
Abstract
Background
Rat liver regeneration (LR) proceeds along a process of highly organized and ordered tissue growth in response to the loss or injury of liver tissue, during which many physiological processes may play important roles. The molecular mechanism of hepatocyte proliferation, energy metabolism and substance metabolism during rat LR had been elucidated. Further, the correlation of circular RNA (circRNA) abundance with proliferation has recently been clarified. However, the regulatory capacity of circRNA in rat LR remains a fascinating topic.
Results
To investigate the regulatory mechanism of circRNA during priming phase of rat LR, high-throughput RNA sequencing technology was performed to unbiasedly profile the expression of circRNA during priming phase of rat LR. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) biological pathway analysis was conducted to predict the functions of differentially expressed circRNAs and their host linear transcripts. Co-expression networks of circRNA-miRNA were constructed based on the correlation analysis between the differentially expressed LR-related circRNAs and the condition of their miRNA binding sites. To excavate the key circRNAs in the early phase of rat LR, we comprehensively evaluated and integrated the relationship of expression level between the circRNAs and the linear transcripts as well as the distribution of miRNA binding sites in circRNA sequences.
Conclusions
This paper is the first to employ the comprehensive circRNA expression profile and to investigate circRNA-miRNA interactions during priming phase of rat LR. Two thousand four hundred twelve circRNAs were detected, and 159 circRNAs deriving from 116 host linear transcripts differentially expressed (p < 0.05). Six significantly changed circRNAs during priming phase of rat LR were screened as key circle molecules, and then were validated by qRT-PCR. This study will lay the foundation for revealing the functional roles of circRNAs during rat LR and help solve the remaining clinical problems.
Similar content being viewed by others


Background
LR is of great clinical significance in various liver-associated diseases [1]. As one of the vital organs in body, liver not only has the ability of detoxification, metabolism, biliation and immune defense, but has a high capacity to regenerate. Following 2/3 partial hepatectomy (PH), hepatocytes can be activated, and then enter cell cycle and proliferate to restore the original mass and function, which is called LR [2, 3]. LR is a complex process regulated by growth factors, cytokines and non-coding RNAs [4, 5].
CircRNAs, a novel type of non-coding RNAs, compose a class of RNA developing covalently closed loop structures without 5’–3’ polarities [6] but with widespread, abundant and tissue-specific expression across animals [7]. CircRNAs are generally formed by ‘backsplicing’ in which an upstream splice acceptor is joined to a downstream splice donor [8–12]. Although there isn’t consistent conclusion as to the function of circRNAs, a number of studies have revealed that circRNAs are classified into three types: circular exonic RNAs (ecircRNAs), circular intronic RNAs (ciRNAs) and exon-intron circRNAs (EiciRNAs) [6].
In recent years, the roles of circRNAs in the organism have been increasingly illuminated. CircRNAs can function as miRNA sponges, as templates for viroid and viral replication, as intermediates in RNA processing reactions, as regulators of transcription in cis, as snoRNAs (small nucleolar RNAs) [13], and so on. Different types of circRNAs may have distinct properties. CiRNAs and EiciRNAs might tend to be enriched in the nucleus and associate with the polymerase II elongation machinery to enhance expression of their corresponding parent genes [14, 15]. However, ecircRNAs have been identified to act as miRNA decoys to regulate the miRNA effects on their coding RNA targets by competing for miRNA binding, which seems like playing a regulation for competing endogenous RNA activities [7]. Hansen et al. [16] attested that circRNAs could act as miRNA sponges to regulate the expression of corresponding genes at the post-transcription level. For example, CDR1as (antisense to the cerebellar degeneration-related protein 1 transcript) with circularization into circRNA harboured 73 conserved binding sites for targeting miR-7, so it interacted with miR-7 and strongly suppressed miR-7 activity with resistant to miRNA-mediated target destabilization, resulting in increased levels of miR-7 targets [8]. Meanwhile, CDR1as was highly complementary to mature miR-671 and could be cleavaged by miR-671 [17]. Similarly, the transcript of sex-determining region Y (SRY) mainly in the form of circRNA in the adult testis [11] contained 18 conserved binding sites for miR-138, therefore it affected the target coding genes expression of miR-138. Nevertheless, some circRNAs had different conserved binding sites of different miRNAs, Yang [18] has verified that circ-FOXO3 could bind to eight miRNAs, effectively changing the translation of the target mRNA. Conclusively, circRNAs function as a sponge to bind a certain miRNA or multiple potential targeting miRNAs, resulting in changed gene expression.
A global reduction of circRNA abundance was reported in colorectal cancer [19], esophageal squamous cell carcinoma (ESCC) [20], gastric cancer [21], rectal cancer and ovarian cancer [19] compared to the peritumoral tissue, indicating that circRNAs might exert their important action in cell proliferation and apoptosis. Cell proliferation was the most important physiological activity during rat LR [22]. Consequently, an intriguing possibility was raised that alternative circRNA formation might constitute a novel regulatory layer in rat LR.
Salmena et al. hypothesized that these ceRNAs (competing endogenous RNAs), such as mRNAs, pseudogenes and long non-coding RNAs (lncRNAs), could communicate with each other through their ability to compete for miRNA binding. The communication generated wide-ranging cis and trans regulatory crosstalk across the transcriptome as a whole [23]. Recently enhancing evidences have indicated that circRNAs could function potent ceRNA molecules [6]. Much work has provided insights into the regulatory mechanism of non-coding RNA in rat LR including miRNA and lncRNA, little is known about the circRNA expression profile and the functions of circRNAs in rat LR. An in-depth study should be carried out to elucidate the potential role of circRNA, one of the ceRNAs, in rat LR. In this paper, high-throughput RNA sequencing technology was applied to detect the classification and expression level of circRNAs during priming phase of rat LR.it was Assumed that the circRNA function would be related to the known function of the host gene, we performed GO and KEGG biological pathway analysis to predict potential functions of circRNAs during priming phase of rat LR. LR-related circRNAs were further screened by analyzing the differentially expressed circRNAs, and then were validated by qRT-PCR.
