
Abstract
Background
Feed efficiency and growth rate have been targets for selection to improve chicken production. The incorporation of genomic tools may help to accelerate selection. We genotyped 529 individuals using a high-density SNP chip (600 K, Affymetrix®) to estimate genomic heritability of performance traits and to identify genomic regions and their positional candidate genes associated with performance traits in a Brazilian F2 Chicken Resource population. Regions exhibiting selection signatures and a SNP dataset from resequencing were integrated with the genomic regions identified using the chip to refine the list of positional candidate genes and identify potential causative mutations.
Results
Feed intake (FI), feed conversion ratio (FC), feed efficiency (FE) and weight gain (WG) exhibited low genomic heritability values (i.e. from 0.0002 to 0.13), while body weight at hatch (BW1), 35 days-of-age (BW35), and 41 days-of-age (BW41) exhibited high genomic heritability values (i.e. from 0.60 to 0.73) in this F2 population. Twenty unique 1-Mb genomic windows were associated with BW1, BW35 or BW41, located on GGA1–4, 6–7, 10, 14, 24, 27 and 28. Thirty-eight positional candidate genes were identified within these windows, and three of them overlapped with selection signature regions. Thirteen predicted deleterious and three high impact sequence SNPs in these QTL regions were annotated in 11 positional candidate genes related to osteogenesis, skeletal muscle development, growth, energy metabolism and lipid metabolism, which may be associated with body weight in chickens.
Conclusions
The use of a high-density SNP array to identify QTL which were integrated with whole genome sequence signatures of selection allowed the identification of candidate genes and candidate causal variants. One novel QTL was detected providing additional information to understand the genetic architecture of body weight traits. We identified QTL for body weight traits, which were also associated with fatness in the same population. Our findings form a basis for further functional studies to elucidate the role of specific genes in regulating body weight and fat deposition in chickens, generating useful information for poultry breeding programs.
Similar content being viewed by others

Background
Poultry breeding programs were developed to increase growth rate, body condition (performance traits) and improve carcass traits in broiler chickens [1, 2]. Nevertheless, selection to increase productivity, reduce production costs and minimize environmental impact remains a challenge for chicken breeders. Feed efficiency and body weight are the two main performance traits with economic importance [3] that may drive increased chicken productivity. The incorporation of genomic tools in breeding programs can increase genetic progress by improving selection accuracy [4, 5]. Additionally, the chicken is considered an important model for animal genomic studies [6]. Thus, the search for genomic regions and positional candidate genes can help to elucidate the molecular mechanisms involved in the regulation of performance traits not only in chickens, but also in other species.
The Chicken QTL database (release 35) [7] hosts 75 quantitative trait loci (QTL) for feeding traits (including feed conversion ratio, feed efficiency, feed intake, and residual feed intake). In contrast, 1637 QTL have been reported for growth traits (including average daily gain and body weight at different days of age). However, many of these QTL have only been coarsely mapped (i.e., they have very broad confidence intervals for location). QTL mapping studies were previously conducted on this Brazilian experimental population (Embrapa F2 Chicken Resource Population) for feeding, growth and carcass traits [8, 9]. However, those studies utilized up to 127 microsatellite markers, which resulted in the identification of QTL that span large regions of the genome. The average length of interval of the QTL reported by Nones et al. [9] and Ambo et al. [8] were 5.46 Mb and 11.9 Mb, respectively.
Recent studies have identified QTL, candidate genes and mutations associated with performance traits in chickens [3, 10, 11]. Yi et al. [11] investigated SNPs associated with performance traits in a Chinese local chicken population and identified two SNPs in the CCKAR gene associated with daily feed intake and daily gain. Mebratie et al. [3] identified 11 QTL and 21 SNPs associated with body weight traits, and 5 QTL and 5 SNPs associated with feed efficiency traits in a commercial broiler chicken population. Despite these recent efforts, the re-visitation of genome-wide association study (GWAS) for performance traits, using a higher density of markers, may enable the identification of genomic regions with smaller intervals [12], thereby facilitating the fine-mapping of novel and known QTL. This can aid in the identification of positional candidate genes and, eventually, the identification of potentially causative mutations [12, 13].
Recently, GWAS for performance traits in Embrapa F2 Chicken Resource Population was performed using 134,528 SNPs generated from a genotyping by sequencing (GBS) approach that used the restriction endonuclease PstI [10]. Although that study identified 21 SNPs that were significantly associated with the performance traits, the use of a high-density SNP array to genotype the same population may provide more uniform coverage of regions across the whole chicken genome.
The aims of this study were to estimate the genomic heritability for performance traits, and to identify genomic regions and positional candidate genes associated with these traits in a Brazilian F2 Chicken Resource population that was derived from a reciprocal cross between a broiler and a layer line. In addition, selection signature regions and a SNP dataset derived from re-sequencing of grandparental individuals were integrated to refine the list of candidate genes and the search for potential causative mutations.
Results
Descriptive statistics
The number of animals, means and standard errors, variance components, and estimated genomic heritabilities are given in Table 1 for: feed intake between 35 and 41 days of age (FI), feed conversion ratio between 35 and 41 days of age (FC), feed efficiency between 35 and 41 days of age (FE), weight gain between 35 and 41 days of age (WG), body weight at hatch (BW1), body weight at 35 days of age (BW35) and body weight at 41 days of age (BW41). Genomic heritability values ranged from 0.0002 for FI to 0.73 for BW41.
Genotyping and genome-wide association studies
As described by Moreira et al. [14], from the 529 genotyped chickens, 12 were removed from the analysis after applying animal DishQC criteria and a sample call rate filter ≥90%. The 28 grandparental chickens and 12 F1 birds did not have phenotypic data and were not considered for GWAS. A total of 489 F2 chickens from seven different families were used in the association analysis.
From the 580,961 SNPs on the SNP chip array, 399,693 segregating SNPs were kept for further analyses. All these SNPs had a call rate ≥ 98%. Among these, 4304 were removed due to minor allele frequency (MAF) criteria (MAF ≤ 0.02), and 23,603 SNPs that were located on the sex chromosomes and linkage groups were also removed, such that 371,558 markers remained for GWAS. An average density of 541 SNPs/Mb per chromosome was observed, with the lowest density on GGA2 (297 SNPs/Mb) and the highest density on GGA21 (816 SNPs/Mb). Missing genotypes were replaced with the average covariate value of that locus as reported by Cesar et al. [15].
The characterization of the 943 1-Mb non-overlapped windows and their respective percentage of the genetic variance explained are available in Additional file 1. The genomic windows associated with performance traits are described in Table 2. Twenty unique 1-Mb windows on GGA1–4, 6, 7, 10, 14, 24, 27 and 28 were associated with the body weight traits. The posterior probability of association (PPA) for these regions ranged from 0.40 to 0.96 and the genetic variance explained by each SNP window ranged from 0.53 to 4.74%. We did not identify any genomic windows associated with FI, FC, FE, or WG.
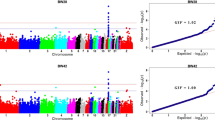
The associated SNP windows cumulatively explained 2.12, 20.59, and 17.31% of the genetic variance for BW1, BW35 and BW41, respectively. Manhattan plots with the percentage of genetic variance explained by all 943 non-overlapped SNP windows for each trait analyzed herein are shown in Fig. 1.

