived peptides exhibit glucose intolerance on a high-fat diet but are resistant to obesity. PLoS One. 2015;10:e0138322.
Sugiyama C, Yamamoto M, Kotani T, Kikkawa F, Murata Y, Hayashi Y. Fertility and pregnancy-associated ß-cell proliferation in mice deficient in proglucagon-derived peptides. PLoS One. 2012;7:e43745.
Saggese G, Baroncelli GI, Vanacore T, Fiore L, Ruggieri S, Federico G. Indications and strategies for continuing GH treatment during transition from late adolescence to early adulthood in patients with GH deficiency: the impact on bone mass. J Endocrinol Invest. 2004;27:596–602.
Bielohuby M, Roemmler J, Manolopoulou J, Johnsen I, Sawitzky M, Schopohl J, et al. Chronic growth hormone excess is associated with increased aldosterone: a study in patients with acromegaly and in growth hormone transgenic mice. Exp Biol Med (Maywood). 2009;234:1002–9.
Lepercq J, Cauzac M, Lahlou N, Timsit J, Girard J, Auwerx J, et al. Overexpression of placental leptin in diabetic pregnancy: a critical role for insulin. Diabetes. 1998;47:847–50.
Bassett JM, Hanson C. Prevention of hypoinsulinemia modifies catecholamine effects in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2000;278:R1171–81.
Smith GD, Jackson LM, Foster DL. Leptin regulation of reproductive function and fertility. Theriogenology. 2002;57:73–86.
Holness MJ, Munns MJ, Sugden MC. Current concepts concerning the role of leptin in reproductive function. Mol Cell Endocrinol. 1999;157:11–20.
Bluher S, Mantzoros CS. Leptin in reproduction. Curr Opin Endocrinol Diabetes Obes. 2007;14:458–64.
Popovic V, Casanueva FF. Leptin, nutrition and reproduction: new insights. Hormones (Athens). 2002;1:204–17.
Song Y, Yu Y, Wang D, Chai S, Liu D, Xiao X, et al. Maternal high-fat diet feeding during pregnancy and lactation augments lung inflammation and remodeling in the offspring. Respir Physiol Neurobiol. 2015;207:1–6.
Khorram O, Keen-Rinehart E, Chuang TD, Ross MG, Desai M. Maternal undernutrition induces premature reproductive senescence in adult female rat offspring. Fertil Steril. 2015;103:291–8. e2
Valleau JC, Sullivan EL. The impact of leptin on perinatal development and psychopathology. J Chem Neuroanat. 2014;61-62:221–32.
El Orabi H, Ghalia AA, Khalifa A, Mahfouz H, El Shalkani A, Shoieb N. Serum leptin as an additional possible pathogenic factor in polycystic ovary syndrome. Clin Biochem. 1999;32:71–5.
Anderson CM, Ren J. Leptin, leptin resistance and endothelial dysfunction in pre-eclampsia. Cell Mol Biol (Noisy-le-Grand). 2002;48:OL323–9.
Kazemian E, Sotoudeh G, Dorosty-Motlagh AR, Eshraghian MR, Bagheri M. Maternal obesity and energy intake as risk factors of pregnancy-induced hypertension among Iranian women. J Health Popul Nutr. 2014;32:486–93.
Luque-Ramirez M, Escobar-Morreale HF. Polycystic ovary syndrome as a paradigm for prehypertension, prediabetes, and preobesity. Curr Hypertens Rep. 2014;16:500.
Taylor PD, Samuelsson AM, Poston L. Maternal obesity and the developmental programming of hypertension: a role for leptin. Acta Physiol (Oxf). 2014;210:508–23.
Perry IJ, Whincup PH, Shaper AG. Environmental factors in the development of essential hypertension. Br Med Bull. 1994;50:246–59.
Bloom SR, Johnston DI. Failure of glucagon release in infants of diabetic mothers. Br Med J. 1972;4:453–4.
Lee PA, Houk CP. Gonadotropin-releasing hormone analog therapy for central precocious puberty and other childhood disorders affecting growth and puberty. Treat Endocrinol. 2006;5:287–96.
Chandrashekar V, Zaczek D, Bartke A. The consequences of altered somatotropic system on reproduction. Biol Reprod. 2004;71:17–27.
Spiliotis BE. Growth hormone insufficiency and its impact on ovarian function. Ann N Y Acad Sci. 2003;997:77–84.
Juul A, Hjortskov N, Jepsen LT, Nielsen B, Halkjaer-Kristensen J, Vahl N, et al. Growth hormone deficiency and hyperthermia during exercise: a controlled study of sixteen GH-deficient patients. J Clin Endocrinol Metab. 1995;80:3335–40.
Grynberg M, Salenave S, Young J, Chanson P. Female gonadal function before and after treatment of acromegaly. J Clin Endocrinol Metab. 2010;95:4518–25.
Karkanias GB, Morales JC, Li CS. Deficits in reproductive behavior in diabetic female rats are due to hypoinsulinemia rather than hyperglycemia. Horm Behav. 1997;32:19–29.
Shvyrkova NA, Aleksandrova EA, Kupets TV. The behavioral development of rats with prenatal hypoinsulinemia. Zh Vyssh Nerv Deiat Im I P Pavlova. 1995;45:1190–7.
Chaffin DG, Clark RM, McCracken D, Philipps AF. Effect of hypoinsulinemia on growth in the fetal rabbit. Biol Neonate. 1995;67:186–93.