Results
Expression pattern of circRNAs during priming phase of rat LR
High-throughput sequencing technology was used to detect the expression profile of circRNAs using the pipeline described by Memczak et al. [8], and the result showed that 2,412 circRNAs were identified in the regenerating rat livers at 0, 2 and 6 h after PH. The size of LR-related circRNA candidates ranged from under 100 nt to over 2000 nt, most of which was not very long in length. About 75% of circRNAs had the predicted spliced length of less than 2000 nt, wherein circRNAs with the length less than 500 nt were 45.56% and the length from 500 nt to 1000 nt were 21.43%. (Fig. 1a). Among the 2,412 circRNAs, 1104 circRNAs were detected at 0 h after PH, 1118 at 2 h, 1095 at 6 h, and 300 at all three time points (Fig. 1b), respectively. Expression abundance of circRNAs at three time-points after PH was measured based on RPM (mapped back-splicing junction reads per million mapped reads), and the result indicated that no abnormal expression was found in the three samples (Fig. 1c).

Expression pattern of circRNAs was detected by high-throughput RNA sequencing during priming phase of rat LR. a The length distribution of circRNAs. b Venn analysis of circRNAs detected at each time points. c Box plots of RPM value of circRNAs in three groups
Annotation of host linear transcripts and identification of differentially expressed circRNAs
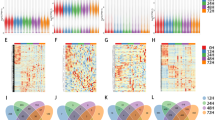
Biogenesis of circRNAs via backsplicing was different from the canonical splicing of linear RNAs, but they all generally formed by alternative splicing of pre-mRNA. To provide a comprehensive landscape of the origination of circRNAs, linear transcripts of circRNAs from the corresponding genes were annotated and the circRNA distribution in the genome was also explored according to the location of the chromosome where the circRNA sequence was overlapped (Additional file 1). It proved that there were several distribution patterns of circRNAs in the gemome (Fig. 2a). One hundred fifty-nine differentially expressed circRNAs were selected during priming phase of rat LR. Among them, compared with control group (CG, 0 h), 100 differentially expressed circRNAs were selected by MA-plot with 72 up-regulated and 28 down-regulated at 2 h after PH (Fig. 2b and c), while 104 were selected with 54 up-regulated and 50 down-regulated at 6 h after PH (Additional file 2, Fig. 2c and d) which elucidated that many circRNAs were down-regulated at 6 h compared to 2 h after PH. Furthermore, at both 2 and 6 h compared with CG after PH, 45 circRNAs were differentially expressed with 28 circRNAs up-regulated and 15 circRNAs down-regulated, while only two circRNAs were regulated oppositely (Fig. 2c and d).

Annotation of Linear transcripts and identification of differentially expressed circRNAs. a The distribution of circular RNAs in the rat genome. b MA-plot of differentially expressed circRNAs at 2 h after PH compared with CG. c Differentially up-expressed circRNAs during priming phase of rat LR. d Differentially down-expressed circRNAs during priming phase of rat LR
The biomathematical predicted biological function of host linear transcripts
Under the assumption that circRNA function would be related to the known function of the host linear transcripts, differentially regulated linear transcripts were further analyzed by DAVID bioinformatics resources 6.7 (http://david.abcc.ncifcrf.gov). GO analysis were made on three different aspects namely biological process (BP), cellular component (CC) and molecular function (MF) for host linear transcripts. Prediction terms with p-value less than 0.05 were selected and ranked by p-value. According to the results, 463 and 340 GO terms were found up regulated (p < 0.05) at 2 and 6 h after PH. In contrast, 107 and 264 GO terms were found down regulated (p < 0.05). Top ten generally changed GO terms in all comparison groups classified by BP, CC, MF and ranked by p-value were listed. The consequence indicated that at 2 h after PH compared to GC, the most enriched and meaningful BP terms were superoxide metabolic process, cell aging, superoxide anion generation, and reactive oxygen species metabolic process. As for CC, the most enriched terms were mostly about focal adhesion, stress fiber and endoplasmic reticulum membrane. The most enriched MF terms were also closely related with the regulatory function of RNA polymerase II and enzyme activity involving in metabolic activity (Table 1). At 6 h after PH compared to GC, the most meaningful BP terms were cellular aldehyde metabolic process, long-chain fatty acid transport and lipid storage. As for CC, the most enriched terms were mostly about lipid particle, apical plasma membrane and basolateral plasma membrane. The most enriched MF terms were also closely related with enzyme activity involving in energy metabolism and substance metabolism, such as L-aspartate:2-oxoglutarate aminotransferase activity, hydroxymethylglutaryl-CoA synthase activity, glyceryl-ether monooxygenase activity and so on (Additional file 3). The results showed that during the early phase of rat LR, highly expressed host linear transcripts were mainly functionally involved in the response to stress, energy metabolism and the regulation of cell proliferation; all of these responses were helpful for the repair of the injured liver. Moreover, KEGG pathway analysis was made, and pathways (p < 0.05) were also selected and ranked by p-value. The top five were listed for differentially changed host linear transcripts at 2 and 6 h compared to GC. KEGG pathway analysis also showed that the top regulated ones were steroid hormone biosynthesis, inflammatory mediator regulation of TRP channels, and biosynthesis of amino acids at 2 h after PH compared to GC (Table 2). The top regulated ones at 6 h after PH were bile secretion, tyrosine metabolism, and fatty acid degradation (Additional file 4).