Manhattan plots of the posterior means of the percentage of genetic variance explained by each 1 Mb SNP window across the 28 autosomal chromosomes for all the performance traits analyzed. The title of each graph indicates the corresponding phenotype: feed intake (FI), feed conversion (FC), feed efficiency (FE), weight gain (WG), body weight at hatch (BW1); body weight at 35 days of age (BW35); body weight at 41 days of age (BW41). The X-axis represents the ordered chromosomes, and Y-axis shows the proportion of genetic variance explained by each window from Bayes B analysis. Red lines indicate the threshold to deem significant SNP windows (0.53%)
From the 20 unique genomic windows (Table 2) associated with body weight traits, two genomic windows were associated exclusively with BW1, eight with BW35, one with BW41 and nine were associated only with BW35 and BW41 (Table 2). Within these latter nine genomic windows associated with BW35 and BW41, SNPs with the highest model frequency were investigated to identify whether the same or different SNPs accounted for the genetic variance explained by the window for BW35 and BW41. The characterization of the SNPs with the highest model frequency are shown in Table 3.
Overlapping with known QTL
From the 20 unique genomic windows (Table 2) associated with body weight traits, 19 overlapped with previously published QTL for body weight traits curated in the Chicken QTL database (release 35). Among those, nine overlapped with QTL associated with exactly the same traits that had been mapped in this same population in previous studies using microsatellite markers [8, 9]. The overlaps are available in the Additional file 2. The genomic window located on GGA24 is a novel QTL, since it did not overlap with any previously published QTL region.
From the 20 unique genomic windows associated with body weight traits (Table 2), five overlapped with QTL previously mapped for fatness traits using the same SNP dataset and the same population (Embrapa F2 Chicken Resource Population) [14] (Table 4).
Positional candidate genes
Our enrichment analysis did not identified MeSH terms associated with any of the categories investigated (i.e, Chemical and Drugs, Diseases, Anatomy, Phenomena and Processes) and also, no enriched clusters of genes were detected. Thus, positional candidate genes within each genomic window were evaluated. From the 386 genes annotated within the associated genomic windows (Additional file 3), 38 were selected as possible candidates for body weight regulation in chickens, based on their respective biological GO terms and literature information (Table 5).
In addition, positional candidate genes that were located within regions under selection previously identified in the grandparental chicken lines used to generate the F2 population [17] were further investigated. The list of genomic windows that harbored positional candidate genes and their overlap with signature selection regions are shown in Table 5.
Search for SNPs in positional candidate genes
Sixteen functional SNPs (13 predicted as deleterious and three as high impact SNPs) were annotated in 11 positional candidate genes (Table 6), integrating the sequence SNPs detected by the re-sequencing of grandparental ancestral chickens used to produce the F2 population.
Discussion
Genomic heritability
The genomic heritability estimates ranged from 0.0002 to 0.13 (Table 1) for FI, WG, FC, and FE. These are complex traits, which are also subject to several environmental factors and, as a result, commonly exhibit low heritability estimates [18]. Moreover, these traits were measured only between 35 and 41 days in an F2 population that exhibits high phenotypic and genotypic variability, and were adjusted using BW35 as a covariate. The short interval between the two measurements could explain the low genomic heritability estimates observed [8]. Nevertheless, FI, FC, FE, and WG are extremely important traits that influence the costs of production and, therefore, should be considered in breeding programs.
Different heritability estimates were found in the literature based on the use of pedigree records to define the genetic variance-covariance among animals, as reported by Gaya et al. [19], who obtained 0.20 for FI and 0.16 for FC (from 5 to 7 weeks of age) using one single sire broiler population. FI, FC (both measured between 35 and 42 days of age) and WG were reported by Aggrey et al. [20] to exhibit values of 0.46, 0.41 and 0.48, respectively, using one random bred population. For body weight traits, Venturini et al. [21] obtained 0.41 for heritability of body weight at 42 days-of-age (BW42) and Grupioni et at [22]. obtained 0.50 for BW42, both using broiler populations.
Using the genomic relationship matrix, Mebratie et al. [23] found heritability estimates of 0.090 for FI, 0.051 for FC and 0.027 for WG (measured in an interval of 7 days of age), in a male broiler population. Abdollahi-Arpanahi et al. [24] obtained heritability of 0.30 for BW35, using a commercial broiler population. It is important to highlight that these studies have used broiler populations that have been under artificial selection. Artificial selection may affect genetic variance and, consequently, genomic heritability [25].
In contrast, this study was performed using an F2 chicken population derived from a cross between divergent lines. Thus, differences in genomic heritability estimates should be expected due to the genetic variability between the lines which introduces segregation variance into the F2. Another study using the same F2 population and a lower density of markers (~ 135 K, Cornell GBS approach), Pértille et al. [10] reported genomic heritability for FC (0.01 ± 0.006), FE (0.11 ± 0.005), FI (0.17 ± 0.094), BW1 (0.45 ± 0.073), BW35 (0.85 ± 0.073) and BW41 (0.75 ± 0.087). Differences in the genomic heritability estimates, even in the same population, are expected due to sampling errors, but can be magnified due to the presence of large effect QTL in the dataset utilized.
Genomic windows identified
The genetic variance explained by each SNP window associated with body weight traits ranged from 0.53 to 4.74%, with windows located on GGA1–4, 6, 7, 10, 14, 24, 27 and 28. Ninety-five percent (19 out of 20) of the genomic windows identified overlapped with at least two known QTL for body weight traits (Additional file 2). Additionally, nine genomic windows on GGA3, 4, 7, 10 and 27 overlapped with known QTL mapped for the same trait in the same population [8] (Additional file 2). Although 95% of the detected genomic windows were already known to be associated with body weight traits as reported in the Chicken QTL database [7], some of them were associated, for the first time, with body weight at the ages analyzed herein. These results provide valuable information to understand the genetic control of body weight, a major factor involved in chicken growth and development.
We also compared genomic window sizes with the span of known QTL previously mapped for the same trait in the same population [8] (Additional file 2). These known QTL were previously mapped using QTL interval mapping (linkage analysis) with up to 127 microsatellite markers, covering 21 linkage groups [8]. Most of our genomic windows had fixed 1 Mb width, with the exception of those windows associated with the same trait that were located in an immediately flanking position, which were merged and the wider combined window was considered as a unique window. Considering this approach, we were able to refine up to 99% of the size of the known QTL (QTL: 7180, 100.4 Mb of size; Additional file 2). Thus, the use of a high density of markers to perform GWAS provided us with much better resolution for QTL detection, facilitating the search for positional candidate genes.
From the 20 unique genomic windows associated with body weight traits (Table 2), five overlapped with QTL previously mapped for fatness traits using the same SNP dataset and the same population (Embrapa F2 Chicken Resource Population) [14] (Table 4). Thus, we suggest that these QTL may exhibit pleiotropic effects, affecting different tissues (such as adipose tissue) and metabolic processes associated with body weight regulation in chickens.
Two genomic windows were associated exclusively with BW1, nine with BW35, one with BW41, while eight were associated with both BW35 and BW41 (Table 2). From these nine genomic windows, four exhibited the same SNP with the highest model frequency for BW35 and BW41. The different percentages explained by the same QTL at different ages may be related to changes in metabolic processes regulating body weight during chicken growth and development. Four genomic windows associated with both BW35 and BW41 exhibited different SNPs with the highest model frequency (Table 3). Among those QTL, the windows on GGA1 at 168 Mb, GGA7 at 36 Mb, and GGA27 at 3 Mb overlapped with QTL previously mapped for fatness traits using the same SNP dataset and the same population (Table 4). Thus, these findings corroborate that different genes, tissues (such as adipose tissue) and metabolic processes can be involved in the regulation of body weight in chickens. Further studies to quantify positional candidate gene expression at different ages could be helpful.