Baaziz N, Curry DL. Synthesis-secretion coupling of insulin: effect of pregnancy and lactation. Pancreas. 1993;8:316–24.
Felig P, Lynch V. Starvation in human pregnancy: hypoglycemia, hypoinsulinemia, and hyperketonemia. Science. 1970;170:990–2.
Fernandez-Twinn DS, Blackmore HL, Siggens L, Giussani DA, Cross CM, Foo R, et al. The programming of cardiac hypertrophy in the offspring by maternal obesity is associated with hyperinsulinemia, AKT, ERK, and mTOR activation. Endocrinology. 2012;153:5961–71.
Gisslen T, Nathan B, Thompson T, Rao R. Hyperinsulinism associated with gestational exposure to bupropion in a newborn infant. J Pediatr Endocrinol Metab. 2011;24:819–22.
Norman RJ, Masters S, Hague W. Hyperinsulinemia is common in family members of women with polycystic ovary syndrome. Fertil Steril. 1996;66:942–7.
Othman NH, Ab Rahman AF. Obstetric and birth outcomes in pregnant women with epilepsy: A hospital-based study. Ann Indian Acad Neurol. 2013;16:534–7.
Katz O, Levy A, Wiznitzer A, Sheiner E. Pregnancy and perinatal outcome in epileptic women: a population-based study. J Matern Fetal Neonatal Med. 2006;19:21–5.
Zhidkova IA, Karlov VA, IuB K'k, Zlatorunskaia MA, Braginskaia SG, Kaznacheeva TV, et al. Reproductive potential of women with epilepsy. Zh Nevrol Psikhiatr Im S S Korsakova. 2009;109:31–6.
Wojcik M, Zieleniak A, Zurawska-Klis M, Cypryk K, Wozniak LA. Increased expression of immune-related genes in leukocytes of patients with diagnosed gestational diabetes mellitus (GDM). Exp Biol Med (Maywood). 2016;241:457–65.
Caron KM, Soo SC, Wetsel WC, Stocco DM, Clark BJ, Parker KL. Targeted disruption of the mouse gene encoding steroidogenic acute regulatory protein provides insights into congenital lipoid adrenal hyperplasia. Proc Natl Acad Sci USA. 1997;94:11540–5.
Roe A, Hillman J, Butts S, Smith M, Rader D, Playford M, et al. Decreased cholesterol efflux capacity and atherogenic lipid profile in young women with PCOS. J Clin Endocrinol Metab. 2014;99:e841–7.
Silliman K, Tall AR, Kretchmer N, Forte TM. Unusual high-density lipoprotein subclass distribution during late pregnancy. Metabolism. 1993;42:1592–9.
Garratt M, Bathgate R, de Graaf SP, Brooks RC. Copper-zinc superoxide dismutase deficiency impairs sperm motility and in vivo fertility. Reproduction. 2013;146:297–304.
Noda Y, Ota K, Shirasawa T, Shimizu T. Copper/zinc superoxide dismutase insufficiency impairs progesterone secretion and fertility in female mice. Biol Reprod. 2012;86:1–8.
Ijiri TW, Vadnais ML, Lin AM, Huang AP, Cao W, Merdiushev T, et al. Male mice express spermatogenic cell-specific triosephosphate isomerase isozymes. Mol Reprod Dev. 2013;80:862–70.
Kirchhoff S, Kim JS, Hagendorff A, Thonnissen E, Kruger O, Lamers WH, et al. Abnormal cardiac conduction and morphogenesis in connexin40 and connexin43 double-deficient mice. Circ Res. 2000;87:399–405.
Anyaegbunam AM, Scarpelli S, Mikhail MS. Chronic hypertension in gestational diabetes: influence on pregnancy outcome. Gynecol Obstet Invest. 1995;39:167–70.
Hagstrom H, Hoijer J, Ludvigsson JF, Bottai M, Ekbom A, Hultcrantz R, et al. Adverse outcomes of pregnancy in women with non-alcoholic fatty liver disease. Liver Int. 2016;36:268–74.
Cheng N, Xiang T, Wu X, Li M, Xie Y, Zhang L. Acute fatty liver of pregnancy: a retrospective study of 32 cases in South China. J Matern Fetal Neonatal Med. 2014;27:1693–7.
Matteo M, Serviddio G, Massenzio F, Scillitani G, Castellana L, Picca G, et al. Reduced percentage of natural killer cells associated with impaired cytokine network in the secretory endometrium of infertile women with polycystic ovary syndrome. Fertil Steril. 2010;94:2222–7. 2227.e1-3
Huber-Buchholz MM, Carey DG, Norman RJ. Restoration of reproductive potential by lifestyle modification in obese polycystic ovary syndrome: role of insulin sensitivity and luteinizing hormone. J Clin Endocrinol Metab. 1999;84:1470–4.
Gunderson EP, Quesenberry CP Jr, Ning X, Jacobs DR Jr, Gross M, Goff DC Jr, et al. Lactation duration and midlife atherosclerosis. Obstet Gynecol. 2015;126:381-390
Skilton MR, Serusclat A, Begg LM, Moulin P, Bonnet F. Parity and carotid atherosclerosis in men and women: insights into the roles of childbearing and child-rearing. Stroke. 2009;40:1152–7.
Belo L, Santos-Silva A, Quintanilha A, Rebelo I. Similarities between pre-eclampsia and atherosclerosis: a protective effect of physical exercise? Curr Med Chem. 2008;15:2223–9.