Construction of the circRNA-miRNA co-expression network
In an attempt to reveal the co-expression pattern of circRNA-miRNA, the circRNA-miRNA co-expression networks were constructed based on the high-throughput RNA sequencing results. A constructed network map contained top 300 relationships between circRNAs and miRNAs ranked by p-value of the hypergeometric distribution (Fig. 3). In the network of the circRNA-miRNA co-expression at 2 h compared with CG, 12 miRNAs interacting with 59 circRNAs among the top 300 relationships were prognosed to have closer connections during rat LR (Fig. 3a). For example, miR-203a-3p was involved in liver regeneration, notably by inhibiting SOCS3, one known regulator of hepatic cell proliferation. Likewise, in the network of the circRNA-miRNA co-expression at 6 h compared with CG, ten miRNAs binding with 71 circRNAs had closer connections among which miR-150-5p had been verified to play a vital part during rat LR (Fig. 3b) [24].

The circRNA-miRNA co-expression network. a Network of the circRNA-miRNA co-expression at 2 h compared with CG during rat LR. b Network of the circRNA-miRNA co-expression at 6 h compared with CG during rat LR. Circle nodes represent circRNAs and triangle nodes represent miRNAs. The size of circle and triangle represents p-value with larger size owing smaller p-value
Screening of key circRNAs during priming phase of rat LR
After all circRNAs were screened during rat LR according to two methods, eight key circRNAs (circ1778, circ1366, circ15, circ3, circ2099, circ1338, circ2077, circ432) were selected according to method I (Table 3), and four circRNAs (circ2270, circ1678, circ137, circ1788) according to method II (Additional file 5). The key miRNAs related with rat LR were suppled to screen key circRNAs and were listed in Table 4. Based on the outcome of RNA high-throughput sequencing technology in rat liver, eight circRNAs selected out by method I were all up-regulated, and three of four circRNAs (circ2270, circ1678 and circ137) identified by method II were down-regulated during priming phase of rat LR.
Verification of the expression changes of key circRNAs during rat LR by qRT-PCR
Twelve selected circRNAs were experimentally tested in 0 h PH group. Both outward-facing primers and opposite-directed primers were designed to detect the circular and linear form. Head-to-tail splicing existed only in circular form was assayed by quantitative polymerase chain reaction (qPCR) after reverse transcription, with outward-facing primers and Sanger sequencing. The result showed that the circular form was only amplified using the outward-facing primers and cDNA as templates, but it couldn’t be amplified using genomic DNA. PCR assays using both cDNA and genomic DNA could produce linear RNA using opposite-directed set (Fig. 4).

Outward-facing primers amplify circRNAs in cDNA but not genomic DNA (gDNA)
To confirm further the circular characteristics and explore the expression changes of 12 key circRNAs during priming phase of rat LR, six circRNAs and four host linear transcripts were randomly selected and amplified using outward-facing primers by qRT-PCR with RNase R digestion at 0, 2 and 6 h after PH. As expected, the circRNAs were more resistant to RNase R treatment than linear control RNAs (Fig. 5). Interestingly, the relative quantitative expression of the circRNAs and their host linear transcripts with or without RNase R digestion had the similar trends during priming phase of rat LR. To further verify the expression changes of six key circRNAs during rat LR, they were also amplified by qRT-PCR. These data demonstrated that circ432, circ2077, circ1366 and circ15 selected by method I significantly changed during rat LR, as well as the consequence showed positive correlation between four circRNAs and their corresponding origination genes, MAPK14, FN1 (fibronectin 1), TNFRSF21 (tumor necrosis factor receptor superfamily member 21) and GOT1 (glutamic-oxaloacetic transaminase 1), respectively (Fig. 6a). In addition, circ137 and circ2270 selected by method II were significantly down-regulated at ten time points during rat LR (Fig. 6b). Therein, four circRNAs (circ432, circ2077, circ1366 and circ15) selected by method I were up-regulated and two circRNAs (circ137 and circ2270) selected by method II were down-regulated during priming phase of rat LR, which had accordance with the high-throughput sequencing result even though the abundance relative expression values of the genes detected by above two methods were not all the same, demonstrating reliability of the RNA high-throughput sequencing technology.

QRT-PCR analysis of differential decay of linear and circRNAs in the early phase of rat LR following the RNase R digestion. Expression levels were shown by CT values. (error bars indicate standard deviation)

The expression changes of key circRNAs during rat LR by qRT-PCR. aThe expression changes of four circRNAs selected by method I and their host linear transcripts during rat LR. b The expression changes of two circRNAs selected by method II during rat LR. Data represent the average value of at least three independent experiments
Discussion
Recently, the phenomenon of circRNA has gone from being perceived as a rare curiosity to having a central regulatory role in RNA metabolism [8, 11, 16, 25]. And the ever-growing discovery of circRNA with functional capacity showed that circRNA was a transcriptional product in various tissues and cell types of archaea [26], human [8, 10], mouse [8], drosophila [27] and nematodes [9] and in hundreds of cases constituted the dominant RNA isoform. Some circRNAs have been shown to affect microRNA regulation. Hansen et al. found that the circular transcripts of SRY had 18 miR-183 binding sites and that CDR1as had 73 miR-7 binding sites [16], so they proposed scientific hypothesis and demonstrated that circRNA could function as miRNA sponges to regulate the expression of target genes indirectly. Li et al. pointed out that cir-ITCH interacted with 3 molecules of miR-7, miR-17 and miR-214 to increase the level of ITCH and suppress the ESCC [20]. Wang et al. proved that HRCR (heart-related circRNA) could protect the heart from pathological hypertrophy and heart failure by targeting miR-223 [28]. Recently, Yang et al. demonstrated that circ-FOXO3 could function as a sponge to bind multiple potential targeting miRNAs, resulting in increased gene expression and inhibition of tumor growth and angiogenesis [18]. Thus, accumulating evidences suggested that circRNAs could function as miRNA sponges, and show distinct expression patterns coupled from their linear counterparts such as cir-ITCH and circ-FOXO3, or uncoupled from host gene expression such as CDR1as and HRCR.
