
Abstract
Background
Entomopathogenic associations between nematodes in the genera Steinernema and Heterorhabdus with their cognate bacteria from the bacterial genera Xenorhabdus and Photorhabdus, respectively, are extensively studied for their potential as biological control agents against invasive insect species. These two highly coevolved associations were results of convergent evolution. Given the natural abundance of bacteria, nematodes and insects, it is surprising that only these two associations with no intermediate forms are widely studied in the entomopathogenic context. Discovering analogous systems involving novel bacterial and nematode species would shed light on the evolutionary processes involved in the transition from free living organisms to obligatory partners in entomopathogenicity.
Results
We report the complete genome sequence of a new member of the enterobacterial genus Serratia that forms a putative entomopathogenic complex with Caenorhabditis briggsae. Analysis of the 5.04 MB chromosomal genome predicts 4599 protein coding genes, seven sets of ribosomal RNA genes, 84 tRNA genes and a 64.8 KB plasmid encoding 74 genes. Comparative genomic analysis with three of the previously sequenced Serratia species, S. marcescens DB11 and S. proteamaculans 568, and Serratia sp. AS12, revealed that these four representatives of the genus share a core set of ~3100 genes and extensive structural conservation. The newly identified species shares a more recent common ancestor with S. marcescens with 99 % sequence identity in rDNA sequence and orthology across 85.6 % of predicted genes. Of the 39 genes/operons implicated in the virulence, symbiosis, recolonization, immune evasion and bioconversion, 21 (53.8 %) were present in Serratia while 33 (84.6 %) and 35 (89 %) were present in Xenorhabdus and Photorhabdus EPN bacteria respectively.
Conclusion
The majority of unique sequences in Serratia sp. SCBI (South African Caenorhabditis briggsae Isolate) are found in ~29 genomic islands of 5 to 65 genes and are enriched in putative functions that are biologically relevant to an entomopathogenic lifestyle, including non-ribosomal peptide synthetases, bacteriocins, fimbrial biogenesis, ushering proteins, toxins, secondary metabolite secretion and multiple drug resistance/efflux systems. By revealing the early stages of adaptation to this lifestyle, the Serratia sp. SCBI genome underscores the fact that in EPN formation the composite end result – killing, bioconversion, cadaver protection and recolonization- can be achieved by dissimilar mechanisms. This genome sequence will enable further study of the evolution of entomopathogenic nematode-bacteria complexes.
Similar content being viewed by others

Background
We present the complete genome sequence and analysis of a novel Serratia species that, in conjunction with the nematode Caenorhabditis briggsae, forms a putative entomopathogenic association lethal to Galleria mellonella larvae [2]. This Serratia species was isolated from C. briggsae nematodes recovered from three separate Galleria traps baited in soil in the Kawa Zulu Natal province in South Africa and resembles the other entomopathogenic nematode (EPN) associations. EPNs are mutualistic associations between a bacterium and a nematode that enables them to kill insects and benefit both partners with nutrients and breeding sites [16, 41]. Although all three players in the EPN life cycle - pathogenic bacteria, nematode and host insect larvae – are ancient and abundant taxa in nature, only two independently evolved entomopathogenic partnerships are well studied. One is the association between bacteria in the genus Photorhabdus and Heterorhabditid nematodes [29, 100] and the other is the association between bacteria in the genus Xenorhabdus with Steinernematid nematodes [46]. EPN associations involve complex interactions between the pathogens and the nematode worms. In typical EPN associations the nematode is responsible for locating suitable host, penetrating the host insect and releasing the bacteria into the hemocoel while the bacteria are responsible for killing the host, bioconversion of complex compounds and protection of the insect cadaver from scavenging competitors thus ensuring nutrition for itself and its nematode partner [29, 100].
Significant bacterial adaptations to the EPN lifestyle include the regulation of the switch between mutualism and pathogenicity, accelerated insect killing, cadaver bioconversion, and re-association with infective juveniles [23, 48]. Recent studies have revealed that in both canonical EPN bacterial species L-proline in the insect hemolymph is the main trigger that initiates a metabolic shift from a quasi-dormant state in the nematode gut to a dramatic increase in secondary metabolite production in the insect hemocoel [33]. Following this L-proline-induced metabolic shift, major regulatory events take place. In the Photorhabdus/Heterorhabdus association, two global regulators, HexA [58] and Ner [69], control the switch between mutualism and pathogenesis, while the phoP/phoQ and the astS/astR two-component systems [38, 39] and the pgbPE operon [12] regulate pathogenicity and mutualism genes. Furthermore, Heterorhabditid nematodes fail to grow and reproduce normally when grown with Photorhabdus mutants defective in ngrA, suggesting that its phosphopantetheinyl (Ppant) transferase product is required for nematode growth and reproduction [27]. Finally, the reassociation of infective juveniles and their cognate bacteria as well as the retention of the bacteria in the nematode gut seems to be mediated by the expression of adhesion fimbriae encoded in one of the many genomic islands rich in phage remnants [46, 68, 86]. By comparison, in the Xenorhabdus/Steinernema association, a similar, but non-homologous, mechanism operates in which the global regulator Lrp and the two component system cpxRA and the lysR regulator lrhA [32] orchestrate all three major stages of the life cycle: infection, reproduction and transmission. Whereas many compounds are implicated in insect killing and sanitization of the insect cadaver [84, 85] transmission in X. nematophila seems to require the nilABC operon which encodes three surface-localized colonization factors whose mutations invariantly lead to defective recolonization of Steinernematid worms by X. nematophila bacteria [77, 78]. The absence of the nilABC genes in X. bovienii and in Photorhabdus species suggest that bacteria-nematode recolonization is realized by different mechanisms in these two well-studied EPN systems. To summarize, the genetic mechanisms by which Photorhabdus and Xenorhabdus achieve entomopathogenicity are quite distinct and evidence of independent evolution of a similar phenotype.
The bacterium we describe here, Serratia sp. SCBI, belongs to the genus Serratia that consists of several species with diverse lifestyles that include free soil dwellers [49], plant associates in the rhizosphere [11, 34, 88, 98], opportunistic pathogens [66, 96, 97] and obligate intracellular endosymbionts [22]. Most Serratia spp. secrete an array of active extracellular enzymes such as nucleases, proteases [17, 25], lipases [64] and hemolysin and have swarming and swimming mobility [4, 65, 70]. These features may enable them to colonize a wide variety of niches and contribute to their success as opportunistic pathogens. Of the sequenced Serratia spp., the closest species to our isolate, S. marcescens DB11, is a confirmed pathogen of invertebrates [49, 63], a function that is likely required for evolution of an EPN complex.
Although Serratia sp. SCBI was initially isolated as an associate of Caenorhabditis briggsae, this bacterium will also associate with strains of the well-studied model eukaryote Caenorhabditis elegans and allow these bacteriovorus nematodes to kill insects [2]. Both C. briggsae and C. elegans are well known associates of invertebrates [79, 83], a likely pre-adaptation to an EPN life cycle. Recent discoveries of Serratia spp. in EPN associations [89, 105] suggest the possibility that Serratia sp. SCBI may belong to a unique lineage within the genus Serratia that has evolved the capacity to confer an EPN lifestyle to diverse insect-associated nematodes. The Serratia sp. SCBI and Caenorhabditid EPN complex represents a unique opportunity to explore the evolution of symbiosis in a third EPN lineage. Here we present the complete genome sequence of Serratia sp. SCBI and explore how it is unique with respect to other closely related Serratia and how the predicted functional proteome compares to features found in the well-studied EPN-associated bacteria, Xenorhabdus and Photorhabdus.