Napoli C, Pignalosa O, Gallo L, Graziano G, Carotenuto F, Fiorito C, et al. Childhood infection and endothelial dysfunction: a potential synergistic link in atherosclerosis. Recenti Prog Med. 2005;96:474–7.
Martínez HR, Marioni SS, Escamilla Ocanas CE, Gonzalez Garza MT, Moreno-Cuevas JE. Amyotrophic lateral sclerosis in pregnancy: clinical outcome during the post-partum period after stem cell transplantation into the frontal motor cortex. Cytotherapy. 2014;16:402–5.
Palomba S, Santagni S, La Sala GB. Progesterone administration for luteal phase deficiency in human reproduction: an old or new issue? J Ovarian Res. 2015;8:77.
Daya S. Efficacy of progesterone support for pregnancy in women with recurrent miscarriage. A meta-analysis of controlled trials. Br J Obstet Gynaecol. 1989;96:275–80.
Nakata K, Yamashita N, Noda Y, Ohsawa I. Stimulation of human damaged sperm motility with hydrogen molecule. Med Gas Res. 2015;5:2.
Frazer KL, Porter S, Goss C. The genetics and implications of neuromuscular diseases in pregnancy. J Perinat Neonatal Nurs. 2013;27:205–14. quiz 215-6
del-Rio-Vellosillo M, Garcia-Medina JJ. Anesthetic considerations in HELLP syndrome. Acta Anaesthesiol Scand. 2016;60:144–57.
Flores JR, Marquez MF. Arrhythmias in pregnancy. How and when to treat? Arch Cardiol Mex. 2007;77:S2-24–31.
Frassica JJ, Orav EJ, Walsh EP, Lipshultz SE. Arrhythmias in children prenatally exposed to cocaine. Arch Pediatr Adolesc Med. 1994;148:1163–9.
Weissgerber TL, Turner ST, Mosley TH Jr, Kardia SL, Hanis CL, Milic NM, et al. Hypertension in pregnancy and future cardiovascular event risk in siblings. J Am Soc Nephrol. 2016;27:894–902.
Matthiesen NB, Henriksen TB, Gaynor JW, Agergaard P, Bach CC, Hjortdal VE, et al. Congenital heart defects and indices of fetal cerebral growth in a nationwide cohort of 924 422 liveborn infants. Circulation. 2016;133:566–75.
Lombo M, Fernandez-Diez C, Gonzalez-Rojo S, Navarro C, Robles V, Herraez MP. Transgenerational inheritance of heart disorders caused by paternal bisphenol A exposure. Environ Pollut. 2015;206:667–78.
Donofrio MT, Duplessis AJ, Limperopoulos C. Impact of congenital heart disease on fetal brain development and injury. Curr Opin Pediatr. 2011;23:502–11.
Martiney JA, Cerami A, Inhibition SAF. of hemozoin formation in Plasmodium falciparum trophozoite extracts by heme analogs: possible implication in the resistance to malaria conferred by the β-thalassemia trait. Mol Med. 1996;2:236–46.
Nalls MA, Wilson JG, Patterson NJ, Tandon A, Zmuda JM, Huntsman S, et al. Admixture mapping of white cell count: genetic locus responsible for lower white blood cell count in the Health ABC and Jackson Heart studies. Am J Hum Genet. 2008;82:81–7.
Michon P, Woolley I, Wood EM, Kastens W, Zimmerman PA, Adams JH. Duffy-null promoter heterozygosity reduces DARC expression and abrogates adhesion of the P. vivax ligand required for blood-stage infection. FEBS Lett. 2001;495:111–4.
Boldt AB, Culpi L, Tsuneto LT, de Souza IR, Kun JF, Petzl-Erler ML. Diversity of the MBL2 gene in various Brazilian populations and the case of selection at the mannose-binding lectin locus. Hum Immunol. 2006;67:722–34.
Sziller I, Babula O, Hupuczi P, Nagy B, Rigo B, Szabo G, et al. Mannose-binding lectin (MBL) codon 54 gene polymorphism protects against development of pre-eclampsia, HELLP syndrome and pre-eclampsia-associated intrauterine growth restriction. Mol Hum Reprod. 2007;13:281–5.
Cervera A, Planas AM, Justicia C, Urra X, Jensenius JC, Torres F, et al. Genetically-defined deficiency of mannose-binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLoS One. 2010;5:e8433.
Starodubtseva NL, Sobolev VV, Soboleva AG, Nikolaev AA, Bruskin SA. Genes expression of metalloproteinases (MMP-1, MMP-2, MMP-9, and MMP-12) associated with psoriasis. Russ J Genet. 2011;47:1117–23.
Manetti M, Ibba-Manneschi L, Fatini C, Guiducci S, Cuomo G, Bonino C, et al. Association of a functional polymorphism in the matrix metalloproteinase-12 promoter region with systemic sclerosis in an Italian population. J Rheumatol. 2010;37:1852–7.
Hunninghake GM, Cho MH, Tesfaigzi Y, Soto-Quiros ME, Avila L, Lasky-Su J, et al. MMP12, lung function, and COPD in high-risk populations. N Engl J Med. 2009;361:2599–608.
Cao A, Galanello R. β-thalassemia. Genet Med. 2010;12:61–76.
Velzing-Aarts FV, van der Dijs FP, Muskiet FA, Duits AJ. The association of pre-eclampsia with the Duffy negative phenotype in women of West African descent. BJOG. 2002;109:453–5.
Kalia N, Singh J, Sharma S, Arora H, Kaur M. Genetic and phenotypic screening of mannose-binding lectin in relation to risk of recurrent vulvovaginal infections in women of North India: a prospective cohort study. Front Microbiol. 2017;8:75.