The genomic window on GGA24 did not overlap with published QTL for body weight traits and was considered as a novel QTL. This QTL might be population specific, thus, further studies in other populations might be helpful to validate the role of this region in body weight regulation.
As mentioned earlier, we did not identify any genomic windows associated with FI, FC, FE or WG. This could be due to the lower genetic variance detected for these traits compared to those reported for the other traits (BW1, BW35 and BW41) (Table 1).
Positional candidate genes for body weight in chickens
Within the associated genomic windows, 38 genes were selected as candidates for body weight regulation in chickens, based on their respective biological GO terms and literature information (Table 5). Three of those have already been associated with carcass and body weight traits in livestock - LCORL [26, 27], PPARGC1A [28, 29] and CHRNA9 [30]. Fifteen genes were previously associated with growth and development processes, such as cell growth and proliferation (HOXB2, 4, 7, 9 and 13, HIPK2 [31], KLB [32,33,34] and PHB [35, 36]); embryo development and early growth (TXNRD1 [37], IGF-1R [38], RBM47 [39] and VEGFA [40, 41]) or muscle hypertrophy and development (LONP1 [42], GRIN2A [43] and BMPR1A [44]). It is important to highlight that HOXB2, 4, 7, 9 and 13 genes belong to the homeobox family, known to be associated with development [45] and stem cell growth and differentiation [33]. In addition, the interaction between VEGF members (such as VEGFA) and their receptors may promote cell differentiation in various tissues, such as skeletal muscle in mammals.
One positional candidate gene was associated with heat stress: HSP90AB1. The HSP90AB1 gene encodes a heat shock protein (HPS), which was associated with the response to heat stress in cattle [46, 47] and in general livestock adaptation [48]. Interestingly, heat stress has a negative impact on performance parameters in chickens [49, 50], corroborating that the HSP90AB1 gene can affect performance traits and, consequently, body weight in chickens.
Three actin A receptor types were identified as positional candidate genes: ACVR1, ACVR1C and ACVR2A. The ACVR1 gene is associated with ossification and its expression can inhibit osteogenesis [51, 52], potentially affecting body weight. The ACVR1C gene was associated with adiposity and body weight in mice [53]. The ACVR2A was associated with breast and carcass weight in chickens [54]. Additionally, the ACVR2A gene overlapped with one signature selection region previously identified in the founders of the Embrapa F2 Chicken Resource Population [17], which indicates that this gene was under positive selection affecting breast and carcass weights and, consequently, body weight in either the broiler or layer line.
Breeders have long been selecting chickens for rapid growth, body weight gain, feed efficiency and breast muscle weight [55,56,57]. Selection has resulted in chickens with higher growth rate and breast meat yield, as well as higher fat deposition [56]. Accordingly, in identifying candidates, we did not limit our consideration to only those genes with roles in cell differentiation and proliferation, skeletal muscle growth and development, but also considered genes with roles in adipose tissue development, or energy and lipid metabolism.
Fifteen positional candidate genes were associated with adipose tissue development, energy and lipid metabolism: SLC41A2, SLC37A3, SLC25A6, SLC29A1, SLC1A6, SLC35B1, AKR1D1, ANGPTL4 [58], RB1 [59, 60], CHST11 [61], PMCH [62], NR4A2 [63], HTR2A [64], KCNJ5 and KCNJ1. It is important to highlight that SLC41A2, SLC37A3, SLC25A6, SLC29A1, SLC1A6 and SLC35B1 belong to the solute carrier family already known to be associated with energy metabolism [36, 65,66,67] and obesity in humans [68, 69]. The AKR1D1 gene is involved with bile acid and steroid hormone homeostasis [70] and, interestingly, effects of dietary supplemental bile acids have already been associated with the activity of intestinal and lipoprotein lipases affecting growth performance in chickens [71]. The KCNJ5 and KCNJ1 genes belong to the potassium channel family, that may affect food intake, energy expenditure and glucose homeostasis [48] and, consequently, body weight.
From all the positional candidate genes identified, six were located within two QTL previously mapped for fatness traits [14] (Table 4), and were selected as candidates by Moreira et al. [14] for fat deposition regulation in the same population studied herein: CHST11, RB1, HTR2A, NR4A2, ANGPTL4 and SLC1A6, suggesting that these genes may have pleiotropic effects. Those regions associated with body weight and fatness traits may help to explain why selection for weight gain is associated with increased fat deposition. Moreover, CHST11 and NR4A2 genes overlapped with a signature selection region previously identified in the founders of the Embrapa F2 Chicken Resource Population [17]. These result indicates that these genes are under positive selection and could help to explain the difference in fat deposition observed in the CC and TT lines used in the study. These findings provide helpful information for poultry breeding programs that aim to select birds with both high body weight and reduced fat deposition.
Predicted deleterious and high impact SNPs
Thirteen predicted deleterious and three high impact SNPs were identified in 11 positional candidate genes from our gene list (Table 6). As previously mentioned, these genes were related to energy homeostasis, lipid metabolism and, consequently, body weight regulation. Moreover, the NR4A2 gene overlapped with a selection signature region, indicating that this gene is under positive selection, may affecting lipid metabolism in one of the founder lines and, consequently, body weight. Thus, predicted deleterious and high impact SNPs in these genes could be causative mutations.
In summary, we identified 20 unique 1-Mb genomic windows associated with body weight traits (19 already known and one novel QTL) and within them, we detected 38 positional candidate genes. Through our integrative approach, we refined our list of candidate genes investigating the overlap between sequence SNPs and signatures of selection detected in the founders of the population. Curiously, three positional candidate genes overlapped with regions exhibiting selection signatures. In addition, thirteen predicted deleterious and three high impact SNPs were annotated in 11 positional candidate genes related to osteogenesis, skeletal muscle development, growth, energy metabolism and lipid metabolism, which may be associated with body weight in chickens. Further functional studies need to be performed to validate the role of these mutations in body weight regulation, thus providing important information for poultry breeding.
Conclusions
The use of a high-density SNP array to identify QTL in an F2 population and the integration of regions exhibiting signatures of selection in their pure line ancestors along with sequence SNPs detected in pure line grandparents allowed the identification of candidate genes and candidate causal variants within those genes. Annotation of candidate genes indicates the importance of osteogenesis, cell growth and differenciation, skeletal muscle development, energy metabolism and lipid metabolism in the control of growth and, consequently, body weight in chicken. Our findings form a basis for further functional studies that can elucidate the role of specific genes in body weight regulation in chickens, generating useful information for poultry breeding programs.
Methods
All experimental protocols related to animal experimentation in this study were performed in agreement with resolution number 010/2012 approved by the Embrapa Swine and Poultry National Research Center Ethics Committee on Animal Utilization to ensure compliance with international guidelines for animal welfare.
Animals, population and phenotypes measured
We used the same population described in Moreira et al. [14]. In addition, the population used in this study is the same previously utilized to map QTL for performance, carcass, chemical components and organs traits using microsatellite markers [8, 9, 72,73,74,75]. Sires that exhibited favorable QTL effects previously mapped for those traits, had their progenies selected for high-density genotyping and genome-wide association.
In summary, 529 chickens from an Embrapa F2 Chicken Resource Population (developed by the Embrapa Swine and Poultry National Research Center) were genotyped (28 grandparental chickens from layer and broiler lines, 5 chickens from F1 and 496 chickens from the F2-TCTC generations) [14] with a high-density SNP array (600 K) [76].