Results and discussion
Overview of the Serratia sp. SCBI Genome Structure and Annotation
The genome of Serratia sp. SCBI is comprised of a single circular 5.04 Mb chromosome with an overall GC content of 59.7 %, 4599 predicted protein coding genes, 84 tRNA genes and seven sets of rRNA genes (Table 1, Fig. 1). In addition, Serratia sp. SCBI contains a single 64.8 Kb conjugative plasmid with 74 putative protein coding genes, 28 of which lack similarity to known proteins. (Additional file 1: Table S1: Plasmid ORFs). Blast analysis (data not shown) has shown that the Serratia sp. SCBI plasmid shows no obvious homology to the other known Serratia plasmid found in S. proteamaculans 568. The annotated genome is available at NCBI under accession numbers CP003424 and CP003425.

Circular representation of the Serratia sp. SCBI genome. Circular representation of Genomic features in Serratia sp. SCBI. From outer to innermost: First and fourth circles, genes in the plus and minus strands, respectively, by COG category (COG category color Scheme A, side panel); second circle, genes shared with other Serratia and EPN species (see color scheme B); third circle, genomic Islands (GIs) (Color Scheme C); fifth circle, GC content, sixth, innermost, circle, GC skew (Color Scheme A Side panel)
Among complete Serratia genomes, the SCBI genome was the smallest in size and encoded fewer genes (Table 1). All the compared Serratia genomes shared a highly conserved genomic architecture as inferred from synteny of protein coding orthologs, tRNA genes, rRNA modules and their origins of replication. These genomes shared 3094 genes by MAUVE progressive alignment [35, 36] at 70 % coverage and 30 % identity. An additional 809 protein coding genes were shared by S. marcescens DB11 and Serratia sp. SCBI (Fig. 2). The Serratia sp. SCBI, S. marcescens DB11, Serratia sp. AS12 and S. proteamaculans 568 genomes had 519, 587, 1015 and 1011 unique genes, respectively (Fig. 2). An additional 1267 genes were shared between two or three species. The patterns of shared and unique genes are consistent with the evolutionary history of the species defined by 16S rDNA phylogeny (Fig. 3).

Venn diagram of shared and unique genes found in four Serratia genomes. The unique and shared genome among the compared genomes was determined by a dual cutoff of 30 % or greater amino acid identity and sequence length coverage of at least 70 %. Analysis was done using the MAUVE genome alignment tool [42]. SCBI: Serratia sp. SCBI; SMAR: S. marcescensDB11; SPRO: S. proteamaculans568, SAS12, Serratia sp. AS12

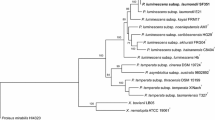
Evolutionary relationships of Serratia and representative bacteria from the entomopathogenic genera Photorhabdus and Xenorhabdus. Evolutionary relationships of Serratia and representative bacteria from the entomopathogenic genera Photorhabdus and Xenorhabdus. a Phylogenetic relationships inferred from the alignment of 1500 bp of 16S rDNA using the Maximum Likelihood [90]; b Phylogenetic relationships inferred from the alignment of 2623 bp of concatenated DNA from four housekeeping genes: atpD (634 bp), gyrB (742 bp), ifnB (613 bp) and rpoB (634 bp) using the Maximum Likelihood [90]. Numbers on internal branches are the results of Bootstrap analysis where the test was done with 1000 replicates [43]. Where applicable the trees are drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. Evolutionary analyses were conducted in MEGA6 [91, 92]
Phylogenetic placement of SCBI
To place Serratia sp. SCBI in an evolutionary framework we compared the near complete 16S rRNA genes from diverse Serratia species and other relevant entomopathogenic bacteria (Fig. 3) as well as concatenated sequences from four housekeeping genes: atpD, gyrB, ifnB and rpoB. The phylogenetic tree from the concatenated housekeeping genes suggest that Serratia sp. SCBI is found in a clade of five Serratia which are all known insect associates and SCBI was closest to S. ureilytica. Among the well-studied and completely sequenced species included in the 16S phylogeny, Serratia sp. SCBI was most closely related to S. marcescens DB11, which is an insect pathogen [49, 50, 63] and antagonist of C. elegans [61, 76, 82]. This close relationship provides an ideal opportunity to identify candidate genes critical to the evolution of the EPN lifestyle. Another closely related species is S. nematodiphila, which was described as part of an EPN association with a Rhabditid nematode [104, 105]. Other close relatives include S. entomophila [50], which is pathogenic to New Zealand grass grubs, and S. ureilytica which is a nickel resistant Serratia isolated from the River Torsa in West Bengal, India [13]. This phylogeny suggests that Serratia sp. SCBI may represent a novel, independently evolving EPN association from within a lineage of Serratia commonly associated with insects.
Genomic alignment and comparative genomic analysis
Whole genome alignment of SCBI with three of the sequenced Serratia genomes show extensive synteny (Fig. 4) where large colinear blocks of genes are interrupted by insertions that resulted in genomic islands, deletions relative to SCBI and a small number of rearrangements involving relatively small genomic regions. In the context of this study, a genomic island (GI) was broadly defined as any stretch of five or more consecutive protein coding genes that: 1) are not present in the other members of the genus, and 2) show a divergent %GC content compared to the core genome. Our analysis predicts at least 29 GIs in Serratia sp. SCBI. Based on these criteria 326 of 519 (62.8 %) of unique genes are located in the genomic islands of SCBI and as discussed below most of these GIs encode functions of potential biological relevance to the EPN lifestyle (Table 2). To ascertain the significance of the Serratia sp. SCBI-C. briggsae association in relation to other Serratia and the established EPN bacteria, we conducted comparative analysis of the predicted proteome against the respective groups using the MAUVE [35] genome alignment tool. The cutoff values of 60 % identity and 70 % coverage, i.e. similarity of 60 % of the compared residues covering 70 % of the total length of the shorter sequence in the comparison, were used to include only hits with high confidence levels and exclude spurious hits (Table 3).

Genomic alignment of the four compared Serratia spp. Alignment statistics were generated and rendered by MAUVE progressive alignment software [35, 36]. SMAR: S. marcescens DB11, SCBI: Serratia sp. SCBI, and SPRO: S. proteamaculans586, SAS12: Serratia sp. AS12. Color schemes represent blocks of contiguous genes interrupted by colorless patches where the genomes differ from each other significantly and identified as GIs are located
COG analysis among Serratia
Analysis of the functional categories and genome wide distribution of all unique genes with assigned Clusters of Orthologous Groups (COG) functions among the sequenced Serratia genomes revealed that the unique genes in Serratia sp. SCBI were biased towards categories that have direct bearing on the symbiosis and/or pathogenesis life style (Fig. 5). Specifically, the gains were in COG categories: [M]- Cell wall/membrane/envelope biogenesis; [N]- Cell motility; [Q]- Secondary metabolites biosynthesis, transport and catabolism; [U] - Intracellular trafficking and secretion and [V]- Defense mechanisms.