Pencovich N, Hantisteanu S, Hallak M, Fainaru O. Gonadotropin stimulation in mice leads to ovarian accumulation of immature myeloid cells and altered expression of proangiogenic genes. Eur J Obstet Gynecol Reprod Biol. 2014;179:75–82.
Huang QT, Chen JH, Hang LL, Liu SS, Zhong M. Activation of PAR-1/NADPH oxidase/ROS signaling pathways is crucial for the thrombin-induced sFlt-1 production in extravillous trophoblasts: possible involvement in the pathogenesis of preeclampsia. Cell Physiol Biochem. 2015;35:1654–62.
Psihogios V, Rodda C, Reid E, Clark M, Clarke C, Bowden D. Reproductive health in individuals with homozygous β-thalassemia: knowledge, attitudes, and behavior. Fertil Steril. 2002;77:119–27.
Le Gouez A, Benachi A, Mercier FJ. Fever and pregnancy. Anaesth Crit Care Pain Med. 2016;35:S5–12.
Hsu WY, Wu CH, Hsieh CT, Lo HC, Lin JS, Kao MD. Low body weight gain, low white blood cell count and high serum ferritin as markers of poor nutrition and increased risk for preterm delivery. Asia Pac J Clin Nutr. 2013;22:90–9.
Wikstrand MH, Hard AL, Niklasson A, Smith L, Lofqvist C, Hellstrom A. Maternal and neonatal factors associated with poor early weight gain and later retinopathy of prematurity. Acta Paediatr. 2011;100:1528–33.
Mendola P, Mumford SL, Mannisto TI, Holston A, Reddy UM, Laughon SK. Controlled direct effects of preeclampsia on neonatal health after accounting for mediation by preterm birth. Epidemiology. 2015;26:17–26.
Allen R, Rogozinska E, Sivarajasingam P, Khan KS, Thangaratinam S. Effect of diet- and lifestyle-based metabolic risk-modifying interventions on preeclampsia: a meta-analysis. Acta Obstet Gynecol Scand. 2014;93:973–85.
Rayman MP, Searle E, Kelly L, Johnsen S, Bodman-Smith K, Bath SC, et al. Effect of selenium on markers of risk of pre-eclampsia in UK pregnant women: a randomised, controlled pilot trial. Br J Nutr. 2014;112:99–111.
Hux VJ, Roberts JM. A potential role for allostatic load in preeclampsia. Matern Child Health J. 2015;19:591–7.
Silva LM, Coolman M, Steegers EA, Jaddoe VW, Moll HA, Hofman A, et al. Low socioeconomic status is a risk factor for preeclampsia: the Generation R Study. J Hypertens. 2008;26:1200–8.
Melchiorre K, Thilaganathan B. Maternal cardiac function in preeclampsia. Curr Opin Obstet Gynecol. 2011;23:440–7.
England L, Zhang J. Smoking and risk of preeclampsia: a systematic review. Front Biosci. 2007;12:2471–83.
Conde-Agudelo A, Belizan JM. Risk factors for pre-eclampsia in a large cohort of Latin American and Caribbean women. BJOG. 2000;107:75–83.
Spinillo A, Capuzzo E, Colonna L, Piazzi G, Nicola S, Baltaro F. The effect of work activity in pregnancy on the risk of severe preeclampsia. Aust N Z J Obstet Gynaecol. 1995;35:380–5.
Gundlapalli AV, Scalchunes C, Boyle M, Hill HR. Fertility, pregnancies and outcomes reported by females with common variable immune deficiency and hypogammaglobulinemia: results from an internet-based survey. J Clin Immunol. 2015;35:125–34.
Brinker KA, Silk HJ. Common variable immune deficiency and treatment with intravenous immunoglobulin during pregnancy. Ann Allergy Asthma Immunol. 2012;108:464–5.
Bereczki D Jr. Pregnancy and acute ischemic stroke. Orv Hetil. 2016;157:763–6.
Savitz DA, Danilack VA, Elston B, Lipkind HS. Pregnancy-induced hypertension and diabetes and the risk of cardiovascular disease, stroke, and diabetes hospitalization in the year following delivery. Am J Epidemiol. 2014;180:41–4.
Reilly CR, Cuesta-Fernandez A, Kayaleh OR. Successful gestation and delivery using clopidogrel for secondary stroke prophylaxis: a case report and literature review. Arch Gynecol Obstet. 2014;290:591–4.
Luo L, Chen D, Qu Y, Wu J, Li X, Mu D. Association between hypoxia and perinatal arterial ischemic stroke: a meta-analysis. PLoS One. 2014;9:e90106.
Mann JR, McDermott S, Pan C, Hardin JW. Maternal hypertension and intrapartum fever are associated with increased risk of ischemic stroke during infancy. Dev Med Child Neurol. 2013;55:58–64.
Tate J, Bushnell C. Pregnancy and stroke risk in women. Womens Health (Lond). 2011;7:363–74.
Le Flem L, Picard V, Emmerich J, Gandrille S, Fiessinger JN, Aiach M, et al. Mutations in promoter region of thrombomodulin and venous thromboembolic disease. Arterioscler Thromb Vasc Biol. 1999;19:1098–104.