Breifly, the layer line (CC) was selected for eight generations for improved egg production, egg weight, feed conversion, viability, sexual maturity, fertility, hatchability, egg quality, and low body weight, prior to the F2 population being created [14]. The broiler line (TT) had been selected for six generations, mainly for improved body weight, feed conversion, carcass and breast yield, viability, fertility, hatchability, lower abdominal fat weight, and reduced metabolic syndromes [14]. More details about the Embrapa F2 Chicken Resource Population are described by Nones et al. [9] and Rosário et al. [77].
Chickens from the F2 population were reared with free access to water and a corn and soybean meal-based diet up to 42 days of age [8]. As described by Ambo et al. [8] and Pértille et al. [10], between 35 and 41 days-of-age, chickens were transferred to individual cages for feed intake (FI) measurement and to compute feed conversion (FC), feed efficiency (FE) and weight gain (WG). Body weight was measured in grams (g) at hatch, 35 and 41 days of age (BW1, BW35, and BW41). The BW41 was collected at the end of the feed conversion test. WG was calculated as the difference between BW41 and BW35. FC was calculated by dividing FI by WG. FE was calculated by dividing WG by FI. Chickens were euthanized by cervical dislocation.
DNA extraction, genotyping and quality control
The DNA extraction, genotyping and quality control have been described in Moreira et al. [14]. Briefly, genomic DNA was extracted from blood with DNAzol® following manufacturer recommendations (Life Technologies Invitrogen). After extraction, DNA integrity was evaluated in agarose gel (1%), quantified in NanoDrop® 2000 spectrophotometer (Thermo Fisher Scientific), then diluted to a final concentration of 20 ng.μL− 1. Diluted genomic DNA was prepared for genotyping following an Affymetrix protocol, and then genotyped with a 600 K Affymetrix Axiom Chicken Genotyping Array (Affymetrix, Inc. Santa Clara, CA, USA). That SNP chip contains segregating SNP for different chicken lines as described by Kranis et al. [76].
Quality control analysis and genotype calling were performed using Affymetrix Power Tools v1.17.0 (APT). Samples that exhibited DishQC ≥0.82 and call rates ≥90% were kept for further analyses. Filtering was performed with the SNPolisher package using R software (http://www.r-project.org/), and SNPs with call rate ≥ 98% and minor allele frequency (MAF) ≥ 2% were kept for further analyses. SNPs monomorphic, located on the sex chromosomes or linkage groups without genomic annotation were removed.
Descriptive statistics, heritability and genome-wide association studies
The SNPs retained after filtering for quality were investigated in GWAS using genomic prediction methodology with a Bayesian approach in GenSel software [78]. In this approach, the genotypes are simultaneously fitted in the model which has been shown to account for any structure in the population [78, 79]. Previous studies have used this approach to perform GWAS and discover QTL and positional candidate genes in chickens based on high density markers [14, 80,81,82,83].
In a first step, BayesC was used to estimate the genetic and residual variances (with π =0). Those values were then used to run a BayesB model, to estimate genomic heritability and perform GWAS, as had been adopted by Cesar et al. [15] and Moreira et al. [14, 83]. The mathematical model presented below was used in the association analyses:
In this model, y represents a vector of phenotypic values, X an incidence matrix for fixed effects, b the vector of fixed effects, k the number of SNP, aj the column vector representing SNP locus j as a covariate coded with the number of B alleles, βj the random substitution effect for locus j assumed normally distributed N (0, σ2β) when δj = 1, with δj being a random indicator variable 0/1, indicating the absence (with probability π) or presence (with probability 1-π) of locus j in the model, and e the vector of the residual effects assumed normally distributed N (0, σ2e). Sex and hatch were included as fixed effects in the model and BW35 was fitted as a covariate for FI, FC, FE and WG.
We adopted π = 0.9988 in the BayesB model to fit approximately 445 SNP per iteration of the Markov chain comprising 41,000 MCMC samples with the first 1000 samples being discarded. A map file was used to allocate the markers to each of 943 1-Mb non-overlapping windows. Based on previous studies that adopted genomic prediction methodology to perform GWAS [14, 15, 80,81,82], we investigated the proportion of genetic variance explained by each and every 1-Mb SNP window across the genome. Due to high linkage disequilibrium between the SNPs fitted simultaneously, the QTL effect can be distributed across nearby markers [78]. These previous studies showed that the 1-Mb windows can capture the effects [14, 80,81,82, 84].
We expect that each window would explain 0.1060% of the genetic variance (100% / 943) in an infinitesimal model as mentioned by Van Goor et al. [80], and windows that explained five times more than expected were considered to be associated with the phenotype. Additionally, we presented the posterior probability of association (PPA) [16] for each associated genomic window, which is the proportion of MCMC samples where the effects of this window were included in the model and accounted for some of the genetic variance [78].
Overlap with previously mapped QTL
All genomic windows detected were compared with published QTL previously mapped to the chicken genome, using the information available at the Chicken QTLdb - release 35 [7]. The search tool in the Chicken QTLdb website was utilized with QTL coordinates based on the Gallus_gallus-5.0 chicken genome assembly. Additionally, to identify possible pleotropic QTL, we compared the genomic windows detected with QTL previously mapped for fatness traits using the same SNP dataset and the same population (Embrapa F2 Chicken Resource Population) [14]. Previously mapped QTL were reported by their respective QTL ID numbers. Genomic windows that did not overlap with previously published QTL regions were considered novel discoveries.
Identification of positional candidate genes and search for potentially causative SNPs
The genes located within every genomic window that had been shown to be associated with a trait, and their corresponding Gene Ontology terms, were retrieved from the Ensembl Genes 92 database available at Ensembl BioMart [85]. A literature search was conducted to increase or decrease support for the selection of a candidate gene. Using the whole gene list, enrichment analyses were performed by two different approaches: MeSH enrichment to identify enriched MeSH terms using the R/Bioconductor package meshr [18, 86] and, Functional Annotation Tool (FAT) in Database for Annotation, Visualization and Integrated Discovery software (DAVID bioinformatics resources v.6.8, [17, 87]) to identify enriched clusters of genes. To identify MeSh terms and genes enriched clusters, we considered the raw p-value < 0.05 and p-value adjusted for multiple testing using the Benjamini & Hochberg [19] procedure (padjusted) < 0.1.
Genes identified in this study were evaluated to determine if they resided within selection signature regions that were previously identified in 28 grandparental chickens (14 TT and 14 CC) that were ancestors of our experimental population [17]. The description of the methods applied to identify the signature selection regions, and the SNP dataset used are available in Boschiero et al. [17].
Additionally, we integrated a sequence SNP dataset from re-sequencing these grandparental animals to identify candidate mutations located in our positional candidate genes. In this study, we only investigated SNPs located in coding regions. To predict whether SNPs that cause changes in amino acids may affect the function of the gene product, we utilized the SIFT (sorting intolerant from tolerant) score to assess the level of conservation in homologous protein sequences using the SIFT algorithm [87] implemented within the VEP tool [86]. High impact SNPs that may cause protein truncation, loss of function or trigger nonsense-mediated decay were also evaluated in the positional candidate genes.
Availability of data and materials
The SNPs reported (identified by sequencing) were submitted to dbSNP- NCBI with the handle “LBA_ESALQ”. The datasets used and/or analyzed during the current study (genotypes and phenotypes) are available from the corresponding author on reasonable request.