Relative COG category abundance in the core proteome in comparison with unique proteins in Serratia. The relative abundance of COG categories between the core and unique gene pools was calculated as follows: the number of proteins in each COG category was determined and the unique pools normalized to their respective total predicted protein numbers. Then the percentage of each COG category in the core proteome was subtracted from the corresponding COG percentage in the unique category and the difference plotted. COG functional categories descriptions are: [A] RNA processing and modification; [B] Chromatin structure and dynamics; [C] Energy production and conversion; [D] Cell cycle control and mitosis; [E] Amino acid metabolism and transport; [F] Nucleotide metabolism and transport; [G] Carbohydrate metabolism and transport; [H] Coenzyme metabolism; [I] Lipid metabolism; [J] Translation; [K] Transcription; [L] Replication and repair; [M] Cell wall/membrane/envelope biogenesis; [N] Cell motility; [O] Post-translational modification] protein turnover] chaperone functions; [P] Inorganic ion transport and metabolism; [Q] Secondary metabolites biosynthesis, transport and catabolism; [T] Signal transduction; [U] Intracellular trafficking and secretion; [R] General functional prediction only ; [S] Function unknown; [V] Defense mechanisms. [X] No cog category
Functional properties of Serratia sp. SCBI shared with other entomopathogens
To evaluate the Serratia sp. SCBI in the context of its role in an EPN complex, we searched the SCBI genome for homologs or functional analogs of all genes previously shown or implicated to be involved in host immune defense, host killing and cadaver protection, pathogenesis and symbiosis and reassociation in the canonical EPN species Xenorhabdus and Photorhabdus (Table 3). Although most of these genes appear in GIs in Serratia SCBI, we focused the comparison on three functions with well documented roles in EPN lifestyle but not confined to putative Genomic Islands: evading host defense, toxicity to host and competitors and recolonization. Functional studies on the Serratia sp. SCBI-C. elegans complex has shown that hemolysin, NRPS proteins (Petersen, LaCourse et al. submitted) and extracellular proteases are crucial to cytotoxicity and virulence in this putative EPN symbiosis [72]. The dynamics of mRNA expression of the alkaline metalloproteases (prtA1-prtA4) compared to the serine metalloproteases which peak before and after the death of the host, respectively, suggest a complex regulatory mechanism in killing the host and bioconversion of the cadaver [72]. Furthermore, inactivation of the hemolysin gene in Serratia sp. SCBI, which resulted in loss of hemolysis, failed to attenuate insecticidal activity but significantly increased motility and antimicrobial activity (Petersen et. al., submitted). Furthermore comparative study on the physiology of three sequenced strains, Serratia sp. SCBI, S. marcescens DB11 and S. proteamaculans 568, revealed that DB11 and SCBI were similar in insect virulence and cytotoxicity consistent with their phylogenetic proximity, but motility and lipase and hemolytic activities differed significantly between them [71]. The implication of these functional studies is that the Serratia sp. SCBI-C. briggsae association is a promising model to study the dynamics of gene expression during the transition from starved, inactive stay in nematode gut to active state during pathogenesis –killing and bioconversion– of insect hosts.
Breaching host defense
Breaching the insect host’s defense –both the humoral and cellular components- is crucial for the establishment of infection. EPNs neutralize humoral response elements like lysozyme and cecropins with serralysin-like proteases such as the PrtA [42, 67] and by haemolysins (XhlA) and lipases (XlpA) and the FhlDC regulator operon [47, 67]. Cellular response evasion is directed mainly at suppressing the phenoloxidase pathway to prevent hemocyte aggregation and melanization. Several mechanisms are employed to suppress the insects’ cellular immune response. These include: inhibition of phospholipase A2 (PLA2) using stilbene and MalPQT operon products in Photorhabdus and expression of LPS in Xenorhabdus, the suppression of phagocytosis by Type III secretion system-mediated deposition of LopT and SctC directly into hemocytes [19, 20] and the production of cytotoxins such as Cif and MrxA that lead to apoptotic cell death. With the exception of the xlpA gene which is absent from Serratia spp. and the cif and stc genes that are absent not only from Serratia spp. but also from Xenorhabdus spp., all other genes identifies as instrumental in the toxicity of EPNS are present in Serratia spp. as well including SCBI. The presence of more serralysins in Serratia spp., by comparison to the EPN genomes, suggests that the SCBI C. briggsae association may heavily rely on the expression of multiple serralysin genes and extracellular lipases and hemolysin production to breach the host immune system.
Toxicity
Toxicity –both for killing the insect host and warding off bacterial and fungal competitors- is mediated by a plethora of insecticidal and bactericidal gene products [44]. These include the Photorhabdus Toxin Complex (TC) operons tcaABCD and their counterparts in Xenorhabdus xptABCD, as well as other toxicity genes like the pirAB, xaxB, sepA, groEL, lopT, mcf, mcf2, rtxA, mptBDE. Various drug efflux systems and numerous nonribosomal peptide synthase (NRPS) and polyketide synthase (PKS) genes [15, 37, 45, 53, 55] are also part of the overall toxicity caused by EPN systems. The redundancy evident in the toxin gene repertoire of Photorhabdus and Xenorhabdus is astounding – at the time of its sequencing in 2003, P. luminescens was described as having more genes encoding toxins than any other genome sequenced to date [40], however, Serratia sp. SCBI also has over 200 genes encoding toxins, NRPS genes, multiple drug efflux systems and assorted virulence factors.
Nematode recolonization
Nematode recolonization involves two key elements: acquisition of partner bacteria by horizontal transmission via endotokia matricida [28], in which eggs hatch within the sacrificial mother to gain access to bacteria before emerging from the maternal corpse and the retention of bacteria in the gut of the infective juvenile. The latter seems to be mediated by ngrA [27, 59], the Type I fimbriae mrxA [24] and at least in X. nematophila, by the products of the nilABC and nilR genes [30, 31, 52]. Both of these requirements seem to be met in SCBI since Caenorhabditids, when grown on SCBI, almost exclusively reproduce by endotokia matricida [1] and SCBI possess homologs of both genes implicated in recolonization. Furthermore, the absence of the nilABC genes of X. nematophila in Xenorhabdus boveinii as well as Photorhabdus spp. show that colonization, like other aspects of EPN biology, can be produced by different mechanisms in EPNs.
From this comparison three patterns emerge: 1) Serratia lack homologs of the toxicity genes lopT, rtxA, prtA, pirB, cif, mcf, and mcf2 and the TC operons but have substantial number of secreted proteases and lipases, and hemolysins and 2) the majority of the Photorhabdus/Xenorhabdus virulence, symbiosis and regulatory genes are equally represented in Serratia (Table 3), and 3) the presence of key genes is not uniform among the different species –nilABC absent in Photorhabdus spp. and cif, stc, cipAB and lopT missing from Xenorhabdus spp.-suggesting that there is no single possible path to pathogenicity, cadaver bioconversion, repelling of competitors and bacterial re-association.