Arnaud E, Barbalat V, Nicaud V, Cambien F, Evans A, Morrison C, et al. Polymorphisms in the 5' regulatory region of the tissue factor gene and the risk of myocardial infarction and venous thromboembolism: the ECTIM and PATHROS studies. Etude Cas-Témoins de l'Infarctus du Myocarde. Paris Thrombosis case-control Study. Arterioscler Thromb Vasc Biol. 2000;20:892–8.
Kavlie A, Hiltunen L, Rasi V, Prydz H. Two novel mutations in the human coagulation factor VII promoter. Thromb Haemost. 2003;90:194–205.
Suzuki K, Hayashi T. Protein C and its inhibitor in malignancy. Semin Thromb Hemost. 2007;33:667–72.
Lay AJ, Liang Z, Rosen ED, Castellino FJ. Mice with a severe deficiency in protein C display prothrombotic and proinflammatory phenotypes and compromised maternal reproductive capabilities. J Clin Invest. 2005;115:1552–61.
Schambeck CM, Grossmann R, Zonnur S, Berger M, Teuchert K, Spahn A, et al. High factor VIII (FVIII) levels in venous thromboembolism: role of unbound FVIII. Thromb Haemost. 2004;92:42–6.
Sood R, Sholl L, Isermann B, Zogg M, Coughlin SR, Weiler H. Maternal Par4 and platelets contribute to defective placenta formation in mouse embryos lacking thrombomodulin. Blood. 2008;112:585–91.
Cocco E, Varughese J, Buza N, Bellone S, Lin KY, Bellone M, et al. Tissue factor expression in ovarian cancer: implications for immunotherapy with hI-con1, a factor VII-IgGF(c) chimeric protein targeting tissue factor. Clin Exp Metastasis. 2011;28:689–700.
Khealani B, Farhat Z, Mozaffar T. Factor XI deficiency-related spontaneous primary intraventricular hemorrhage. South Med J. 2000;93:1017–8.
Witzke G, Bork K, Benes P, Bockers M. Hereditary angioneurotic oedema and blood-coagulation: interaction between C1-esterase-inhibitor and the activation factors of the proteolytic enzyme systems. Klin Wochenschr. 1983;61:1131–5.
Lin HF, Maeda N, Smithies O, Straight DL, Stafford DW. A coagulation factor IX-deficient mouse model for human hemophilia B. Blood. 1997;90:3962–6.
Ameri A, Kurachi S, Sueishi K, Kuwahara M, Kurachi K. Myocardial fibrosis in mice with overexpression of human blood coagulation factor IX. Blood. 2003;101:1871–3.
Dumas G, Dufresne M, Asselin E, Girouard J, Carrier C, Reyes-Moreno C. CD40 pathway activation reveals dual function for macrophages in human endometrial cancer cell survival and invasion. Cancer Immunol Immunother. 2013;62:273–83.
Lyons TR, Borges VF, Betts CB, Guo Q, Kapoor P, Martinson HA, et al. Cyclooxygenase-2-dependent lymphangiogenesis promotes nodal metastasis of postpartum breast cancer. J Clin Invest. 2014;124:3901–12.
Maller O, Hansen KC, Lyons TR, Acerbi I, Weaver VM, Prekeris R, et al. Collagen architecture in pregnancy-induced protection from breast cancer. J Cell Sci. 2013;126:4108–10.
Rana K, Thaper D, Prabha V. Is there a role for Serratia marcescens in male infertility: An experimental study? Microb Pathog. 2017;105:13–8.
Moretti E, Figura N, Campagna MS, Gonnelli S, Iacoponi F, Collodel G. Sperm parameters and semen levels of inflammatory cytokines in Helicobacter pylori-infected men. Urology. 2015;86:41–6.
Burnazian RA, Oganesian MN, Burshtein GE. The biochemical indices of the sperm in chronic inflammatory diseases of the accessory genital glands. Urol Nefrol (Mosk). 1992:36–8.
I B. Inflammatory gynecological diseases as a social problem in women of reproductive age. Akush Ginekol (Sofiia). 1999;38:33–5.
Sun Y, Cui M, Zhu W, Xu W, Li N. A case report of a pregnancy-related death caused by primary antiphospholipid antibody syndrome. Int Med Case Rep J. 2014;7:159–63.
Lopez-Pedrera C, Buendía P, Barbarroja N, Siendones E, Velasco F, Cuadrado MJ. Antiphospholipid-mediated thrombosis: interplay between anticardiolipin antibodies and vascular cells. Clin Appl Thromb Hemost. 2006;12:41–5.
Hughes G. Hughes syndrome (antiphospholipid syndrome) and myocardial infarction. Int J Clin Pract. 2010;64:1183–4.
Soderberg LH, Johansen PP, Herning M, Berg SK. Women's experiences of sexual health after first-time myocardial infarction. J Clin Nurs. 2013;22:3532–40.
Rosa GM, Bauckneht M, Ferrero S, Leone Roberti Maggiore U, Brunelli C. Acute myocardial infarction in pregnancy. G Ital Cardiol (Rome). 2013;14:126–34.
Bush N, Nelson-Piercy C, Spark P, Kurinczuk JJ, Brocklehurst P, Knight M, et al. Myocardial infarction in pregnancy and postpartum in the UK. Eur J Prev Cardiol. 2013;20:12–20.
La Vecchia C, Decarli A, Franceschi S, Gentile A, Negri E, Parazzini F. Menstrual and reproductive factors and the risk of myocardial infarction in women under fifty-five years of age. Am J Obstet Gynecol. 1987;157:1108–12.