Abbreviations
- BW1:
-
Body weight at hatch
- BW35:
-
Body weight at 35 days of age
- BW41:
-
Body weight at 41 days of age
- FC:
-
Feed conversion ratio
- FE:
-
Feed efficiency
- FI:
-
Feed intake
- GWAS:
-
Genome-Wide Association Study
- MAF:
-
Minor Allele Frequency
- MCMC:
-
Markov chain Monte Carlo
- PPA:
-
Posterior Probability of Association
- QTL:
-
Quantitative Trait Locus
- SNP:
-
Single Nucleotide Polymorphism
- WG:
-
Weight gain
References
Berri C, Wacrenier N, Millet N, Le Bihan-Duval E. Effect of selection for improved body composition on muscle and meat characteristics of broilers from experimental and commercial lines. Poult Sci. 2001;80:833–8.
Moreira GCM, Godoy TF, Boschiero C, Gheyas A, Gasparin G, Andrade SCS, et al. Variant discovery in a QTL region on chromosome 3 associated with fatness in chickens. Anim. Genet.. 2015 [cited 2018 may 5];46:141–147. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25643900.
Mebratie W, Reyer H, Wimmers K, Bovenhuis H, Jensen J. Genome wide association study of body weight and feed efficiency traits in a commercial broiler chicken population, a re-visitation. Sci. Rep. Nature Publishing Group; 2019 [cited 2019 Sep 8];9:922 Available from: http://www.nature.com/articles/s41598-018-37216-z
Gao N, Li J, He J, Xiao G, Luo Y, Zhang H, et al. Improving accuracy of genomic prediction by genetic architecture based priors in a Bayesian model. BMC Genet. [Internet]. BioMed Central; 2015 [cited 2019 Jun 3];16:120. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26466667.
Wolc A, Kranis A, Arango J, Settar P, Fulton JE, O’Sullivan NP, et al. Implementation of genomic selection in the poultry industry. Anim. Front. [Internet]. Narnia; 2016 [cited 2019 Jun 3];6:23–31 Available from: https://academic.oup.com/af/article/6/1/23/4638780
CROOIJMANS RPMA, VAN OERS PAM, STRIJK JA, VAN DER POEL JJ, GROENEN MAM. Preliminary Linkage Map of the Chicken (Gallus domesticus) Genome Based on Microsatellite Markers: 77 New Markers Mapped. Poult. Sci. [Internet]. 1996 [cited 2018 Aug 17];75:746–754. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8737840.
Hu Z-L, Park CA, Wu X-L, Reecy JM. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. [Internet]. 2013 [cited 2017 Nov 7];41:D871–D879. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23180796.
Ambo M, MT MASA, Ledur MC, LFB P, Baron EE, Ruy DC, et al. Quantitative trait loci for performance traits in a broiler x layer cross. Anim. Genet. 2009;40:200–8.
Nones K, Ledur MC, Ruy DC, Baron EE, Melo CMR, Moura ASAMT, et al. Mapping QTLs on chicken chromosome 1 for performance and carcass traits in a broiler x layer cross. Anim. Genet. [Internet]. 2006 [cited 2017 Nov 7];37:95–100. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16573522.
Pértille F, Moreira GCM, Zanella R, Nunes JR, Boschiero C, Rovadoscki GA, et al. Genome-wide association study for performance traits in chickens using genotype by sequencing approach. Sci. Rep. [Internet]. Nature Publishing Group; 2017 [cited 2018 Jul 14];7:41748. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28181508.
Yi Z, Li X, Luo W, Xu Z, Ji C, Zhang Y. et al. Feed conversion ratio, residual feed intake and cholecystokinin type A receptor gene polymorphisms are associated with feed intake and average daily gain in a Chinese local chicken population. J. Anim. Sci. Biotechnol. BioMed Central Ltd. 2018;9.
Georges M. Mapping, fine mapping, and molecular dissection of quantitative trait Loci in domestic animals. Annu. Rev. Genomics Hum. Genet. [Internet]. 2007 [cited 2012 Jul 25];8:131–162. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17477823.
Georges M, Charlier C, Hayes B. Harnessing genomic information for livestock improvement. Nat. Rev. Genet. [Internet]. Nature Publishing Group; 2019 [cited 2019 3];20:135–156 Available from: http://www.nature.com/articles/s41576-018-0082-2
Moreira GCM, Boschiero C, Cesar ASM, Reecy JM, Godoy TF, Pértille F, et al. Integration of genome wide association studies and whole genome sequencing provides novel insights into fat deposition in chicken. Sci. Rep. [Internet]. Nature Publishing Group; 2018 [cited 2018 Nov 10];8:16222. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30385857.
Cesar AS, Regitano LC, Mourão GB, Tullio RR, Lanna DP, Nassu RT, et al. Genome-wide association study for intramuscular fat deposition and composition in Nellore cattle. BMC Genet. [Internet]. BioMed Central; 2014 [cited 2017 Nov 7];15:39 Available from: http://bmcgenet.biomedcentral.com/articles/10.1186/1471-2156-15-39
Onteru SK, Gorbach DM, Young JM, Garrick DJ, Dekkers JCM, Rothschild MF. Whole Genome Association Studies of Residual Feed Intake and Related Traits in the Pig. Liu Z, editor. PLoS One [Internet]. Public Library of Science; 2013 [cited 2017 Nov 7];8:e61756. Available from: http://dx.plos.org/10.1371/journal.pone.0061756
Boschiero C, Moreira GCM, Gheyas AA, Godoy TF, Gasparin G, Mariani PDSC, et al. Genome-wide characterization of genetic variants and putative regions under selection in meat and egg-type chicken lines. BMC Genomics [Internet]. 2018 [cited 2018 mar 19];19:83. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29370772.
Reyer H, Hawken R, Murani E, Ponsuksili S, Wimmers K. The genetics of feed conversion efficiency traits in a commercial broiler line. Sci. Rep. [Internet]. Nature Publishing Group; 2015 [cited 2018 Oct 5];5:16387 Available from: http://www.nature.com/articles/srep16387
Gaya LG, Ferraz JBS, Rezende FM, Mourao GB, Mattos EC, Eler JP, et al. Heritability and Genetic Correlation Estimates for Performance and Carcass and Body Composition Traits in a Male Broiler Line. Poult. Sci. [Internet]. 2006 [cited 2018 Jul 20];85:837–43. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16673760.
Aggrey SE, Karnuah AB, Sebastian B, Anthony NB. Genetic properties of feed efficiency parameters in meat-type chickens. Genet. Sel. Evol. [Internet]. 2010 [cited 2018 Oct 5];42:25. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20584334.
Venturini GC, Cruz VAR, Rosa JO, Baldi F, El Faro L, Ledur MC, et al. Genetic and phenotypic parameters of carcass and organ traits of broiler chickens. Genet. Mol. Res. [Internet]. 2014 [cited 2017 Nov 10];13:10294–300. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25501241.
Grupioni N V., Cruz VAR, Stafuzza NB, Freitas LA, Ramos SB, Savegnago RP, et al. Phenotypic, genetic and environmental parameters for traits related to femur bone integrity and body weight at 42 days of age in a broiler population. Poult. Sci. Oxford University Press; 2015;94:2604–2607.
Mebratie W, Bovenhuis H, Jensen J. Estimation of genetic parameters for body weight and feed efficiency traits in a broiler chicken population using genomic information.
Abdollahi-Arpanahi R, Pakdel A, Nejati-Javaremi A, Moradi Shahrbabak M, Morota G, Valente BD, et al. Dissection of additive genetic variability for quantitative traits in chickens using SNP markers. J Anim Breed Genet. 2014;131:183–93.