Unique genes and Genomic Islands (GI) of SCBI
Apart from those summarized in Table 3, many other genes with well-documented pathogenicity/symbiosis functions in other microbial systems are found in the genome of SCBI more than half of which are located on genomic islands. Of the 29 genomic islands identified in Serratia sp. SCBI genome comparison (Fig. 1, third circle, Additional file 2: Table S2), many were enriched in defense and virulence genes and in - phage remnants which, in many bacteria including those in EPN associations, have been diverted into novel toxin/virulence factor delivery vehicles [8, 21, 56, 62].
Perhaps the most notable feature among the genomic islands is the urea pathway genes located on GI-4. The hoxN/hupN/nixA family nickel/cobalt transporter and the urea metabolism pathway proteins found in GI-4, are absent from other members of the genus Serratia but interestingly present in all the EPN bacteria. The hoxN transporter is necessary for urea hydrolysis [60, 103] making it an integral part of the pathway. While there is no evidence so far of a direct role for urea metabolism in the EPN lifestyle, a potential role for these pathways can be exemplified by current research on diatoms where the urea pathway is a key to facilitating rapid recovery from prolonged nitrogen limitation followed by rapid growth under nutrient rich conditions [6]. The EPN life cycle has a similar pattern in that the symbionts that survive in nutrient-limited conditions while in transit to the next cycle of infection face a sudden abundance of organic compounds upon entering the insect hemocoel, thus requiring rapid transitioning from starved state to exponential growth of the bacteria. Furthermore, the regulation of the urea pathway in the diatoms has been linked to proline [5] which has also been identified as the cue for the transition from starved to metabolically active state in Photorhabdus and Xenorhabdus.
Among the unique genes of SCBI that are relevant to its association with C. briggsae as a putative EPN are the O-antigen biosynthesis protein(SCBI_1044), NRPS proteins(SCBI_1017, SCBI_1055-1059, SCBI_2978), colicins (SCBI_1883-1887), Type IV secretion systems (SCBI_2992, SCBI_2984, SCBI_2994), and iron acquisition proteins(SCBI_1973-1975). These genes have been shown to be required for both symbiotic and pathogenic properties [12, 101, 102] and may play a similar role in the Serratia-Caenorhabditis EPN association.
The list of genes unique to SCBI also include efflux system proteins (SCBI_1013, SCBI_1208) that are specific for macrolide- a class of antibiotics that inhibit the growth of bacteria- and the resistance-nodulation- division (RND) efflux proteins (SCBI_3788-3791) that are known to actively scavenge antimicrobial compounds released by competitors or the host immune system [3, 14, 73, 74, 87, 93, 94], a predicted HtpX protease, a heat shock/stress inducible membrane bound zinc metalloprotease [81], the DinI family protein also known as MsgA (macrophage survival protein) (SCBI_3186) and prophage lysozyme proteins (SCBI_2756-2758) an endolysin/autolysin system known to degrade the peptidoglycan structures in bacterial cell walls [18, 51, 99]; Microcin H47(SCBI_2968, SCBI_3536), a bactericidal peptide antibiotic related to the Colicin V family secretion protein [10] and associated secretion ATPase required for its export [95]; several fimbrial proteins with functional annotations ranging from pilus assembly to anchoring which, in light of recent findings that implicate Photorhabdus and Xenorhabdus adherence to the gut walls of their respective worm associates via fimbrial proteins, [46, 68, 86] and two homologs of the HigB toxin protein/HigA protein (antitoxin to HigB)( SCBI_0907-0908), which in Vibrio, have been shown to inhibit cell growth in Escherichia coli upon ectopic expression [26].
Conclusions
EPN associations are complex tripartite interactions between bacterial pathogens, symbiotic entomophagous nematodes and susceptible insect/insect larval hosts. Thus far our understanding of the mechanisms of EPN associations is limited to two superficially similar and convergent systems. Here we report the complete genome sequence of the bacterium involved in a novel independently evolving putative EPN association between a species of Serratia and nematodes in the genus Caenorhabditis. This Serratia is most closely related to another recently discovered EPN association, Serratia nematodiphila, and the well-studied insect pathogen Serratia marcescens DB11. Based on a comparison to the two well studies EPN systems, the genome of Serratia sp. SCBI contains a large number of genes that are potential candidates for EPN adaptations. Among the most notable shared functions with other EPN associates are the O-antigen, the syrP protein, several non-ribosomal peptide synthetases, bacteriocins, many fimbrial biogenesis and ushering proteins, secondary metabolite and toxin secretion systems and multiple drug resistance/efflux systems. However, the SCBI genome carries neither the TC complexes nor the mcf genes, which implies its use of different mechanisms of insect killing. The presence of several ORFs encoding putative virulence factors in horizontally acquired genomic islands suggest that EPN associations can be established by dissimilar sets of mechanisms for killing, bioconversion, sanitization and colonization. The presence of these sets of genes in many bacteria further suggests that the major hurdle in EPN complex formation may be the initial development of co-tolerance between potential partners. This complete genome sequence of one of the partners in a nascent EPN association should enable future analysis of the Serratia/Caenorhabditis EPN complex.
Methods
Bacterial isolation and identification
The bacterium was isolated from a Galleria mellonela trap laid in soil in the Kawa Zulu Natal province of South Africa. The detailed procedure has been previously described elsewhere [2]. Following isolation bacterial identity was determined by 16S rDNA PCR which shows 99 % identity with Serratia marcescens rDNA sequences at NCBI (CP003959.1).
Genomic DNA isolation
Genomic DNA (gDNA) was isolated from overnight cultures grown in LB medium [10 g Bacto-tryptone, 5 g Bacto-yeast, 5 g NaCL, H2 O to 1 liter, pH 7.5] (BD, Sparks, MD) using the Qiagen Genomic DNA isolation kit (Qiagen, Germantown, MD) and following the procedure outlined for bacterial gDNA isolation. Precipitated DNA was collected by spooling the DNA using flame-sterilized and cooled glass rod. The spooled gDNA was immediately transferred to a microcentrifuge tube containing 1.5 ml sterile, nuclease free water. The DNA was dissolved on a shaker at 55 °C for 2 h. The yield, purity, and length of the DNA was determined for 1-5 μl samples by electrophoresis on 0.8 % agarose gel and by spectrophotometry on a NanoDrop 1000 (Thermo Scientific, USA).