Letourneau J, Chan J, Salem W, Chan SW, Shah M, Ebbel E, et al. Fertility sparing surgery for localized ovarian cancers maintains an ability to conceive, but is associated with diminished reproductive potential. J Surg Oncol. 2015;112:26–30.
Crawford NM, Pritchard DA, Herring AH, Steiner AZ. Prospective evaluation of the impact of intermenstrual bleeding on natural fertility. Fertil Steril. 2016;105:1294–300.
Papp Z, Toth-Pal E, Papp C, Sziller I, Silhavy M, Gavai M, et al. Bilateral hypogastric artery ligation for control of pelvic hemorrhage, reduction of blood flow and preservation of reproductive potential. Experience with 117 cases. Orv Hetil. 2005;146:1279–85.
Fotopoulou C, Dudenhausen JW. Uterine compression sutures for preserving fertility in severe postpartum haemorrhage: an overview 13 years after the first description. J Obstet Gynaecol. 2010;30:339–49.
Adams Hillard PJ. Menstruation in adolescents: what's normal, what's not. Ann N Y Acad Sci. 2008;1135:29–35.
den Tonkelaar I, Oddens BJ. Preferred frequency and characteristics of menstrual bleeding in relation to reproductive status, oral contraceptive use, and hormone replacement therapy use. Contraception. 1999;59:357–62.
Koeberle P, Pequenot-Jeannin C, Douge C, Barale F. Angioneurotic edema and anesthesia: preparation and perioperative monitoring. Ann Fr Anesth Reanim. 1994;13:417–20.
Hardy F, Ngwingtin L, Bazin C, Babinet P. Hereditary angioneurotic edema and pregnancy. J Gynecol Obstet Biol Reprod (Paris). 1990;19:65–8.
Hopkinson RB, Sutcliffe AJ. Hereditary angioneurotic oedema. Anaesthesia. 1979;34:183–6.
Yu F, Zheng A, Qian J, Li Y, Wu L, Yang J, et al. Prenatal nicotine exposure results in the myocardial fibrosis in the adult male offspring rats. Exp Toxicol Pathol. 2016;68:445–50.
Chen X, Tang Y, Gao M, Qin S, Zhou J, Li X. Prenatal exposure to lipopolysaccharide results in myocardial fibrosis in rat offspring. Int J Mol Sci. 2015;16:10986–96.
Lowenstine LJ, McManamon R, Terio KA. Comparative pathology of aging great apes: Bonobos, Chimpanzees, Gorillas, and Orangutans. Vet Pathol. 2016;53:250–76.
James AH, Hoots K. The optimal mode of delivery for the haemophilia carrier expecting an affected infant is caesarean delivery. Haemophilia. 2010;16:420–4.
Kadir RA, Sabin CA, Goldman E, Pollard D, Economides DL, Lee CA. Reproductive choices of women in families with haemophilia. Haemophilia. 2000;6:33–40.
Yang YC, Fu HC, Wu CY, Wei KT, Huang KE, Kang HY. Androgen receptor accelerates premature senescence of human dermal papilla cells in association with DNA damage. PLoS One. 2013;8:e79434.
Perez-Sepulveda A, Espana-Perrot PP, Fernandez XB, Ahumada V, Bustos V, Arraztoa JA. Levels of key enzymes of methionine-homocysteine metabolism in preeclampsia. Biomed Res Int. 2013;2013:731962.
Mikael LG, Pancer J, Jiang X, Wu Q, Caudill M, Rozen R. Low dietary folate and methylenetetrahydrofolate reductase deficiency may lead to pregnancy complications through modulation of ApoAI and IFN-γ in spleen and placenta, and through reduction of methylation potential. Mol Nutr Food Res. 2013;57:661–70.
Zhou R, Chen F, Chang F, Bai Y, Chen L. Persistent overexpression of DNA methyltransferase 1 attenuating GABAergic inhibition in basolateral amygdala accounts for anxiety in rat offspring exposed perinatally to low-dose bisphenol A. J Psychiatr Res. 2013;47:1535–44.
Matrisciano F, Tueting P, Dalal I, Kadriu B, Grayson DR, Davis JM, et al. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology. 2013;68:184–94.
Cannuyer J, Van Tongelen A, Loriot A, De Smet C. A gene expression signature identifying transient DNMT1 depletion as a causal factor of cancer-germline gene activation in melanoma. Clin Epigenetics. 2015;7:114.
Xu B, Gao L, Cui Y, Gao L, Dai X, Li M, et al. SET protein up-regulated testosterone production in the cultured preantral follicles. Reprod Biol Endocrinol. 2013;11:9.
Marsh CA, Auchus RJ. Fertility in patients with genetic deficiencies of cytochrome P450c17 (CYP17A1): combined 17-hydroxylase/17,20-lyase deficiency and isolated 17,20-lyase deficiency. Fertil Steril. 2014;101:317–22.
Ferraz-de-Souza B, Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, NR5A1) and human disease. Mol Cell Endocrinol. 2011;336:198–205.
Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak SM, et al. Variation in transcription factor binding among humans. Science. 2010;328:232–5.
1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, MA DP, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65.
Pugh BF. Purification of the human TATA-binding protein, TBP. Methods Mol Biol. 1995;37:359–67.
Savinkova L, Drachkova I, Arshinova T, Ponomarenko P, Ponomarenko M, Kolchanov N. An experimental verification of the predicted effects of promoter TATA box polymorphisms associated with human diseases on interactions between the TATA boxes and TATA-binding protein. PLoS One. 2013;8:e54626.