Flisar T, Malovrh Š, Terčič D, Holcman A, Kovač M. Thirty-four generations of divergent selection for 8-week body weight in chickens. Poult Sci. 2014;93:16–23.
Metzger J, Schrimpf R, Philipp U, Distl O. Expression Levels of LCORL Are Associated with Body Size in Horses. Cameron EZ, editor. PLoS One [Internet]. Public Library of Science; 2013 [cited 2018 Sep 28];8:e56497. Available from: http://dx.plos.org/10.1371/journal.pone.0056497
Lindholm-Perry AK, Sexten AK, Kuehn LA, Smith TP, King DA, Shackelford SD, et al. Association, effects and validation of polymorphisms within the NCAPG - LCORL locus located on BTA6 with feed intake, gain, meat and carcass traits in beef cattle. BMC Genet. [Internet]. 2011 [cited 2018 Sep 28];12:103. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22168586.
Pértille F, Zanella R, Felício AM, Ledur MC, Peixoto JO, Coutinho LL. Identification of polymorphisms associated with production traits on chicken (Gallus gallus) chromosome 4. Genet. Mol. Res. [Internet]. 2015 [cited 2017 Nov 10];14:10717–28. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26400301.
Nassar MK, Goraga ZS, Brockmann GA. Quantitative trait loci segregating in crosses between New Hampshire and White Leghorn chicken lines: IV. Growth performance. Anim. Genet. [Internet]. 2015 [cited 2018 Oct 11];46:441–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25908024.
Liu R, Sun Y, Zhao G, Wang F, Wu D, Zheng M, et al. Genome-wide association study identifies loci and candidate genes for Body Composition and meat quality Traits in Beijing-you chickens. PLoS One. 2013;8.
Luo C, Qu H, Wang J, Wang Y, Ma J, Li C, et al. Genetic parameters and genome-wide association study of hyperpigmentation of the visceral peritoneum in chickens. BMC Genomics [Internet]. BioMed Central; 2013 [cited 2018 Oct 11];14:334. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23679099.
Mallo M, Alonso CR. The regulation of Hox gene expression during animal development. Development [Internet]. 2013 [cited 2018 Oct 10];140:3951–63. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24046316.
Morgan R, Pettengell R, Sohal J. The double life of HOXB4. FEBS Lett. [Internet]. No longer published by Elsevier; 2004 [cited 2018 Oct 10];578:1–4 Available from: https://www.sciencedirect.com/science/article/pii/S0014579304013481
Antonchuk J, Sauvageau G, Humphries RK. HOXB4-Induced Expansion of Adult Hematopoietic Stem Cells Ex Vivo. Cell [Internet]. Cell Press; 2002 [cited 2018 Oct 10];109:39–45 Available from: https://www.sciencedirect.com/science/article/pii/S0092867402006979?via%3Dihub
He L, Vasiliou K, Nebert DW. Analysis and update of the human solute carrier (SLC) gene superfamily. Hum. Genomics [Internet]. 2009 [cited 2017 Nov 7];3:195–206. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19164095.
Chou JY, Mansfield BC. The SLC37 family of sugar-phosphate/phosphate exchangers. Curr. Top. Membr. [Internet]. NIH Public Access; 2014 [cited 2018 Oct 11];73:357–82. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24745989.
Jakupoglu C, Przemeck GKH, Schneider M, Moreno SG, Mayr N, Hatzopoulos AK, et al. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol. Cell. Biol. [Internet]. American Society for Microbiology (ASM); 2005 [cited 2018 Oct 11];25:1980–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15713651.
Lei M, Peng X, Zhou M, Luo C, Nie Q, Zhang X. Polymorphisms of the IGF1R gene and their genetic effects on chicken early growth and carcass traits. BMC Genet. [Internet]. BioMed Central; 2008 [cited 2018 Oct 11];9:70 Available from: http://bmcgenet.biomedcentral.com/articles/10.1186/1471-2156-9-70
Fossat N, Radziewic T, Jones V, Tourle K, Tam PPL. Conditional restoration and inactivation of R bm47 reveal its tissue-context requirement for viability and growth. genesis [Internet]. 2016 [cited 2018 Oct 11];54:115–22. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26789794.
Flamme I, Breier G, Risau W. Vascular Endothelial Growth Factor (VEGF) and VEGF Receptor 2(flk-1) Are Expressed during Vasculogenesis and Vascular Differentiation in the Quail Embryo. Dev. Biol. [Internet]. 1995 [cited 2018 Oct 11];169:699–712. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7781909.
Arsic N, Zacchigna S, Zentilin L, Ramirez-Correa G, Pattarini L, Salvi A, et al. Vascular endothelial growth factor stimulates skeletal muscle regeneration in Vivo. Mol. Ther. [Internet]. 2004 [cited 2018 Oct 11];10:844–54. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15509502.
Dikoglu E, Alfaiz A, Gorna M, Bertola D, Chae JH, Cho T-J, et al. Mutations in LONP1 , a mitochondrial matrix protease, cause CODAS syndrome. Am. J. Med. Genet. Part A [Internet]. Wiley-Blackwell; 2015 [cited 2018 Oct 11];167:1501–9 Available from: http://doi.wiley.com/10.1002/ajmg.a.37029
Du YF, Ding QL, Li YM, Fang WR. Identification of Differentially Expressed Genes and Pathways for Myofiber Characteristics in Soleus Muscles between Chicken Breeds Differing in Meat Quality. Anim. Biotechnol. [Internet]. 2017 [cited 2018 Oct 11];28:83–93 Available from: https://www.tandfonline.com/doi/full/10.1080/10495398.2016.1206555
Verbrugge SAJ, Schönfelder M, Becker L, Yaghoob Nezhad F, Hrabě de Angelis M, Wackerhage H. Genes Whose Gain or Loss-Of-Function Increases Skeletal Muscle Mass in Mice: A Systematic Literature Review. Front. Physiol. [Internet]. Frontiers; 2018 [cited 2018 Oct 11];9:553 Available from: https://www.frontiersin.org/article/10.3389/fphys.2018.00553/full
Richardson MK, Crooijmans RPMA, Groenen MAM. Sequencing and genomic annotation of the chicken <i>(Gallus gallus)</i> Hox clusters, and mapping of evolutionarily conserved regions. Cytogenet. Genome Res. [Internet]. 2007 [cited 2018 Oct 10];117:110–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17675851.
Charoensook R, Gatphayak K, Sharifi AR, Chaisongkram C, Brenig B, Knorr C. Polymorphisms in the bovine HSP90AB1 gene are associated with heat tolerance in Thai indigenous cattle. Trop. Anim. Health Prod. [Internet]. Springer Netherlands; 2012 [cited 2018 Oct 11];44:921–8 Available from: http://link.springer.com/10.1007/s11250-011-9989-8
Sajjanar B, Deb R, Singh U, Kumar S, Brahmane M, Nirmale A, et al. Identification of SNP in HSP90AB1 and its Association with the Relative Thermotolerance and Milk Production Traits in Indian Dairy Cattle. Anim. Biotechnol. [Internet]. 2015 [cited 2018 Oct 11];26:45–50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25153455.
