A compendium of human genes regulating feeding behavior and body weight, its functional characterization and identification of GWAS genes involved in brain-specific PPI network
A compendium of human genes regulating feeding behavior and body weight, its functional characterization and identification of GWAS genes involved in brain-specific PPI network
Obesity is heritable. It predisposes to many diseases. The objectives of this study were to create a compendium of genes relevant to feeding behavior (FB) and/or body weight (BW) regulation; to construct and to analyze networks formed by associations between genes/proteins; and to identify the most significant genes, biological processes/pathways, and tissues/organs involved in BW regulation.
Results
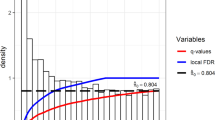
The compendium of genes controlling FB or BW includes 578 human genes. Candidate genes were identified from various sources, including previously published original research and review articles, GWAS meta-analyses, and OMIM (Online Mendelian Inheritance in Man). All genes were ranked according to knowledge about their biological role in body weight regulation and classified according to expression patterns or functional characteristics. Substantial and overrepresented numbers of genes from the compendium encoded cell surface receptors, signaling molecules (hormones, neuropeptides, cytokines), transcription factors, signal transduction proteins, cilium and BBSome components, and lipid binding proteins or were present in the brain-specific list of tissue-enriched genes identified with TSEA tool. We identified 27 pathways from KEGG, REACTOME and BIOCARTA whose genes were overrepresented in the compendium. Networks formed by physical interactions or homological relationships between proteins or interactions between proteins involved in biochemical/signaling pathways were reconstructed and analyzed. Subnetworks and clusters identified by the MCODE tool included genes/proteins associated with cilium morphogenesis, signal transduction proteins (particularly, G protein–coupled receptors, kinases or proteins involved in response to insulin stimulus) and transcription regulation (particularly nuclear receptors). We ranked GWAS genes according to the number of neighbors in three networks and revealed 22 GWAS genes involved in the brain-specific PPI network. On the base of the most reliable PPIs functioning in the brain tissue, new regulatory schemes interpreting relevance to BW regulation are proposed for three GWAS genes (ETV5, LRP1B, and NDUFS3).
Conclusions
A compendium comprising 578 human genes controlling FB or BW was designed, and the most significant functional groups of genes, biological processes/pathways, and tissues/organs involved in BW regulation were revealed. We ranked genes from the GWAS meta-analysis set according to the number and quality of associations in the networks and then according to their involvement in the brain-specific PPI network and proposed new regulatory schemes involving three GWAS genes (ETV5, LRP1B, and NDUFS3) in BW regulation. The compendium is expected to be useful for pathology risk estimation and for design of new pharmacological approaches in the treatment of human obesity.
The pathogenesis of obesity is apparently complex. It involves multiple interactions among behavioral, environmental, and genetic factors. According to twin studies, 40–70% of inter-individual variability in body mass index (BMI), commonly used to assess obesity, is attributed to genetic factors [1–3].
Monogenic forms of obesity account for approximately 5% of severe obesity cases [4]. For most of the monogenic forms of obesity in murine models, human counterparts have been found. Eleven monogenic obesity genes have been identified to date [5]. Monogenic obesity results from mutations in genes involved in the hypothalamic appetite regulation system. Loss-of-function mutations causing deficiencies of appetite-regulating hormones or their receptors, such as leptin [6], leptin receptor [7], pro-opiomelanocortin (POMC) [8], and melanocortin 4 receptor (MC4R) [9], are examples of such monogenic syndromes. Furthermore, mutations in the proprotein convertase subtilisin/kexin type 1 (PCSK1) gene, a key component in the proteolytic processing of POMC, cause extreme childhood obesity and abnormal glucose homeostasis [10]. In addition, patients with chromosomal aberrations resulting in disruption or deletion of the single-minded homologue 1 gene (SIM1), which is essential for proper development of the paraventricular nucleus of hypothalamus, have early-onset obesity [11, 12]. Monogenic obesity also results from mutations in some other genes involved in eating behavior and energy balance regulation: (1) brain-derived neurotrophic factor (BDNF) [13]; (2) its receptor, tyrosine receptor kinase B (NTRK2) [14]; (3) SH2B adaptor protein 1 (SH2B1), involved in the regulation of leptin signaling [15, 16]; (4) KSR2, which encodes a scaffolding protein kinase suppressor of Ras 2, participating in signaling pathways relevant to glucose homoeostasis and food intake control [17]; (5) TUB, encoding Tubby bipartite transcription factor [18]. Although not formally defined as a syndrome, the clinical features of TUB deficiency in humans may be consistent with a novel ciliopathy [5]. Most monogenic obesity cases were investigated only in individual families; thus, their true contribution to “common” obesity in the general population is poorly known and, probably, underestimated [4].