To analyze the comprehensive circRNA expression profile by high-throughput RNA sequencing technology, 2,412 circRNAs were defined during priming phase of rat LR. One hundred fifty-nine differentially expressed circRNAs of them originated from 116 host linear transcripts. On one hand, 17 of 159 circRNAs were intergenic indicating that they did not have the corresponding linear transcripts. On the other hand, a gene could be spliced into one or more circRNAs [29]. Nineteen genes could produce more than one circRNAs during priming phase of rat LR, such as four circRNAs expressed from Cpamd8. The similar case had reported that ANRIL (antisense noncoding RNA in the INK4 Locus) encoded multiple, non-abundant linear and circular species and the expression of circular ANRIL isoforms (cANRIL) was correlated with INK4/ARF transcription and atherosclerotic vascular disease (ASVD) risk [30]. Furthermore, various circRNA isoforms from the same host linear transcripts expressed at different time points during priming phase of rat LR, implying the important function of temporal regulation of LR-related circRNAs.
To investigate the circRNA regulatory role in rat LR further, GO analysis was performed to annotate the biological functions of host linear transcripts during priming phase of rat LR, the result showed obviously that a significant amount of GO terms were related with the response to stress, energy metabolism and regulation of cell proliferation, which had been acknowledged as the important activities in the early stage of rat LR [1, 22, 31, 32]. The most enriched CC terms were about different kinds of plasma membranes. The plasma membranes were embedded with many proteins which could monitor and maintain the cell’s chemical climate and assist in the transfer of molecules across the membrane. The most enriched MF terms were closely related with diverse enzymes participating in energy metabolism and protein binding which was similar with circRNA function. Circ-FOXO3 interacted with the anti-senescent protein ID-1 and the transcription factor E2F1, as well as the anti-stress proteins FAK and HIF1a and retained them in the cytoplasm. As a result, these proteins could no longer exert their anti-senescent and anti-stress roles, resulting in increased cellular senescence [33]. The ciRNA, ci-ankrd52, largely accumulated to its sites of transcription, was associated with elongation Pol II machinery, and acted as a positive regulator of Pol II transcription [14]. In this way, circRNA might function as a scaffold for RNA binding proteins exerting its biological functions including regulating tanscription and so on. Coinciding well with the results of GO analysis, KEGG pathway analysis showed that linear transcripts were related with the signaling pathways closely linked to energy metabolism and substance metabolism including amino acid metabolism, fatty acid metabolism and bile secretion. All of them could provide a multitude of energy and materials to make preparation to trigger LR.
The amounting evidences have indicated that circRNAs might regulate the function of miRNAs acting as competing ceRNAs [8, 16, 20, 34]. In order to explore the role of the huge amount of circRNAs in rat LR, circRNA-miRNA co-expression networks were established to predict the relationships between circRNAs and miRNAs. Hitherto, there were no annotation of circRNAs in the circRNA-miRNA co-expression network, but some of the co-expressed miRNAs had already been proved functional crucially in rat LR. Exemplification showed that miR-150 regulated cell proliferation by targeting VEGF [25], besides miR-382 negatively regulated PTEN expression and increased Akt phosphorylation during LR [35]. From the co-expression network, the obvious conclusion was that miR-150 was related with nine circRNAs, and miR-382 was related with 22 circRNAs. Collectively, these results suggested that circRNAs might regulate rat LR by binding up to miRNAs that could regulate the associated gene expression.
Examining the roles of individual circRNAs will be the subject of future investigations. We screened the key circRNAs by two methods, and 12 key circRNAs during priming phase of rat LR were confirmed. Then, six of them were randomly selected to examine by qRT-PCR and sequenced correctly. Moreover, the expression of four circRNAs, circ432, circ2077, circ1366 and circ15 selected by method I was consistent with their linear transcripts MAPK14, FN1, TNFRSF21 and GOT1, respectively, affirming that our methods were creditable. Simultaneously, the host genes of these four circRNAs have been interestingly substantiated to be vital for cell proliferation and energy metabolism. For example, MAPK14, one member of the four p38 MAPKs, played an important place in the cascades of cellular responses evoked by extracellular stimuli such as proinflammatory cytokines or physical stress. FN1 was a glycoprotein component of the extracellular matrix which functioned in embryogenesis, development, wound healing and metastasis. Blocking the adhesion function of FN1 by inhibiting its expression with miR-204-3p could promote the growth of hepatocellular carcinoma tumor endothelial cells [36]. Tumor necrosis factor (TNF) receptors were key factors in inflammation and immune regulation. Like other death receptors (DR), TNFRSF21, also known as DR6, could interact with TRADD, which had previously been shown to associate with TNFR1. Furthermore, ectopic expression of TNFRSF21 in mammalian cells induced apoptosis and activation of both NF-κB and JNK [37]. GOT1 existed in cytoplasmic and mitochondrial forms and played a role in amino acid metabolism and the urea and tricarboxylic acid cycles. Hence, MAPK14 was involved in a wide variety of cellular processes such as proliferation, differentiation, and development. TNFRSF21 was strongly connected to tumorigenesis and apoptosis. FN1 encoded structural protein involving in cell adhesion and migration processes. GOT1 participated in energy and substance metabolism. Based on these investigation, A hypothesis was raised that circ432, circ2077, circ1366 and circ15 were novel and valuable parts in the process of hepatocyte proliferation, apoptosis, energy metabolism and substance metabolism during rat LR by altering the expression of their linear transcripts MAPK14, FN1, TNFRSF21 and GOT1, respectively. In addition, circ137 and circ2270 selected by method II were both significantly down-regulated during rat LR. The outcome of biomathematical prediction manifested that circ137 and circ2270 could both couple with miR-127. Some experiments have demonstrated that miR-127 was capable of suppressing the hepatocyte proliferation, notably by inhibiting BCL6 and SETD8 [38]. Thus, circ137 and circ2270 might regulate the hepatocyte proliferation to control rat LR process by interacting with miR-127. Taken together, this paper provided the comprehensive landscape of circRNA expression profile during priming phase of rat LR and determined six representative circRNA candidates.