Fosmid Library construction
The fosmid library was constructed from the genomic DNA (gDNA) using the CopyControl pCC1FOS™ Vector that contains both the E. coli F-factor single-copy origin of replication and the inducible high-copy oriV according to the manufacturer’s protocol (Epicentre, Madison, U.S.A.). Briefly, gDNA was mechanically sheared by passing through a narrow gage sterile syringe then it was separated by pulse field gel electrophoresis (PFGE) overnight. A gel slice was excised from the 36-40 KB size window and the DNA extracted by gel extraction. The gel extracted gDNA was subsequently ligated into the fosmid vector exactly as described in the Epicenter protocol. The ligated mixture was then packaged into lambda phages using MaxPlax Lambda Packaging Extracts (Epicentre, Madison, U.S.A.). The packaged library was then transduced into E. coli EPI300™ Plating Strain, and transformants were selected on LB agar supplemented with 34 mg/ml Chloramphenicol. The library clones were picked by the Genetix Q-bot colony picking robot (Genetix Ltd, UK) and inoculated into 384 well plates and allowed to grow for 24 h at 37oC in humidified chamber. Colonies were stored in -80oC freezers until they were retrieved for downstream processing. To isolate fosmid DNA, randomly selected individual clones were grown overnight in Chloramphenicol supplemented LB broth and plasmid copy number were amplified by adding 5ul induction solution and incubating further for 4 hours. Fosmids were isolated by alkaline lysis method using Qiagen plasmid isolation buffers. The presence of recombinant DNA in the isolated fosmids and the polymorphism of the insert DNA were evaluated by agarose gel electrophoresis of BamHI (NEB, USA) digestion of the purified plasmid DNA.
PCR primer design and amplicon sequencing
The genome sequencing gaps were closed using PCR based amplicon sequencing. To this end batches of PCR primer were designed by the Primer3 software driven by an in-house Perl script. Primers were synthesized by IDG (IDG Inc., MA, USA) and they were used to both amplify and sequence the amplicons after cleaning the PCR product from any unused primers. PCR fragments were purified using magnetic beads and SPRI solution. The primers were designed to have a melting point of 60 °C or above for increased specificity and simultaneous amplification at a single annealing temperature. Cycling conditions were as follows: Initial denaturation of plasmid, 5 min at 95 °C; 30 cycles or denaturation, annealing and primer extension at 95 °C 30 s, 60 °C 30 s and 2 min at 72 °C, respectively; a final synthesis hold at 72 °C for 10 min and a 4 °C hold until reactions were removed from the iCycler PCR machine (Biorad Inc. CA, USA). Long PCR for amplification of 3-8 KB fragments were performed the same way except that the 10X buffer was supplemented with additional MgCl2 to bring the final concentration to 25 mM and the extension time was increased to 6 min. All PCR amplifications were done using Finnzymes PCR kit (NEB, MA, USA) and the 15 mM 10X buffer was used for fragments between 100 to 2000 bases long.
Fosmid end sequencing and quality monitoring
DNA isolated from 209 plasmids was end sequenced using the pCC1FOS™ forward and reverse primers designed by Epicenter. Half of the clones were sent to a commercial sequencing facility (Genewiz Inc, NJ, USA) the other half were sequenced in house by the ABI 3130 genetic analyzer. Efficiency of Fosmid library construction and the quality of the library were monitored by selecting at random 209 clones and subjecting them to different analyses. A subset of these (46 clones) were tested by Bam HI digestion and, except for a failed plasmid isolation in one clone, all 45 (100 %) of them gave distinct digestion patterns and one common band at 8 KB, representing the plasmid backbone. BLAST results showed that except for 4 clones that failed to sequence, probably due to sample cross over contamination, all fosmid end sequences hit the assembled genome at 34 to 45 KB apart in the right orientation giving an overall high efficiency of library construction and quality.
Genome sequencing by 454 technology and assembly of contigs
An aliquot of the Serratia sp. SCBI gDNA was sent to the genome sequencing center at the University of Indiana for genome sequencing by 454 pyrosequencing technology. Two plates of genomic sequencing and one plate of 2 KB paired end sequencing were done to generate the complete genomic sequence of one circular chromosome and one large, circular plasmid of 68 KB size. The 454 sequencing resulted in ten contigs ranging in size from 2 KB to 3.5 MB and harbored 236 gaps of ranging from 4 nucleotides to 2 kb in length which were closed by PCR amplicon sequencing as detailed in the PCR primer design and amplicon sequencing section. The genome sequencing output from the 2 plates of 454 genomic reads and one plate of 2 KB paired end sequencing yielded 40 and 21 MB data, giving an overall 12X coverage of the genome. The main chromosome was resolved in seven genomic regions separated by seven sets of rDNA assemblies of 5 KB length as outlined below.
Sequence gap filling, genome assembly and bioinformatics analysis
The 10 scaffolds assembled by the Newbler Assembler sequence initially included 236 gaps ranging from 4 to 2050 bases long. PCR based amplicon end sequencing resolved all the gaps bringing the sequence assembly to a satisfactory level of completion. PCR primers were designed on regions flanking gaps by the Primer3 program [80] after orientation and order of scaffolds were determined by a combination of bioinformatics and laboratory based methods. Briefly, truncated scaffold sequences consisting of 500 bp from the start and end of each scaffold were generated by a Perl script. These truncated sequences were then blasted against the S. marcescens DB11 genome which identified their counterparts unambiguously showing a great deal of synteny between the two genomes. This finding was then confirmed by long PCR performed using primers pointing outward from the pairs of gap ends predicted by the bioinformatics analysis. All of the predicted combinations resulted in the amplification of the expected 6 KB fragment confirming both the order and orientation of the clones as well as their size. The remaining two fragments were resolved into a circular plasmid by blast hit with two fosmid clones which hit the two scaffolds unambiguously. This was also confirmed by PCR that amplified predicted size of amplicons from primers designed at the ends of the two scaffolds and individual fosmids used as template. Long PCR and amplicon sequencing were performed as described above and the resultant amplicon sequences were then added to the 454 reads and reassembled by the Newbler to generate the complete genome sequence. Comparative genomic analysis of Serratia sp. SCBI genome with the two other completed genomic sequences in the genus, S. marcescensDB11 and S. proteamaculans568 was done to determine the genes shared between the three genomes and the three way paired genomes (Serratia sp. SCBI-DB11, Serratia sp. SCBI-SPRO, DB11-SPRO) and the genes unique to each of them using MAUVE [35, 36], MURASAKI [75] and Blast [7, 57] bioinformatics tools, the RAST Annotation Server [9] and in house developed Perl scripts. A comparison was also done with P. luminescens and X. nematophilia to shed light on the entomopathogenic aspects of the Serratia sp. SCBI-Caenorhabditis relationship and to determine whether it resembles the Photorhabdus/Heterorhabdus or the Xenorhabdus/Steinernema type association or it constitutes a novel class of EPN association with a different entomopathogenic signature driven by a novel set of genes and pathways.
Accession numbers
The genome sequence and its annotations are available at NCBI under the accession numbers CP003424 and CP003425.
References
Abebe E, Akele FA, Morrison J, Cooper V, Thomas WK. An insect pathogenic symbiosis between a Caenorhabditis and Serratia. Virulence. 2011;2(2):158–61.
Abebe E, Jumba M, Bonner K, Gray V, Morris K, Thomas WK. An entomopathogenic Caenorhabditis briggsae. J Exp Biol. 2010;213(18):3223–9.
Aeschlimann JR. The role of multidrug efflux pumps in the antibiotic resistance of Pseudomonas aeruginosa and other gram-negative bacteria - Insights from the society of infectious diseases pharmacists. Pharmacotherapy. 2003;23(7):916–24.
Alberti L, Harshey RM. Differentiation of Serratia-Marcescens 274 into Swimmer and Swarmer Cells. J Bacteriol. 1990;172(8):4322–8.