Arkova O, Kuznetsov N, Fedorova O, Savinkova L. A real-time study of the interaction of TBP with a TATA box-containing duplex identical to an ancestral or minor allele of human gene LEP or TPI. J Biomol Struct Dyn. 2016; https://doi.org/10.1080/07391102.2016.1241190.
Wolfe A, Kim HH, Tobet S, Stafford DE, Radovick S. Identification of a discrete promoter region of the human GnRH gene that is sufficient for directing neuron-specific expression: a role for POU homeodomain transcription factors. Mol Endocrinol. 2002;16:435–49.
Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–8.
Waardenberg AJ, Basset SD, Bouveret R, Harvey RP. CompGO: an R package for comparing and visualizing Gene Ontology enrichment differences between DNA binding experiments. BMC Bioinformatics. 2015;16:275.
Ponomarenko PM, Savinkova LK, Drachkova IA, Lysova MV, Arshinova TV, Ponomarenko MP, et al. A step-by-step model of TBP/TATA box binding allows predicting human hereditary diseases by single nucleotide polymorphism. Dokl. Biochem. Biophys. 2008;419:88–92.
Hahn S, Buratowski S, Sharp PA, Guarente L. Yeast TATA-binding protein TFIID binds to TATA elements with both consensus and nonconsensus DNA sequences. Proc Natl Acad Sci USA. 1989;86:5718–22.
Ponomarenko JV, Ponomarenko MP, Frolov AS, Vorobyev DG, Overton GC, Kolchanov NA. Conformational and physicochemical DNA features specific for transcription factor binding sites. Bioinformatics. 1999;15:654–68.
Karas H, Knuppel R, Schulz W, Sklenar H, Wingender E. Combining structural analysis of DNA with search routines for the detection of transcription regulatory elements. Comput Appl Biosci. 1996;12:441–6.
Bucher P. Weight matrix descriptions of four eukaryotic RNA polymerase II promoter elements derived from 502 unrelated promoter sequences. J Mol Biol. 1990;212:563–78.
Ponomarenko MP, Ponomarenko JV, Frolov AS, Podkolodny NL, Savinkova LK, Kolchanov NA, et al. Identification of sequence-dependent DNA features correlating to activity of DNA sites interacting with proteins. Bioinformatics. 1999;15:687–703.
Sokolenko AA, Sandomirskii II, Savinkova LK. Interaction of yeast TATA-binding protein with short promotor segments. Mol Biol (Mosk). 1996;30:279–85.
Drachkova I, Savinkova L, Arshinova T, Ponomarenko M, Peltek S, Kolchanov N. The mechanism by which TATA box polymorphisms associated with human hereditary diseases influence interactions with the ТАТА-binding protein. Hum Mutat. 2014;35:601–8.
Drachkova IA, Ponomarenko PM, Arshinova TV, Ponomarenko МP, Suslov VV, Savinkova LK, et al. In vitro examining the existing prognoses how TBP binds to TATA with SNP associated with human diseases. Health. 2011;3:577–83.
Ponomarenko PM, Ponomarenko MP, Drachkova IA, Lysova MV, Arshinova TV, Savinkova LK, et al. Prediction of the affinity of the TATA-binding protein to TATA boxes with single nucleotide polymorphisms. Mol Biol (Mosk). 2009;43:327–36.
Arkova OV, Kuznetsov NA, Fedorova OS, Kolchanov NA, Savinkova LK. Real-time interaction between ТВР and the TATA box of the human triosephosphate isomerase gene promoter in the norm and pathology. Acta Naturae. 2014;6:36–40.
Gunbin KV, Ponomarenko MP, Suslov VV, Gusev F, Fedonin GG, Rogaev EI. Evolution of brain active gene promoters in human lineage towards the increased plasticity of gene regulation. Mol Neurobiol. 2017; https://doi.org/10.1007/s12035-017-0427-4.
Ponomarenko PM, Ponomarenko MP. Sequence-based prediction of transcription up-regulation by auxin in plants. J Bioinform Comput Biol. 2015;13:1540009.
Drachkova IA, Shekhovtsov SV, Peltek SE, Ponomarenko PM, Arshinova TV, Ponomarenko MP, et al. Surface plasmon resonance study of the interaction between the human TATA box binding protein and the TATA element of the NOS2A gene promoter. Vavilov. J Genet Breed. 2012;16:391–6.
Suslov VV, Ponomarenko PM, Ponomarenko MP, Drachkova IA, Arshinova TV, Savinkova LK, et al. TATA box polymorphisms in genes of commercial and laboratory animals and plants associated with selectively valuable traits. Russ J Genet. 2010;46:394–403.
Suslov VV, Ponomarenko PM, Efimov VM, Savinkova LK, Ponomarenko MP, Kolchanov NA. SNPs in the HIV-1 TATA box and the AIDS pandemic. J Bioinform Comput Biol. 2010;8:607–25.
Mironova VV, Omelyanchuk NA, Ponomarenko PM, Ponomarenko MP, Kolchanov NA. Specific/nonspecific binding of TBP to promoter DNA of the auxin response factor genes in plants correlated with ARFs function on gene transcription (activator/repressor). Dokl Biochem Biophys. 2010;433:191–6.
Ponomarenko MP, Suslov VV, Gunbin KV, Ponomarenko PM, Vishnevsky OV, Kolchanov NA. Identification of the relationship between the variability of the expression of signaling pathway genes in the human brain and the affinity of TATA-binding protein to their promoters. Russ J Genet Appl Res. 2015;5:626–34.
NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2015;43:D6–17.