Conclusions
In this work, 2,412 circRNAs were detected and 159 of them were preliminarily determined changing remarkably during priming phase of rat LR indicating their potential roles in rat LR. GO and KEGG biological pathway analysis indicated that host linear transcripts of circRNAs during the early phase of rat LR critically contributed to the process of energy metabolism, substance metabolism and hepatocyte proliferation. circ432, circ2077, circ1366 and circ15 might regulate the rat LR by controlling the expression level of MAPK14, FN1, TNFRSF21 and GOT1, respectively, involved in networks critical for hepatocyte proliferation, energy metabolism and substance metabolism. It was also scientifically predicted that circ137 and circ2270 could regulate the hepatocyte proliferation by interacting with miR-127. So we suggest that expression of circRNAs sponging up miRNAs might play an important regulatory role as well. The findings will undoubtedly facilitate the future analyses of the regulatory mechanism of circRNA in rat LR, which may provide further insights into the comprehension of the regenerative process and have great promise to solve clinical problems.
Methods
Preparation of rat LR model
Adult Sprague–Dawley rats weighing 200 ~ 220 g were obtained from the Animal Center of Henan Normal University. They were kept in a controlled temperature room at 19 ~ 23 °C with a relative humidity of 50 ~ 70%, illumination of a 12 h period (8:00–20:00), and free access to water and standard rodent chow for two months. Experiments were performed among 18 rats with six animals per group: two PH groups and one normal (CG). The rats in PH groups were performed 2/3 PH according to the methods of Higgins [39]. The rats were anesthetized and sacrificed at 0, 2, and 6 h after surgery, and the right lobe of liver were instantly removed and stored at −80 °C in RNA Later. All experiments complied with the Animal Protection Law of China and animal Ethics.
High-throughput RNA sequencing of circRNA related to rat LR
Liver tissues were harvested and pooled for total RNA extraction using TRIzol reagent. The quality of total RNA was tested by AGE (agarose gel electrophoresis) (100 V, 10 min). Mass, concentration, and purity of total RNA were measured by Spectrophotometer at 260 and 280 nm wavelengths. The samples with a ratio of OD260/280 of approximately 2.0 were used. CircRNAs were quantitatively analyzed by Shanghai OE Biotech (Shanghai, China). After removal ribosomal RNA and then building a library, a high-throughput RNA sequencing was performed. According to Memczak et al.’s methods [8], the clean reads were aligned to the reference genome by Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml). For unmapped reads, the junctions were picked out using back-splice algorithm. Finally, circRNAs were verified with a software developed by OE which were considered as the reference sequence for further analysis. The expression level of circRNAs was measured by "Mapped back-splicing junction reads per million mapped reads" (RPM).
Annotation of host linear transcripts and identification of differentially expressed circRNAs
Differentially expressed circRNAs were detected by the negative binomial distribution test based on the DESeq package. Two criteria were chosen: one was the foldchange of the same circRNA in two PH groups using 0 h group as control, and the other was p-value (p < 0.05) or FDR (false discovery rate) which was used to control filtration upon the statistics of alignment quality scores. Linear transcripts were annotated according to the location of the chromosome where the circRNA sequence was overlapped. Comparing the circRNA with genetic elements, the circRNA distribution in the genome could be explored.
GO & KEGG pathway analysis of linear transcripts
DAVID (Database for Annotation, Visualization and Integrated Discovery) was used to analyze the potential functions of linear transcripts. Gene functions were classified into three subgroups namely BP, CC and MF. The top enriched GO terms among the two PH groups ranked by enrichment score were presented, and the top 10 enriched GO terms during priming phase of rat LR were ranked by the number of differentially expressed linear transcripts. KEGG pathway analysis was performed to determine the involvement of linear transcripts in different biological pathways.
miRNA target prediction
Putative interactions between the miRNAs and circRNAs were evaluated using miRanda, investigating only perfect seed matching without gap of Wooble pairing (‘strict’ parameter). A hit between any expressed miRNA (including the new predicted miRNA) and a target circRNA was considered for a miRanda score of 140 or higher, corresponding to at least a perfect seed match.
CircRNA-miRNA co-expression network analysis during priming phase of rat LR
Evidences have showed that circRNAs could bind with miRNAs and function as natural miRNA sponges to influence related miRNAs’ activities. CircRNA-miRNA co-expression network was built based on the prediction of miRNA binding sites and the correlations between circRNA and miRNA that was ranked by miRanda according to p-value of the hypergeometric distribution. The top 300 circRNA-miRNA were selected to generate a network map with cytoscape software (V. 3.2.1). Circle nodes represented circRNAs and triangle nodes represented miRNAs. The size of circle and triangle represented p-value with larger size stand for smaller p-value.
The screening criteria of the key circRNAs during priming phase of rat LR
The correlation of circRNAs and origination genes was inconclusive. Some papers revealed the high correlation between circRNA abundance and expression levels of linear host genes across some cell lines [18], while others held the different view [19] that circRNA expression was often uncoupled with host gene expression [40, 41]. As a consequence, two methods were applied to screen the key circRNAs during rat LR.
Method I: Firstly, circRNAs were selected with foldchange ≥ 2 or foldchange ≤ 0.5 in expression levels and p < 0.05 in PH groups compared with CG. Then, the mRNA level of the host genes at ten time points of 0, 2, 6, 12, 24, 30, 36, 72, 120, and 168 h during rat LR was profiled using Rat Genome 230 2.0 array. Meanwhile the level of proteins coded by the host genes at ten time-points was investigated by two-dimensional differential gel electrophoresis (2D) and mass spectrometry (MS). The genes with mRNA expression change over 3 times and corresponding proteins change over twice were defined as significantly expression genes. If circRNAs and their host genes could both comply with the above standards, circRNAs were regarded as key circRNAs in rat LR.