Allen AE, CL Dupont, M Obornik, A Horak, A Nunes-Nesi, JP McCrow, et al. Evolution and metabolic significance of the urea cycle in photosynthetic diatoms. Nature. 2011;473(7346):203−+.
Allen AE, Moustafa A, Montsant A, Eckert A, Kroth PG, Bowler C. Evolution and Functional Diversification of Fructose Bisphosphate Aldolase Genes in Photosynthetic Marine Diatoms. Mol Biol Evol. 2012;29(1):367–79.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic Local Alignment Search Tool. J Mol Biol. 1990;215(3):403–10.
Asadulghani M, Ogura Y, Ooka T, Itoh T, Sawaguchi A, Iguchi A, et al. The Defective Prophage Pool of Escherichia coli O157: Prophage-Prophage Interactions Potentiate Horizontal Transfer of Virulence Determinants. Plos Pathogens. 2009;5(5).
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST server: Rapid annotations using subsystems technology. Bmc Genomics. 2008;9.
Azpiroz MF, Rodriguez E, Lavina M. The structure, function, and origin of the microcin H47 ATP-binding cassette exporter indicate its relatedness to that of colicin V. Antimicrob Agents Chemother. 2001;45(3):969–72.
Bai YM, Souleimanov A, Smith DL. An inducible activator produced by a Serratia proteamaculans strain and its soybean growth-promoting activity under greenhouse conditions. J Exp Bot. 2002;53(373):1495–502.
Bennett HPJ, Clarke DJ. The pbgPE operon in Photorhabdus luminescens is required for pathogenicity and symbiosis. J Bacteriol. 2005;187(1):77–84.
Bhadra B, Roy P, Chakraborty R. Serratia ureilytica sp nov., a novel urea-utilizing species. Int J Syst Evol Microbiol. 2005;55:2155–8.
Bina XR, Provenzano D, Nguyen N, Bina JE. Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect Immun. 2008;76(8):3595–605.
Bode HB. Entomopathogenic bacteria as a source of secondary metabolites. Curr Opin Chem Biol. 2009;13(2):224–30.
Bovien P. Some types of association between nematodes and insects. Videnskabelige Meddelelser Fra Dansk Naturhistorisk Forening, ed. P. Bovien, Ed. . Vol. København 101. 1932, Copenhagen.
Braun V, Schmitz G. Excretion of a protease by Serratia marcescens. Arch Microbiol. 1980;124(1):55–61.
Briers Y, Volckaert G, Cornelissen A, Lagaert S, Michiels CW, Hertveldt K, et al. Muralytic activity and modular structure of the endolysins of Pseudomonas aeruginosa bacteriophages phiKZ and EL. Mol Microbiol. 2007;65(5):1334–44.
Brugirard-Ricaud K, Duchaud E, Givaudan A, Girard PA, Kunst F, Boemare N, et al. Site-specific antiphagocytic function of the Photorhabdus luminescens type III secretion system during insect colonization. Cell Microbiol. 2005;7(3):363–71.
Brugirard-Ricaud K, Givaudan A, Parkhill J, Boemare N, Kunst F, Zumbihl R, et al. Variation in the effectors of the type III secretion system among Photorhabdus species as revealed by genomic analysis. J Bacteriol. 2004;186(13):4376–81.
Brussow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev. 2004;68(3):560−+.
Burke GR, Moran NA. Massive Genomic Decay in Serratia symbiotica, a Recently Evolved Symbiont of Aphids. Genome Biol Evol. 2011;3:195–208.
Burnell AM, Stock SP. Heterorhabditis, Steinernema and their bacterial symbionts - lethal pathogens of insects. Nematology. 2000;2:31–42.
Chandra H, Khandelwal P, Khattri A, Banerjee N. Type 1 fimbriae of insecticidal bacterium Xenorhabdus nematophila is necessary for growth and colonization of its symbiotic host nematode Steinernema carpocapsiae. Environ Microbiol. 2008;10(5):1285–95.
Chen YC, Shipley GL, Ball TK, Benedik MJ. Regulatory Mutants and Transcriptional Control of the Serratia-Marcescens Extracellular Nuclease Gene. Mol Microbiol. 1992;6(5):643–51.
Christensen-Dalsgaard M, Gerdes K. Two higBA loci in the Vibrio cholerae superintegron encode mRNA cleaving enzymes and can stabilize plasmids. Mol Microbiol. 2006;62(2):397–411.
Ciche TA, Bintrim SB, Horswill AR, Ensign JC. A phosphopantetheinyl transferase homolog is essential for Photorhabdus luminescens to support growth and reproduction of the entomopathogenic nematode Heterorhabditis bacteriophora. J Bacteriol. 2001;183(10):3117–26.
Ciche TA, Kim KS, Kaufmann-Daszczuk B, Nguyen KCQ, Hall DH. Cell invasion and matricide during Photorhabdus luminescens transmission by Heterorhabditis bacteriophora nematodes. Appl Environ Microbiol. 2008;74(8):2275–87.
Clarke DJ. Photorhabdus: a model for the analysis of pathogenicity and mutualism. Cell Microbiol. 2008;10(11):2159–67.
Cowles CE, Goodrich-Blair H. Characterization of a lipoprotein, NilC, required by Xenorhabdus nematophila for mutualism with its nematode host. Mol Microbiol. 2004;54(2):464–77.
Cowles CE, Goodrich-Blair H. nilR is necessary for co-ordinate repression of Xenorhabdus nematophila mutualism genes. Mol Microbiol. 2006;62(3):760–71.
Cowles KN, Cowles CE, Richards GR, Martens EC, Goodrich-Blair H. The global regulator Lrp contributes to mutualism, pathogenesis and phenotypic variation in the bacterium Xenorhabdus nematophila. Cell Microbiol. 2007;9(5):1311–23.
Crawford JM, Kontnik R, Clardy J. Regulating Alternative Lifestyles in Entomopathogenic Bacteria. Curr Biol. 2010;20(1):69–74.
Cycon M, Wojcik M, Piotrowska-Seget Z. Biodegradation of the organophosphorus insecticide diazinon by Serratia sp and Pseudomonas sp and their use in bioremediation of contaminated soil. Chemosphere. 2009;76(4):494–501.
Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–403.
Darling AE, Mau B, Perna NT. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. Plos One. 2010;5(6).
Derzelle S, Duchaud E, Kunst F, Danchin A, Bertin P. Identification, characterization, and regulation of a cluster of genes involved in carbapenem biosynthesis in Photorhabdus luminescens. Appl Environ Microbiol. 2002;68(8):3780–9.
Derzelle S, Ngo S, Turlin E, Duchaud E, Namane A, Kunst F, et al. AstR-AstS, a new two-component signal transduction system, mediates swarming, adaptation to stationary phase and phenotypic variation in Photorhabdus luminescens. Microbiology-Sgm. 2004;150:897–910.
Derzelle S, Turlin E, Duchaud E, Pages S, Kunst F, Givaudan A, et al. The PhoP-PhoQ two-component regulatory system of Photorhabdus luminescens is essential for virulence in insects. J Bacteriol. 2004;186(5):1270–9.
Duchaud E, Rusniok C, Frangeul L, Buchrieser C, Givaudan A, Taourit S, et al. The genome sequence of the entomopathogenic bacterium Photorhabdus luminescens. Nat Biotechnol. 2003;21(11):1307–13.