A number of syndromes have been identified in addition to monogenic forms of obesity. The clinical features of these syndromes include obesity and developmental delay [5]. Examples are the Bardet–Biedl [19], Prader–Willi [20], and Alström [21] syndromes. These syndromes were earlier viewed as monogenic, but later studies pointed to a heterogeneous genetic background [4, 19, 20].
Genetic variants associated with “common polygenic obesity” were extensively sought in the pre-GWAS era using genome-wide linkage studies and candidate gene approaches. Unfortunately, none of the SNP markers discovered to have associations with common obesity had meaningful predictive power [4]. Many of them broadly varied across ethnic groups, and they were found difficult to replicate [22].
The development of high-throughput genotyping techniques in conjunction with the progress of statistical and computational methods and the completion of the HapMap and Human Genome Projects enable scientists to carry out large-scale genome-wide association studies, in which a large number of genetic variants are tested for association with a trait of interest [22]. Genome-wide association studies have identified multiple genetic variants associated with the risk of obesity or elevated BMI [23]. However, replication efforts very often yield very inconsistent results [24, 25]. It is important to determine how genetic variants influence body weight, but most of them are non-coding, and there is little understanding of how these variants contribute to BW control.
Recent publications on GWAS meta-analysis present current lists of lead SNPs (lead signals) and candidate genes (secondary signals), which were mostly revealed according to two main criteria: (1) the gene was the nearest to the index SNP; or (2) the gene was found in the vicinity of the lead SNP and was biologically related to obesity, a related metabolic disorder, or energy expenditure according to the results of manual literature mining [26–28]. In some other advanced GWAS meta-analysis reports [29, 30], additional and more sophisticated supportive data are considered, namely: (1) genes contain a cis-expression quantitative trait locus (eQTL) in linkage disequilibrium (LD) with the index SNP; (2) genes contain missense, or nonsense, or copy number variants; (3) genes have been prioritized by connections in published GRAIL (Gene Relationships Across Implicated Loci) abstracts; (4) genes have been prioritized by integrative methods implemented in the DEPICT tool [31]. Thus, GWAS papers register candidate genes, but biological functions of many of them revealed so far (especially for nearest genes or genes containing eQTL) remain unknown or poorly understood. Therefore, the explanation of biological functions of genes noted in GWAS in the context of BW regulation is a separate and essential task.
Thus, in spite of the huge body of information obtained by various experimental approaches, including genome-wide ones, the knowledge of the genetic prerequisites of obesity is insufficient.
The creation of the compendium of genes presumably involved in obesity can be based on the analysis of physiologic systems controlling BW, including basal metabolism, which is regulated by the nervous, endocrine, and immune systems.
The system controlling feeding behavior (FB) is among the most important ones, determining BW. It involves proteins and genes expressed in the brain [32] and in peripheral organs and tissues: the stomach, intestine, pancreas, and fat tissue. The central core of the system is formed by two types of neurons in the hypothalamic arcuate nucleus. They secrete neuropeptide Y (NPY) and the Agouti-related peptide (AgRP) (NPY/AgRP-expressing neurons) or α-melanocyte-stimulating hormone (α-MSH), which is produced from proopiomelanocortin (POMC) by proprotein convertases PCSK1 and PCSK2 (POMC-expressing neurons) [33]. The function of neurons of the arcuate nucleus is controlled by hormones (leptin, insulin, ghrelin, polypeptide YY (PYY), glucocorticoids, adrenocorticotropin, and the corticotropin-releasing hormone), as well as neurotransmitter systems of the brain (serotonergic, dopaminergic, adrenergic, and GABAergic), and neurotrophic factors (BDNF and others) [33, 34].