Method II: This criterion was provided as following. Firstly circRNAs were selected that both conformed to p < 0.05 and foldchange ≥ 2 or foldchange ≤ 0.5 in PH groups compared with CG. Secondly, ecircRNAs were prioritized. Thirdly, the circRNAs with high expression levels may have the much possibility of developing more important functions. Last, the distribution of miRNA binding sites in circRNA sequence was predicted by miRanda (http://www.microrna.org/) software. The key miRNAs in rat LR confirmed in other papers such as miR-221 and miR-34a had the priority to be picked out, then circRNAs harbouring more than one above-mentioned miRNA binding sites were sorted out.
RNase R digestion
Rat LR DNase-treated total RNA (6 mg) was incubated 1 h at 37 °C with zero units (mock treatment) or 20 units of RNase R (Epicentre Biotechnologies). RNA was subsequently purified by phenol-chloroform extraction, retro-transcribed and used in qRT-PCR.
The expression level of circRNAs verification by qRT-PCR assay
To validate the reliability of high-throughput RNA sequencing and explore the expression trend of circRNAs during rat LR, the expression level of circRNAs was examined by qRT-PCR.
Reference to Memczak’s method [8], two sets of primers for each circRNAs were designed using the Primer Express software version 5.0 (Table 5): a outward-facing set which was expected to amplify only the circRNAs, and an opposite-directed set to amplify the linear forms.
Then total RNA was extracted, digested using RNase R and purified, cDNA was synthesized using the AMV reverse transcription kit (Promega, USA). Outward-facing primers were designed to amplify the fragment across the junction from cDNA, then the fragment was sequenced by Sangon Biological Engineering Company (Shanghai China). QRT-PCR was performed using Q-SYBR green Supermix (BioRad), and PCR-specific amplification was conducted in Rotor-Gene 3000 (Corbett Robotics, Australia). The expression of circRNAs was defined based on the threshold cycle (Ct), and relative expression levels were calculated via the 2-ΔΔCt method. GAPDH was served as internal standard control, and all reactions were performed in triplicate.
Abbreviations
- 2D:
-
Two-dimensional differential gel electrophoresis
- AGE:
-
Agarose gel electrophoresis
- ANRIL:
-
Antisense noncoding RNA in the INK4 Locus
- ASVD:
-
Atherosclerotic vascular disease
- BP:
-
Biological process
- cANRIL:
-
Circular ANRIL
- CC:
-
Cellular component
- CDR1as:
-
Antisense to the cerebellar degeneration-related protein 1 transcript
- ceRNAs:
-
Competing endogenous RNAs
- CG:
-
Control group
- DAVID:
-
Database for annotation, visualization and integrated discovery
- DR:
-
Death receptors
- ESCC:
-
Esophageal squamous cell carcinoma
- FDR:
-
False discovery rate
- FN1:
-
Fibronectin 1
- GO:
-
Gene ontology
- GOT:
-
Glutamic-oxaloacetic transaminase
- HRCR:
-
Heart-related circRNA
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- lncRNA:
-
Long non-coding RNAs
- LR:
-
Liver regeneration
- MF:
-
Molecular function
- MS:
-
Mass spectrometry
- PH:
-
Partial hepatectomy
- RPM:
-
Mapped back-splicing junction reads per million mapped reads
- SRY:
-
Sex-determining region Y
- TNF:
-
Tumor necrosis factor
- TNFRSF21:
-
Tumor necrosis factor receptor superfamily member 21
References
Yuan X, Miao M. Lipid metabolism and peroxisome proliferator-activated receptor signaling pathways participate in late-phase liver regeneration. J Proteome Res. 2011;10(3):1179–90.
Ave. SSNS. Heterogeneity and plasticity of hepatocyte lineage cells. Hepatol (Baltimore, Md). 2001;33(3):738–50.
Fausto N, Laird AD, Webber EM. Liver regeneration. 2. Role of growth factors and cytokines in hepatic regeneration. Faseb J. 1995;9(15):1527–36.
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt Phosphorylation of BAD Couples Survival Signals to the Cell-Intrinsic Death Machinery. Cell. 1997;91(2):231–41.
Kalimuthu S, Sekwon K. Cell survival and apoptosis signaling as therapeutic target for cancer: marine bioactive compounds. Int J Mol Sci. 2013;14(2):2334–54.
Qu S, Yang X, Li X, Wang J, Gao Y, Shang R, Sun W, Dou K, Li H. Circular RNA: A new star of noncoding RNAs. Cancer Lett. 2015;365(2):141–8.
Chen I, Chen CY, Chuang TJ. Biogenesis, identification, and function of exonic circular RNAs. Wiley Interdisciplinary Rev-RNA. 2015;6(5):563–79.
Jens M. Circular RNAs Are a Large Class of Animal RNAs with Regulatory Potency. Nature. 2013;495(7441):1439–49.
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141–57.
Stoffelen R, Jimenez MI, Dierckxsens C. Circular RNAs Are the Predominant Transcript Isoform from Hundreds of Human Genes in Diverse Cell Types. PLoS One. 2012;7(2):e30733.
Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P, Goodfellow P, Lovell-Badge R. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell. 1993;73(5):1019–30.
Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, Kinzler KW, Vogelstein B. Scrambled exons. Cell. 1991;64(3):607–13.
Lasda E, Parker R. Circular RNAs: diversity of form and function. RNA. 2014;20(12):1829–42.
Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L, Chen LL. Circular Intronic Long Noncoding RNAs. Mol Cell. 2013;51(6):792–806.
Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22(3):256–64.
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–8.
Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, Kjems J. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011;30(21):4414–22.