PR A, J A, P P, MK V, PA AN, M B, et al. Role of Heat Shock Proteins in Livestock Adaptation to Heat Stress. J. Dairy, Vet. Anim. Res. [Internet]. MedCrave Online; 2017 [cited 2018 Oct 11];5:1–0 Available from: https://medcraveonline.com/JDVAR/JDVAR-05-00127.php
Quinteiro-Filho WM, Ribeiro A, Ferraz-de-Paula V, Pinheiro ML, Sakai M, Sa LRM, et al. Heat stress impairs performance parameters, induces intestinal injury, and decreases macrophage activity in broiler chickens. Poult. Sci. [Internet]. Oxford University Press; 2010 [cited 2018 Oct 11];89:1905–14 Available from: https://academic.oup.com/ps/article-lookup/doi/10.3382/ps.2010-00812
Mashaly MM, Hendricks GL, Kalama MA, Gehad AE, Abbas AO, Patterson PH. Effect of Heat Stress on Production Parameters and Immune Responses of Commercial Laying Hens. Poult. Sci. [Internet]. Oxford University Press; 2004 [cited 2018 Oct 11];83:889–94 Available from: https://academic.oup.com/ps/article-lookup/doi/10.1093/ps/83.6.889
Kamiya N. The role of BMPs in bone anabolism and their potential targets SOST and DKK1. Curr. Mol. Pharmacol. [Internet]. 2012 [cited 2018 Oct 11];5:153–63. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21787290.
Kamiya N, Kaartinen VM, Mishina Y. Loss-of-function of ACVR1 in osteoblasts increases bone mass and activates canonical Wnt signaling through suppression of Wnt inhibitors SOST and DKK1. Biochem. Biophys. Res. Commun. [Internet]. 2011 [cited 2018 Oct 11];414:326–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21945937.
Yogosawa S, Mizutani S, Ogawa Y, Izumi T. Activin receptor-like kinase 7 suppresses lipolysis to accumulate fat in obesity through downregulation of peroxisome proliferator-activated receptor γ and C/EBPα. Diabetes [Internet]. American Diabetes Association; 2013 [cited 2018 Oct 11];62:115–23. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22933117.
Bhattacharya TK, Chatterjee RN, Dushyanth K, Paswan C, Guru Vishnu P. Activin receptor 2A and activin receptor 2B genes in chicken: effect on carcass traits. J. Appl. Anim. Res. [Internet]. Taylor & Francis; 2016 [cited 2018 Oct 11];44:480–6 Available from: http://www.tandfonline.com/doi/full/10.1080/09712119.2015.1091321
Schmidt G, Figueiredo E, Ledur M. Genetic gain for body weight, feed conversion and carcass traits in selected broiler strains. Rev. Bras. Ciência Avícola [Internet]. Fundação APINCO de Ciência e Tecnologia Avícolas; 2006 [cited 2018 Oct 10];8:29–32 Available from: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S1516-635X2006000100004&lng=en&tlng=en
Baéza E, Le Bihan-Duval E. Chicken lines divergent for low or high abdominal fat deposition: a relevant model to study the regulation of energy metabolism. Animal [Internet]. 2013 [cited 2013 Nov 12];7:965–73. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23433003.
Wang SZ, Hu XX, Wang ZP, Li XC, Wang QG, Wang YX, et al. Quantitative trait loci associated with body weight and abdominal fat traits on chicken chromosomes 3, 5 and 7. Genet. Mol. Res. [Internet]. 2012 [cited 2017 Nov 7];11:956–65. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22576922.
Robciuc MR, Skrobuk P, Anisimov A, Olkkonen VM, Alitalo K, Eckel RH, et al. Angiopoietin-like 4 mediates PPAR delta effect on lipoprotein lipase-dependent fatty acid uptake but not on beta-oxidation in myotubes PLoS One [Internet]. Public Library of Science; 2012 [cited 2017 Nov 7];7:e46212. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23056264.
Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene [Internet]. 1999 [cited 2017 Nov 7];18:7873–82. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10630640.
Moreno-Navarrete JM, Petrov P, Serrano M, Ortega F, Garcia-Ruiz E, Oliver P, et al. Decreased RB1 mRNA, Protein, and Activity Reflect Obesity-Induced Altered Adipogenic Capacity in Human Adipose Tissue. Diabetes [Internet]. 2013 [cited 2017 Nov 7];62:1923–31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23315497.
Tasdelen I, Berger R, Kalkhoven E. PPARγ Regulates Expression of Carbohydrate Sulfotransferase 11 (CHST11/C4ST1), a Regulator of LPL Cell Surface Binding. Najmanovich RJ, editor. PLoS One [Internet]. Public Library of Science; 2013 [cited 2017 Nov 7];8:e64284. Available from: http://dx.plos.org/10.1371/journal.pone.0064284
Sun G, Li M, Li H, Tian Y, Chen Q, Bai Y, et al. Molecular cloning and SNP association analysis of chicken PMCH gene. Mol. Biol. Rep. [Internet]. 2013 [cited 2018 Oct 11];40:5049–55. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23670042.
Han Y-F, Cao G-W. Role of nuclear receptor NR4A2 in gastrointestinal inflammation and cancers. World J. Gastroenterol. [Internet]. Baishideng Publishing Group Inc; 2012 [cited 2017 Nov 7];18:6865–73. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23322982.
Halder I, Muldoon MF, Ferrell RE, Manuck SB. Serotonin Receptor 2A (HTR2A) Gene Polymorphisms Are Associated with Blood Pressure, Central Adiposity, and the Metabolic Syndrome. Metab. Syndr. Relat. Disord. [Internet]. NIH Public Access; 2007 [cited 2017 Nov 7];5:323–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18370802.
Pan C-J, Chen S-Y, Jun HS, Lin SR, Mansfield BC, Chou JY. SLC37A1 and SLC37A2 Are Phosphate-Linked, Glucose-6-Phosphate Antiporters. Zilberstein D, editor. PLoS One [Internet]. Public Library of Science; 2011 [cited 2018 Oct 11];6:e23157. Available from: http://dx.plos.org/10.1371/journal.pone.0023157
Newsholme P, Lima MMR, Procopio J, Pithon-Curi TC, Doi SQ, Bazotte RB, et al. Glutamine and glutamate as vital metabolites. Brazilian J. Med. Biol. Res. = Rev. Bras. Pesqui. medicas e Biol. [Internet]. 2003 [cited 2017 Nov 7];36:153–63. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12563517.
Collin M, Bäckberg M, Ovesjö M-L, Fisone G, Edwards RH, Fujiyama F, et al. Plasma membrane and vesicular glutamate transporter mRNAs/proteins in hypothalamic neurons that regulate body weight. Eur. J. Neurosci. [Internet]. 2003 [cited 2017 Nov 7];18:1265–78. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12956725.
Durand E, Boutin P, Meyre D, Charles MA, Clement K, Dina C, et al. Polymorphisms in the amino acid transporter solute carrier family 6 (neurotransmitter transporter) member 14 gene contribute to polygenic obesity in French Caucasians. Diabetes [Internet]. 2004 [cited 2017 Nov 7];53:2483–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15331564.
Suviolahti E, Oksanen LJ, Ohman M, Cantor RM, Ridderstrale M, Tuomi T, et al. The SLC6A14 gene shows evidence of association with obesity. J. Clin. Invest. [Internet]. American Society for Clinical Investigation; 2003 [cited 2017 Nov 7];112:1762–72. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14660752.
Valanejad L, Ghareeb M, Shiffka S, Nadolny C, Chen Y, Guo L, et al. Dysregulation of Δ 4–3-oxosteroid 5β-reductase in diabetic patients: Implications and mechanisms. Mol. Cell. Endocrinol. [Internet]. 2018 [cited 2018 Oct 11];470:127–41. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29024782.