Dutky SR, Hough WS. Note on parasitic nematode from codling moth larvae, Carpocapsa pomonella. Proc Entomol Soc Wash. 1955;57.
Felfoldi G, Marokhazi J, Kepiro M, Venekei I. Identification of natural target proteins indicates functions of a serralysin-type metalloprotease, PrtA, in anti-immune mechanisms. Appl Environ Microbiol. 2009;75(10):3120–6.
Felsenstein J. Confidence-Limits on Phylogenies - an Approach Using the Bootstrap. Evolution. 1985;39(4):783–91.
ffrench-Constant RH, Bowen DJ. Novel insecticidal toxins from nematode-symbiotic bacteria. Cell Mol Life Sci. 2000;57(5):828–33.
Ffrench-Constant RH, Waterfield N, Burland V, Perna NT, Daborn PJ, Bowen D, et al. A genomic sample sequence of the entomopathogenic bacterium Photorhabdus luminescens W14: Potential implications for virulence. Appl Environ Microbiol. 2000;66(8):3310–29.
Forst S, Dowds B, Boemare N, Stackebrandt E. Xenorhabdus and Photorhabdus spp.: Bugs that kill bugs. Annu Rev Microbiol. 1997;51:47–72.
Givaudan A, Lanois A. flhDC, the flagellar master operon of Xenorhabdus nematophilus: Requirement for motility, lipolysis, extracellular hemolysis, and full virulence in insects. J Bacteriol. 2000;182(1):107–15.
Goodrich-Blair H, Clarke DJ. Mutualism and pathogenesis in Xenorhabdus and Photorhabdus: two roads to the same destination. Mol Microbiol. 2007;64(2):260–8.
Grimont PA, Grimont F. The genus Serratia. Annu Rev Microbiol. 1978;32:221–48.
Grimont PAD, Jackson TA, Ageron E, Noonan MJ. Serratia-Entomophila Sp-Nov Associated with Amber Disease in the New-Zealand Grass Grub Costelytra-Zealandica. Int J Syst Bacteriol. 1988;38(1):1–6.
Grundling A, Smith DL, Blasi U, Young R. Dimerization between the holin and holin inhibitor of phage lambda. J Bacteriol. 2000;182(21):6075–81.
Heungens K, Cowles CE, Goodrich-Blair H. Identification of Xenorhabdus nematophila genes required for mutualistic colonization of Steinernema carpocapsae nematodes. Mol Microbiol. 2002;45(5):1337–53.
Hinchliffe SJ, Hares MC, Dowling AJ, ffrench-Constant RH. Insecticidal Toxins from the Photorhabdus and Xenorhabdus Bacteria. Open Toxinol J. 2010;3:18.
Hu K, Webster JM. Antibiotic production in relation to bacterial growth and nematode development in Photorhabdus Heterorhabditis infected Galleria mellonella larvae. Fems Microbiol Lett. 2000;189(2):219–23.
Hu KJ, Li JX, Li B, Webster JM, Chen GH. A novel antimicrobial epoxide isolated from larval Galleria mellonella infected by the nematode symbiont, Photorhabdus luminescens (Enterobacteriaceae). Bioorg Med Chem. 2006;14(13):4677–81.
Hurst MRH, Glare TR, Jackson TA. Cloning Serratia entomophila antifeeding genes - A putative defective prophage active against the grass grub Costelytra zealandica (vol 186, pg 5116, 2004). J Bacteriol. 2004;186(20):7023–4.
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36(Web Server issue):W5–9.
Joyce SA, Clarke DJ. A hexA homologue from Photorhabdus regulates pathogenicity, symbiosis and phenotypic variation. Mol Microbiol. 2003;47(5):1445–57.
Joyce SA, Watson RJ, Clarke DJ. The regulation of pathogenicity and mutualism in Photorhabdus. Curr Opin Microbiol. 2006;9(2):127–32.
Komeda H, Kobayashi M, Shimizu S. A novel transporter involved in cobalt uptake. Proc Natl Acad Sci U S A. 1997;94(1):36–41.
Kurz CL, Ewbank JJ. Caenorhabditis elegans for the study of host-pathogen interactions. Trends Microbiol. 2000;8(3):142–4.
Lang AS, Beatty JT. Genetic analysis of a bacterial genetic exchange element: the gene transfer agent of Rhodobacter capsulatus. Proc Natl Acad Sci U S A. 2000;97(2):859–64.
Lauzon CR, Bussert TG, Sjogren RE, Prokopy RJ. Serratia marcescens as a bacterial pathogen of Rhagoletis pomonella flies (Diptera : Tephritidae). Eur J Entomol. 2003;100(1):87–92.
Li XY, Tetling S, Winkler UK, Jaeger KE, Benedik MJ. Gene Cloning, Sequence-Analysis, Purification, and Secretion by Escherichia-Coli of an Extracellular Lipase from Serratia-Marcescens. Appl Environ Microbiol. 1995;61(7):2674–80.
Lin CS, Horng JT, Yang CH, Tsai YH, Su LH, Wei CF, et al. RssAB-FlhDC-ShlBA as a Major Pathogenesis Pathway in Serratia marcescens. Infect Immun. 2010;78(11):4870–81.
Maragakis LL, Winkler A, Tucker MG, Cosgrove SE, Ross T, Lawson E, et al. Outbreak of multidrug-resistant Serratia marcescens infection in a neonatal intensive care unit. Infect Control Hosp Epidemiol. 2008;29(5):418–23.
Marokhazi J, Lengyel K, Pekar S, Felfoldi G, Patthy A, Graf L, et al. Comparison of proteolytic activities produced by entomopathogenic Photorhabdus bacteria: Strain- and phase-dependent heterogeneity in composition and activity of four enzymes. Appl Environ Microbiol. 2004;70(12):7311–20.
Meslet-Cladiere LM, Pimenta A, Duchaud E, Holland IB, Blight MA. In vivo expression of the mannose-resistant fimbriae of Photorhabdus temperata K122 during insect infection. J Bacteriol. 2004;186(3):611–22.
O'Neill KH, Roche DM, Clarke DJ, Dowds BC. The ner gene of Photorhabdus: effects on primary-form-specific phenotypes and outer membrane protein composition. J Bacteriol. 2002;184(11):3096–105.
Orear J, Alberti L, Harshey RM. Mutations That Impair Swarming Motility in Serratia-Marcescens-274 Include but Are Not Limited to Those Affecting Chemotaxis or Flagellar Function. J Bacteriol. 1992;174(19):6125–37.
Petersen LM, Tisa LS. Influence of temperature on the physiology and virulence of the insect pathogen Serratia sp. Strain SCBI. Appl Environ Microbiol. 2012;78(24):8840–4.
Petersen LM, Tisa LS. Molecular characterization of protease activity in Serratia sp. strain SCBI and its importance in cytotoxicity and virulence. J Bacteriol. 2014;196(22):3923–36.
Piddock LJ. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin Microbiol Rev. 2006;19(2):382–402.
Piddock LJV. Multidrug-resistance efflux pumps - not just for resistance. Nat Rev Microbiol. 2006;4(8):629–36.