Yang W, Du WW, Li X, Yee AJ, Yang BB. Foxo3 activity promoted by non-coding effects of circular RNA and Foxo3 pseudogene in the inhibition of tumor growth and angiogenesis. Oncogene. 2016;35(30):3919–31.
Bachmayrheyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitnerhofmann T, Mesteri I, Grunt TW, Zeillinger R, Pils D. Correlation of circular RNA abundance with proliferation--exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep. 2015;5:8057.
Li F, Zhang L, Li W, Deng J, Zheng J, An M, Lu J, Zhou Y. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/beta-catenin pathway. Oncotarget. 2015;6(8):6001–13.
Li P, Chen S, Chen H, Mo X, Li T, Shao Y, Xiao B, Guo J. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin Chimica Acta. 2015;444:132–6.
Chen X, Zhao Y, Wang F, Bei Y, Xiao J, Yang C. MicroRNAs in Liver Regeneration. Cell Physiol Biochem. 2015;37(2):615–28.
Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-coding RNAs: A new frontier in the study of human diseases. Cancer Lett. 2013;339(2):159–66.
Yu ZY, Bai YN, Luo LX, Wu H, Zeng Y. Expression of microRNA-150 targeting vascular endothelial growth factor-A is downregulated under hypoxia during liver regeneration. Mol Med Rep. 2013;8(1):287–93.
Hansen TB, Kjems J, Damgaard CK. Circular RNA and miR-7 in cancer. Cancer Res. 2013;73(18):5609–12.
Danan M, Schwartz S, Edelheit S, Sorek R. Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res. 2012;40(7):3131–42.
Westholm J, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P, Celniker S, Graveley B, Lai E. Genome-wide Analysis of Drosophila Circular RNAs Reveals Their Structural and Sequence Properties and Age-Dependent Neural Accumulation. Cell Rep. 2014;9(5):1966–80.
Wang K, Long B, Liu F, Wang JX, Liu CY, Zhao B, Zhou LY, Sun T, Wang M, Yu T, et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J. 2016;37(33):2602–11.
Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell. 2014;159(1):134–47.
Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genetics. 2010;6(12):e1001233.
Toshima T, Shirabe K, Fukuhara T, Ikegami T, Yoshizumi T, Soejima Y, Ikeda T, Okano S, Maehara Y. Suppression of autophagy during liver regeneration impairs energy charge and hepatocyte senescence in mice. Hepatology (Baltimore, Md). 2014;60(1):290–300.
Ding L, Yang Y, Qu Y, Yang T, Wang K, Liu W, Xia W. Bile acid promotes liver regeneration via farnesoid X receptor signaling pathways in rats. Mol Med Rep. 2015;11(6):4431–7.
Du WW, Yang W, Chen Y, Wu ZK, Foster FS, Yang Z, Li X, Yang BB. Foxo3 circular RNA promotes cardiac senescence by modulating multiple factors associated with stress and senescence responses. Eur Heart J. 2016;ehw001.
Taulli R, Loretelli C, Pandolfi PP. From pseudo-ceRNAs to circ-ceRNAs: a tale of cross-talk and competition. Nat Struct Mol Biol. 2013;20(5):541–3.
Bei Y, Yang S, Fei W, Dimitrova-Shumkovska J, Yang X, Zhao Y, Liu J, Xiao J, Yang C. miR-382 targeting PTEN-Akt axis promotes liver regeneration. Oncotarget. 2015;7(2):1584–97.
Cui ZH, Shen SQ, Chen ZB, Hu C. Growth inhibition of hepatocellular carcinoma tumor endothelial cells by miR-204-3p and underlying mechanism. World J Gastroenterol. 2014;20(18):5493–504.
Pan G, Bauer JH, Haridas V, Wang S, Liu D, Yu G, Vincenz C, Aggarwal BB, Ni J, Dixit VM. Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett. 1998;431(3):351–6.
Pan C, Chen H, Wang L, Yang S, Fu H, Zheng Y, Miao M, Jiao B. Down-regulation of MiR-127 facilitates hepatocyte proliferation during rat liver regeneration. PLoS One. 2012;7(6):e39151.
Higgins GM, Anderson RM. Experimental pathology of the liver: restoration of the liver of the white rat following partial surgical removal. Arch Pathol. 1931;12:186–202.
Venø MT, Hansen TB, Venø ST, Clausen BH, Grebing M, Finsen B, Holm IE, Kjems J. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol. 2015;16(1):1–17.
Barrett SP, Salzman J. Circular RNAs: analysis, expression and potential functions. Development. 2016;143(11):1838–47.
Marquez RT, Wendlandt E, Galle CS, Keck K, McCaffrey AP. MicroRNA-21 is upregulated during the proliferative phase of liver regeneration, targets Pellino-1, and inhibits NF-kappaB signaling. Am J Physiol Gastrointest Liver Physiol. 2010;298(4):G535–41.
Yuan B, Dong R, Shi D, Zhou Y, Zhao Y, Miao M, Jiao B. Down-regulation of miR-23b may contribute to activation of the TGF-beta1/Smad3 signalling pathway during the termination stage of liver regeneration. FEBS Lett. 2011;585(6):927–34.
Song G, Sharma AD, Roll GR, Ng R, Lee AY, Blelloch RH, Frandsen NM, Willenbring H. MicroRNAs control hepatocyte proliferation during liver regeneration. Hepatology. 2010;51(5):1735–43.
John K, Hadem J, Krech T, Wahl K, Manns MP, Dooley S, Batkai S, Thum T, Schulze-Osthoff K, Bantel H. MicroRNAs play a role in spontaneous recovery from acute liver failure. Hepatology. 2014;60(4):1346–55.
Best J, Dollé L, Manka P, Coombes J, van Grunsven LA, Syn WK. Role of liver progenitors in acute liver injury. Front Physiol. 2013;4(4):258.