The objectives of this study were: (1) to compile a compendium of genes controlling human body weight and feeding behavior; (2) to construct and analyze networks formed by associations between genes/proteins from compendium; (3) to reveal tissues or organs, signaling or biochemical pathways, biological processes and physiological systems of the human body associated with genes from compendium; (4) to examine associations between genes identified by GWAS meta-analysis and other genes from compendium and to prioritize GWAS genes according to the number and quality of associations.
We compiled a compendium of 578 human genes collected from: (1) research and review articles, (2) OMIM, and (3) publications presenting GWAS meta-analysis results. We ranked all genes according to our knowledge about the biological roles of particular genes in BW and/or FB regulation. We found that considerable and overrepresented numbers of genes from the compendium encoded cell surface receptors, signaling molecules (hormones, neuropeptides, and cytokines), transcription factors, signal transduction proteins, and cilium and BBSome components or were present in the brain-specific list of genes expressed in a tissue-enriched manner. By using DAVID, we identified 27 pathways enriched in genes from the compendium that might be classified into the following categories: (1) signaling molecules and interaction; (2) signal transduction; (3) endocrine system; (4) excretory system; (5) development; (6) endocrine and metabolic diseases; (7) tumors. Module network analysis of the network involving homology or physical interactions between genes/proteins from compendium revealed notable clusters formed by G protein-coupled receptors and nuclear receptors, as well as clusters associated with cilium morphogenesis, transcription regulation, and insulin signaling.
We prioritized genes collected from the GWAS meta-analysis papers according to the number and quality of associations in the networks. We selected three most reliable PPIs that involved GWAS genes and proved to be functional in the brain tissue according to expression data. These PPIs involved three GWAS genes (ETV5, LRP1B, and NDUFS3) and four other proteins with known biological roles in BW and/or FB regulation. Then we manually reviewed literature related to these genes/proteins and constructed putative regulatory pathways implicating three selected PPIs. We hypothesized that physical interactions between ETV5 and AR, LRP1B and SERPINE1, NDUFS3, and ADRB2 might be regarded as potential mechanisms involving GWAS genes (ETV5, LRP1B, and NDUFS3) in the central regulation of body weight.
Methods
Extracting genes from diverse data sources. Scoring schemes
Genes were extracted from three data sources: (a) scientific publications (research papers and review articles), (b) the OMIM database, and (c) GWAS meta-analysis results presented in scientific papers (Table 1).
Table 1 The sources of data for creating the compendium of genes controlling human body weight and feeding behavior and characteristics of the corresponding gene sets
The first data source included research papers and review articles describing genes that regulate FB in humans or in other mammalian species (mice or rats). This data source and the corresponding set of genes are designated below as Publications. In case the publication described a non-human mammalian gene, the homologous human gene was found and included into the compendium (provided with a special comment).
The second data source was An Online Catalog of Human Genes and Genetic Disorders, OMIM [35]. Three query terms (“obesity”, “hyperphagia”, or “anorexia”) were used. A total of 336 genes were extracted from OMIM. They were divided into two categories. The first category included 73 genes for which at least one of the query terms was found in the chapter dedicated to allelic variants. This data source and the corresponding set of genes are designated below as OMIM_allelic_variants. The second category included 263 genes for which at least one of query terms was found in any other chapter (outside the chapter on allelic variants). This data source and the corresponding set of genes are designated below as OMIM_all_text.
Since genes from the OMIM_all_text set were revealed in cases when query terms were sought through the whole text present in an OMIM entry, we consider such search process to be very similar to the text-mining approach. So genes from OMIM_all_text lacked data on allelic variants associated with pathological states, and they may be characterized as potential regulators of FB or BW.
OMIM was also used to extract genes implicated in the Bardet-Biedl and other syndromes associated with obesity. They are designated below as Syndromes. The list of syndromes associated with obesity and fitting genes was completed according to review articles devoted to the genetics of obesity as well [5, 36].
The third data source was scientific papers presenting GWAS meta-analysis results. We extracted 164 loci with genome-wide significant associations (p-value < 5 × 10−8) with BMI from nine articles. This data source and the corresponding set of genes are designated below as GWAS meta-analysis.