Popendorf K, Tsuyoshi H, Osana Y, Sakakibara Y. Murasaki: a fast, parallelizable algorithm to find anchors from multiple genomes. PLoS One. 2010;5(9):e12651.
Pradel E, Ewbank JJ. Genetic models in pathogenesis. Annu Rev Genet. 2004;38:347–63.
Richards GR, Goodrich-Blair H. Examination of Xenorhabdus nematophila lipases in pathogenic and mutualistic host interactions reveals a role for xlpA in nematode progeny production. Appl Environ Microbiol. 2010;76(1):221–9.
Richards GR, Vivas EI, Andersen AW, Rivera-Santos D, Gilmore S, Suen G, et al. Isolation and characterization of Xenorhabdus nematophila transposon insertion mutants defective in lipase activity against Tween. J Bacteriol. 2009;191(16):5325–31.
Richter S. Phoretic Association between the Dauerjuveniles of Rhabditis-Stammeri (Rhabditidae) and Life-History Stages of the Burying Beetle Nicrophorus-Vespilloides (Coleoptera, Silphidae). Nematologica. 1993;39(3):346–55.
Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86.
Sakoh M, Ito K, Akiyama Y. Proteolytic activity of HtpX, a membrane-bound and stress-controlled protease from Escherichia coli. J Biol Chem. 2005;280(39):33305–10.
Schulenburg H, Ewbank JJ. Diversity and specificity in the interaction between Caenorhabditis elegans and the pathogen Serratia marcescens. Bmc Evolution Biol. 2004;4.
Schulte F. The Association between Rhabditis-Necromena Sudhaus and Schulte, 1989 (Nematoda, Rhabditidae) and Native and Introduced Millipedes in South-Australia. Nematologica. 1989;35(1):82–9.
Sergeant M, Jarrett P, Ousley M, Morgan JA. Interactions of insecticidal toxin gene products from Xenorhabdus nematophilus PMFI296. Appl Environ Microbiol. 2003;69(6):3344–9.
Sheets JJ, Hey TD, Fencil KJ, Burton SL, Ni W, Lang AE, et al. Insecticidal toxin complex proteins from Xenorhabdus nematophilus: structure and pore formation. J Biol Chem. 2011;286(26):22742–9.
Somvanshi VS, Kaufmann-Daszczuk B, Kim KS, Mallon S, Ciche TA. Photorhabdus phase variants express a novel fimbrial locus, mad, essential for symbiosis. Mol Microbiol. 2010;77(4):1021–38.
Stavri M, Piddock LJ, Gibbons S. Bacterial efflux pump inhibitors from natural sources. J Antimicrob Chemother. 2007;59(6):1247–60.
Taghavi S, Garafola C, Monchy S, Newman L, Hoffman A, Weyens N, et al. Genome survey and characterization of endophytic bacteria exhibiting a beneficial effect on growth and development of poplar trees. Appl Environ Microbiol. 2009;75(3):748–57.
Tambong JT. Phylogeny of bacteria isolated from Rhabditis sp. (Nematoda) and identification of novel entomopathogenic Serratia marcescens strains. Curr Microbiol. 2013;66(2):138–44.
Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10(3):512–26.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011;28(10):2731–9.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Tenover FC. Mechanisms of antimicrobial resistance in bacteria. Am J Infect Control. 2006;34(5):S3–S10.
Tenover FC. Rapid detection and identification of bacterial pathogens using novel molecular technologies: Infection control and beyond. Clin Infect Dis. 2007;44(3):418–23.
Trujillo M, Rodriguez E, Lavina M. ATP synthase is necessary for microcin H47 antibiotic action. Antimicrob Agents Chemother. 2001;45(11):3128–31.
Van Houdt R, Givskov M, Michiels CW. Quorum sensing in Serratia. Fems Microbiol Rev. 2007;31(4):407–24.
Voelz A, Muller A, Gillen J, Le C, Dresbach T, Engelhart S, et al. Outbreaks of Serratia marcescens in neonatal and pediatric intensive care units: Clinical aspects, risk factors and management. Int J Hyg Environ Health. 2010;213(2):79–87.
von Reuss SH, Kai M, Piechulla B, Francke W. Octamethylbicyclo[3.2.1]octadienes from the Rhizobacterium Serratia odorifera. Angewandte Chemie Int Edition. 2010;49(11):2009–10.
Wang IN, Smith DL, Young R. Holins: The protein clocks of bacteriophage infections. Annu Rev Microbiol. 2000;54:799–825.
Waterfield NR, Ciche T, Clarke D. Photorhabdus and a host of hosts. Annu Rev Microbiol. 2009;63:557–74.
Watson RJ, Millichap P, Joyce SA, Reynolds S, Clarke DJ. The role of iron uptake in pathogenicity and symbiosis in Photorhabdus luminescens TT01. Bmc Microbiol. 2010;10.
Wilkinson P, Waterfield NR, Crossman L, Corton C, Sanchez-Contreras M, Vlisidou I, et al. Comparative genomics of the emerging human pathogen Photorhabdus asymbiotica with the insect pathogen Photorhabdus luminescens. Bmc Genomics. 2009;10.
Wolfram L, Friedrich B, Eitinger T. The Alcaligenes-Eutrophus Protein Hoxn Mediates Nickel Transport in Escherichia-Coli. J Bacteriol. 1995;177(7):1840–3.
Yi YK, Park HW, Shrestha S, Seo J, Kim YO, Shin CS, et al. Identification of two entomopathogenic bacteria from a nematode pathogenic to the oriental beetle, Blitopertha orientalis. J Microbiol Biotechnol. 2007;17(6):968–78.
Zhang CX, Yang SY, Xu MX, Sun J, Liu H, Liu JR, et al. Serratia nematodiphila sp nov., associated symbiotically with the entomopathogenic nematode Heterorhabditidoides chongmingensis (Rhabditida: Rhabditidae). Int J Syst Evolution Microbiol. 2009;59:1603–8.
Acknowledgements
This work was supported in part by Hatch grant NH496, and by the College of Life Sciences and Agriculture, The University of New Hampshire, Durham, NH. Partial funding was provided by the New Hampshire Agricultural Experiment Station. This is Scientific Contribution Number 2571. We thank Heidi-Goodrich-Blair and Jonathan Ewbank for access to the Xenorhabdus and Serratia genome databases, respectively. We thank Jobriah Anderson for technical help.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
FAA, WKT, VSC, LST and EA conceived of and provided project oversight. FAA conducted the molecular biology and comparative genomics analysis and manuscript draft. PJH provided key scripts for the analysis. All authors edited, read and approved manuscript.
Additional files
Additional file 1: Table S1.
Serratia sp. SCBI predicted plasmid encoded genes.
Additional file 2: Table S2.
Serratia sp. SCBI Genomic Island genes with putative/Predicted functions relevant to EPN life style.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Abebe-Akele, F., Tisa, L.S., Cooper, V.S. et al. Genome sequence and comparative analysis of a putative entomopathogenic Serratia isolated from Caenorhabditis briggsae . BMC Genomics 16, 531 (2015). https://doi.org/10.1186/s12864-015-1697-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-015-1697-8