Michalopoulos GK. Liver regeneration after partial hepatectomy: critical analysis of mechanistic dilemmas. Am J Pathol. 2010;176(1):2–13.
Ng R, Song G, Roll GR, Frandsen NM, Willenbring H. A microRNA-21 surge facilitates rapid cyclin D1 translation and cell cycle progression in mouse liver regeneration. J Clin Invest. 2012;122(3):1097–108.
da Silva CG, Studer P, Skroch M, Mahiou J, Minussi DC, Peterson CR, Wilson SW, Patel VI, Ma A, Csizmadia E, et al. A20 promotes liver regeneration by decreasing SOCS3 expression to enhance IL-6/STAT3 proliferative signals. Hepatology. 2013;57(5):2014–25.
da Silva CG, Cervantes JR, Studer P, Ferran C. A20--an omnipotent protein in the liver: prometheus myth resolved? Adv Exp Med Biol. 2014;809:117–39.
Longo CR, Patel VI, Shrikhande GV, Scali ST, Csizmadia E, Daniel S, Sun DW, Grey ST, Arvelo MB, Ferran C. A20 protects mice from lethal radical hepatectomy by promoting hepatocyte proliferation via a p21waf1-dependent mechanism. Hepatology. 2005;42(1):156–64.
Zhou X, Liu W, Gu M, Zhou H, Zhang G. Helicobacter pylori infection causes hepatic insulin resistance by the c-Jun/miR-203/SOCS3 signaling pathway. J Gastroenterol. 2015;50(10):1027–40.
Chu F, Barkinge J, Hawkins S, Gudi R, Salgia R, Kanteti PV. Expression of Siva-1 protein or its putative amphipathic helical region enhances cisplatin-induced apoptosis in breast cancer cells: effect of elevated levels of BCL-2. Cancer Res. 2005;65(12):5301–9.
Yan-nan B, Zhao-yan Y, Li-xi L, Jiang Y, Qing-jie X, Yong Z. MicroRNA-21 accelerates hepatocyte proliferation in vitro via PI3K/Akt signaling by targeting PTEN. Biochem Biophys Res Commun. 2014;443(3):802–7.
Li JJ, Chan WH, Leung WY, Wang Y, Xu CS. MicroRNA-21 promotes proliferation of rat hepatocyte BRL-3A by targeting FASLG. Genet Mol Res. 2015;14(2):4150–60.
Fornari F, Gramantieri L, Ferracin M, Veronese A, Sabbioni S, Calin GA, Grazi GL, Giovannini C, Croce CM, Bolondi L, et al. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene. 2008;27(43):5651–61.
Chen X, Murad M, Cui YY, Yao LJ, Venugopal SK, Dawson K, Wu J. miRNA regulation of liver growth after 50% partial hepatectomy and small size grafts in rats. Transplantation. 2011;91(3):293–9.
Yuan Q, Loya K, Rani B, Mobus S, Balakrishnan A, Lamle J, Cathomen T, Vogel A, Manns MP, Ott M, et al. MicroRNA-221 overexpression accelerates hepatocyte proliferation during liver regeneration. Hepatology. 2013;57(1):299–310.
Zhou J, Ju W, Wang D, Wu L, Zhu X, Guo Z, He X. Down-regulation of microRNA-26a promotes mouse hepatocyte proliferation during liver regeneration. PLoS One. 2012;7(4):e33577.
Chen J, Wang M, Guo M, Xie Y, Cong YS. miR-127 regulates cell proliferation and senescence by targeting BCL6. PLoS One. 2013;8(11):e80266.
Khrapenko LI, Huang N, Swenson E, Braconi C, Patel T. 3 The MicroRNA miR-181b Is Involved in Extracellular Matrix Remodeling During Hepatic Regeneration Following Partial Hepatectomy. Gastroenterology. 2009;136(5):A-789.
Cirera-Salinas D, Pauta M, Allen RM, Salerno AG, Ramirez CM, Chamorro-Jorganes A, Wanschel AC, Lasuncion MA, Morales-Ruiz M, Suarez Y, et al. Mir-33 regulates cell proliferation and cell cycle progression. Cell Cycle. 2012;11(5):922–33.
Chen H, Sun Y, Dong R, Yang S, Pan C, Xiang D, Miao M, Jiao B. Mir-34a is upregulated during liver regeneration in rats and is associated with the suppression of hepatocyte proliferation. PLoS One. 2011;6(5):e20238.
Acknowledgements
The study was financially supported by the National Natural Science Foundation of China (No. 31572270) and Research Program of Foundation and Advanced Technology of Henan (No. 162300410144). We thank Shanghai OE Biotech Co., Ltd for data analysis.
Funding
The study was financially supported by the National Natural Science Foundation of China (No. 31572270) and Research Program of Foundation and Advanced Technology of Henan (No. 162300410144).
Authors’ contributions
LL wrote the manuscript and analyzed data. CX participated in its design and interpretation. LL and YC conducted the experiments. JG and CC critically revised the manuscript, and all authors have read and given approval of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
The results of annotating circRNA host linear transcripts and analysing differentially expressed circRNAs at 2 h and 6 h after PH compared with CG. (XLSX 71 kb)
Additional file 2:
MA-plot of differentially expressed circRNAs at 6 h after PH compared with CG. (PNG 14 kb)
Additional file 3:
GO annotations of host linear transcripts at 6 h after PH compared to GC. (DOCX 17 kb)
Additional file 4:
KEGG analysis of host linear transcripts at 6 h after PH compared to GC. (DOCX 13 kb)
Additional file 5:
8 circRNAs selected by method Method II. (XLSX 10 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Li, L., Guo, J., Chen, Y. et al. Comprehensive CircRNA expression profile and selection of key CircRNAs during priming phase of rat liver regeneration. BMC Genomics 18, 80 (2017). https://doi.org/10.1186/s12864-016-3476-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-016-3476-6