Missala I, Kassner U, Steinhagen-Thiessen E. A systematic literature review of the association of lipoprotein(a) and autoimmune diseases and atherosclerosis. Int J Rheumatol. 2012;2012:480784.
Burgner D, Rockett K, Ackerman H, Hull J, Usen S, Pinder M, et al. Haplotypic relationship between SNP and microsatellite markers at the NOS2A locus in two populations. Genes Immun. 2003;4:506-14
Casal AJ, Sinclair VJ, Capponi AM, Nicod J, Huynh-Do U, Ferrari P. A novel mutation in the steroidogenic acute regulatory protein gene promoter leading to reduced promoter activity. J Mol Endocrinol. 2006;37:71–80.
Matsunaga A, Sasaki J, Han H, Huang W, Kugi M, Koga T, et al. Compound heterozygosity for an apolipoprotein A1 gene promoter mutation and a structural nonsense mutation with apolipoprotein A1 deficiency. Arterioscler Thromb Vasc Biol. 1999;19:348–55.
Plengpanich W, Le Goff W, Poolsuk S, Julia Z, Guerin M, Khovidhunkit W. CETP deficiency due to a novel mutation in the CETP gene promoter and its effect on cholesterol efflux and selective uptake into hepatocytes. Atherosclerosis. 2011;216:370–3.
Niemann S, Broom WJ, Brown RH Jr. Analysis of a genetic defect in the TATA box of the SOD1 gene in a patient with familial amyotrophic lateral sclerosis. Muscle Nerve. 2007;36:704–7.
Watanabe M, Zingg BC, Mohrenweiser HW. Molecular analysis of a series of alleles in humans with reduced activity at the triosephosphate isomerase locus. Am J Hum Genet. 1996;58:308–16.
Vives-Corrons JL, Rubinson-Skala H, Mateo M, Estella J, Feliu E, Dreyfus JC. Triosephosphate isomerase deficiency with hemolytic anemia and severe neuromuscular disease: familial and biochemical studies of a case found in Spain. Hum Genet. 1978;42:171–80.
Wirka RC, Gore S, van Wagoner DR, Arking DE, Lubitz SA, Lunetta KL, et al. A common connexin-40 gene promoter variant affects connexin-40 expression in human atria and is associated with atrial fibrillation. Circ Arrhythm Electrophysiol. 2011;4:87–93.
Firouzi M, Ramanna H, Kok B, Jongsma HJ, Koeleman BP, Doevendans PA, et al. Association of human connexin40 gene polymorphisms with atrial vulnerability as a risk factor for idiopathic atrial fibrillation. Circ. Res. 2004;95:e29–33.
Clark IA, Rockett KA, Burgner D. Genes, nitric oxide and malaria in African children. Trends Parasitol. 2003;19:335–7.
Gonzalez-Martinez JA, Moddel G, Ying Z, Prayson RA, Bingaman WE, Najm IM. Neuronal nitric oxide synthase expression in resected epileptic dysplastic neocortex. J Neurosurg. 2009;110:343–9.
Acknowledgments
We are grateful to Shevchuk Editing (Brooklyn, NY, USA; URL: http://www.shevchuk-editing.com) for English editing.
Funding
The publication costs for this article were funded by grant #14.B25.31.0033 from the government of the Russian Federation, Resolution No. 220 (to Professor E.I. Rogaev as principal investigator, PI). The data compilation was supported by Russian Ministry of Science and Education under 5-100 Excellence Programme (for IVC and MPP). The experiment ex vivo was supported by project #0324-2016-0002 (for EVK, LVO, and AVO), the experiments in vitro were supported by project #0324-2016-0003 (for LKS, IAD, EBS, OVA, and DAZ), the software development was financed by projects #0324-2016-0008 (for DAR and NAK).
Availability of data and materials
Web service SNP_TATA_Comparator is public available (URL=http://beehive.bionet.nsc.ru/cgi-bin/mgs/tatascan/start.pl).
About this supplement
This article has been published as part of BMC Genomics Volume 19 Supplement 3, 2018: Selected articles from Belyaev Conference 2017: genomics. The full contents of the supplement are available online at https://bmcgenomics.biomedcentral.com/articles/supplements/volume-19-supplement-3.
Author information
Authors and Affiliations
Contributions
NAK conceived of and supervised the study. LKS, IAD, EBS, OVA, and DAZ conducted the in vitro experiments. EVK conducted the ex vivo experiments. PMP and DAR designed, developed, maintained, adapted, and tuned the software for sequence analysis. IVC analyzed data in silico. LVO and AVO interpreted the computer-based predictions in comparison with experimental data. MPP wrote the manuscript. All the coauthors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Supplementary method. A sequence-based statistical estimate of the SNP-caused alteration in the affinity of TATA box binding protein (TBP) for the human gene promoter containing this SNP within its region [-70; -20]. (PDF 220 kb)
Additional file 2:
Supplementary method. Keyword search in the PubMed database. (PDF 221 kb)
Additional file 3:
Table S1. Clinically known dependences between reproductive potential and hereditary diseases whose SNP markers were predicted in this work. (PDF 198 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chadaeva, I.V., Ponomarenko, P.M., Rasskazov, D.A. et al. Candidate SNP markers of reproductive potential are predicted by a significant change in the affinity of TATA-binding protein for human gene promoters. BMC Genomics 19 (Suppl 3), 0 (2018). https://doi.org/10.1186/s12864-018-4478-3
Published:
DOI: https://doi.org/10.1186/s12864-018-4478-3