Lai W, Huang W, Dong B, Cao A, Zhang W, Li J, et al. Effects of dietary supplemental bile acids on performance, carcass characteristics, serum lipid metabolites and intestinal enzyme activities of broiler chickens. Poult. Sci. [Internet]. 2018 [cited 2018 Oct 11];97:196–202. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29136214.
Nones K, Ledur MC, Zanella EL, Klein C, Pinto LFB, Moura ASAMT, et al. Quantitative trait loci associated with chemical composition of the chicken carcass. Anim. Genet. [Internet]. 2012 [cited 2017 Nov 7];43:570–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22497237.
Campos RLR, Nones K, Ledur MC, Moura AS a MT, Pinto LFB, Ambo M, et al. Quantitative trait loci associated with fatness in a broiler-layer cross. Anim. Genet. [Internet]. 2009 [cited 2012 Jul 25];40:729–736. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19466938.
>Baron EE, Moura ASAMT, Ledur MC, Pinto LFB, Boschiero C, Ruy DC, et al. QTL for percentage of carcass and carcass parts in a broiler x layer cross. Anim. Genet. [Internet]. John Wiley & Sons, Ltd (10.1111); 2011 [cited 2019 3];42:117–124 Available from: http://doi.wiley.com/10.1111/j.1365-2052.2010.02105.x
Moura ASAMTAMT, Ledur MC, Boschiero C, Nones K, Pinto LFBB, Jaenisch FRFF, et al. Quantitative trait loci with sex-specific effects for internal organs weights and hematocrit value in a broiler-layer cross. J. Appl. Genet. [Internet]. 2016 [cited 2018 Aug 22];57:215–224. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26496990.
Kranis A, Gheyas A a, Boschiero C, Turner F, Yu L, Smith S, et al. Development of a high density 600K SNP genotyping array for chicken. BMC Genomics 2013;14:59.
Rosário MF Do, Ledur MC, Moura ASAMT, Coutinho LL, Garcia AAF. Genotypic characterization of microsatellite markers in broiler and layer selected chicken lines and their reciprocal F1s. Sci. Agric. [Internet]. Scientia Agricola; 2009 [cited 2017 7];66:150–158 Available from: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0103-90162009000200002&lng=en&tlng=en
Garrick DJ, Fernando RL. Implementing a QTL detection study (GWAS) using genomic prediction methodology. Methods Mol. Biol. [Internet]. 2013 [cited 2017 Nov 7]. p. 275–298. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23756895.
Toosi A, Fernando RL, Dekkers JCM. Genomic selection in admixed and crossbred populations. J Anim Sci. 2010;88:32–46.
Van Goor A, Ashwell CM, Persia ME, Rothschild MF, Schmidt CJ, Lamont SJ. Quantitative trait loci identified for blood chemistry components of an advanced intercross line of chickens under heat stress. BMC Genomics [Internet]. BioMed Central; 2016 [cited 2017 Nov 7];17:287. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27076351.
Van Goor A, Bolek KJ, Ashwell CM, Persia ME, Rothschild MF, Schmidt CJ, et al. Identification of quantitative trait loci for body temperature, body weight, breast yield, and digestibility in an advanced intercross line of chickens under heat stress. Genet. Sel. Evol. [Internet]. BioMed Central; 2015 [cited 2018 mar 25];47:96. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26681307.
Wolc A, Arango J, Jankowski T, Dunn I, Settar P, Fulton JE, et al. Genome-wide association study for egg production and quality in layer chickens. J. Anim. Breed. Genet. 2014 [cited 2018 Jul 14];131:173–182. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24628796.
Moreira GCM, Boschiero C, Cesar ASM, Reecy JM, Godoy TF, Trevisoli PA, et al. A genome-wide association study reveals novel genomic regions and positional candidate genes for fat deposition in broiler chickens. BMC Genomics [Internet]. BioMed Central; 2018 [cited 2018 Jun 7];19:374. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29783939.
Wolc A, Arango J, Settar P, Fulton JE, O’Sullivan NP, Preisinger R, et al. Genome-wide association analysis and genetic architecture of egg weight and egg uniformity in layer chickens. Anim. Genet. [Internet]. 2012 [cited 2018 Aug 27];43:87–96. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22742506.
Kinsella RJ, Kahari A, Haider S, Zamora J, Proctor G, Spudich G, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database . 2011 [cited 2018 Jul 17];2011:bar030–bar030. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21785142.
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics [Internet]. 2010 [cited 2013 Oct 21];26:2069–70 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2916720&tool=pmcentrez&rendertype=abstract
Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. [Internet]. 2003 [cited 2017 Nov 7];31:3812–4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12824425.
Acknowledgements
The authors are grateful to Ricardo Brassaloti (University of Sao Paulo) for help in genotyping the chickens. The authors are also grateful for the support from the FAPESP, CAPES and CNPq mentioned in funding statement. The authors would like to acknowledge the collaborative efforts among EMBRAPA, University of São Paulo and Iowa State University.
Funding
This study was supported by the thematic project (2014/08704–0) from São Paulo Research Foundation (FAPESP). The Embrapa F2 Chicken Resource Population was funded by the Embrapa/PRODETAB project no. 038–01/01 (Brazilian Government). G.C.M. Moreira received fellowships from CAPES (This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil – Finance Code 001), FAPESP grants 14/21380–9 (in cooperation agreement with CAPES) and 16/00569–1, and from CNPq grant 153113/2018–9. C. Boschiero received a fellowship from the program Science without Borders – CNPq, grant 370620/2013–5. T.F. Godoy received fellowship from FAPESP, grant 15/00616–7. F. Pértille received fellowship from FAPESP, grant 16/20440–3. M.D. Poleti received fellowships from CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil - Finance Code 001) and from CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, grant 150914/2017–2). L.L. Coutinho and M.C. Ledur are recipients of productivity fellowship from CNPq. FAPESP, CAPES, CNPq and the Brazilian Government through Embrapa/PRODETAB provided financial support to generate the data, however, they do not participate on the design of the study, sample collection, analysis, data interpretation and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
GCMM, CB, MCL, JMR and LLC conceived the idea of this research and participated in the experimental design. GCMM, CB, TFG, DJG performed data analysis. GCMM, MDP and FP drafted the manuscript. GCMM, MDP, FP, CB, ASMC, JMR, TFG, MCL, DJG and LLC collaborated with interpretation, discussion and writing of the manuscript. MCL, JMR and LLC provided the experimental environment, phenotype and data analysis support. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental protocols related to animal experimentation in this study were performed in agreement with the resolution number 010/2012 approved by the Embrapa Swine and Poultry National Research Center Ethics Committee on Animal Utilization to ensure compliance with international guidelines for animal welfare.
Consent for publication
Not applicable.
Competing interests
Dr. James Reecy is a member of the editorial board (Associate Editor) of BMC Genetics. The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
The characterization of all the 943 1-Mb SNP windows analyzed including the percentage of the genetic variance explained by each one.
Additional file 2.
– Genomic windows, associated traits and published overlapping QTL for body weight traits, available at the Chicken QTL database (release 35). The underlined published QTL were previously mapped with microsatellite markers for the same traits in the same population (Embrapa F2 Chicken Resource Population).
Additional file 3.
List with the Ensembl Gene ID and their respective gene names of the 386 genes annotated within the QTL detected.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Moreira, G.C.M., Poleti, M.D., Pértille, F. et al. Unraveling genomic associations with feed efficiency and body weight traits in chickens through an integrative approach. BMC Genet 20, 83 (2019). https://doi.org/10.1186/s12863-019-0783-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-019-0783-3