According to GWAS meta-analysis papers, each locus was characterized by a lead SNP and one or more genes located in the vicinity of the lead SNP. Consequently, we collected all genes mentioned in papers and provided each gene with comments regarding its status or potential significance: (1) biological candidate; (2) gene notable for biological relevance to obesity; (3) the BMI-associated variant is in strong linkage disequilibrium (LD; r2 ≥ 0.75) with a missense variant in the indicated gene; (4) eQTL; (5) nearest gene; (6) other nearby gene; etc. A total of 184 genes were obtained from the GWAS meta-analysis papers.
The procedure of ranking was performed on the base of our knowledge of the biological role of a gene in BW regulation. At this step, we divided genes into two groups. The first group (Rank_1: genes with biological interpretation) included all genes from the following data sources: (1) Publications, (2) OMIM-allelic variant, (2) OMIM_all_text, (3) Syndromes. Rank_1 also included some genes from the GWAS meta-analysis data source for which the biological role in FB or BW regulation had been described or explained in GWAS meta-analysis papers. The second group of genes (Rank_2: genes without biological interpretation) accumulated the rest of the genes from the GWAS meta-analysis gene set, whose biological functions had not been explained in the context of FB or BW regulation.
Assignment of genes to functional categories and pathway analysis
To reveal protein-coding genes we used Gene_info table_2016_03_06 from EntrezGene (ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/). To characterize the biological roles of protein-coding genes from the compendium, we divided them into nonoverlapping groups. Some groups (transmembrane receptors, signaling molecules (i.e. hormones, cytokines and neuropeptides), enzymes, etc.) were created manually. To select genes encoding transcription factors we used TFClass database [37]. To classify genes as a transcriptional regulators we used the list of 167 genes encoding proteins with chromatin-modifying activities that was compiled previously [38] from three databases: EntrezGene (http://www.ncbi.nlm.nih.gov/gene), CREMOFAC [39], and CR Cistrome [40]. To annotate genes encoding proteins related to the cilium or BBSome, we used genes extracted from EntrezGene utilizing the GO terms “BBSome” or “cilium” as a query. To confirm that some functional groups of genes were overrepresented in the compendium, we applied the web-based functional annotation tool known as the DAVID (Database for Annotation, Visualization and Integrated Discovery) tool [41]. The significance of GO terms was estimated through the adjusted p-values based on the Benjamini-Hochberg procedure (BH adjusted p-value), presented in the functional annotation chart (a built-in function of DAVID). The standard significance level 0.05 for BH adjusted p-value was applied.
Our second analysis was aimed at the identification of canonical pathways enriched in genes belonging to the compendium of genes regulating FB/BW. At this step, we also applied the DAVID tool [41]. Pathway enrichment analyses were undertaken for 459 genes from the list Rank_1: genes with biological interpretation and for four sets of genes — Publications, OMIM_allelic_variants, OMIM_all-text, Syndromes, GWAS meta-analysis (Table 1) - using DAVID against the ‘whole genome’ background. The enriched biological pathways from KEGG Pathway, Reactome and Biocarta databases are considered in our study. The significance of biological pathways was estimated through the BH adjusted p-value. Pathways with fold enrichments 1.5 or more and the BH adjusted p-values of at most 0.05 were considered interesting.
Gene expression analysis
We invoked freely available data from the Human Protein Atlas [42] version 14 (http://www.proteinatlas.org/about/downloadrna_tissue.csv.zip) to classify genes from the compendium according to the tissue specificity of RNA and protein expression. This resource presents a classification of 19,709 protein-coding genes according to their tissue-specific expression into six categories (Tissue enriched, Group enriched, Tissue enhanced, Expressed in all, Mixed, Not detected), which are defined on the base of transcript levels in 32 human tissues. Our analysis included an additional category, “Tissue elevated”, comprising all genes assigned to the first three categories (Tissue enriched, Group enriched, and Tissue enhanced). The statistical significance of differences between the observed fractions of genes from the compendium classified into particular expression categories and the expected fraction was estimated by the Chi-square test. In each case, the expected fraction of genes calculated for a certain expression category was the same as the fraction of all protein-coding genes from the human genome that were ascribed to this expression category by [42].
At the next step, we used the TSEA (Tissue Specific Expression Analysis) tool (http://genetics.wustl.edu/jdlab/tsea/) to evaluate the significance of the overlap between genes from the compendium and the cell-specific lists of transcripts expressed in a tissue-enriched manner within a particular human organ or tissue. The TSEA tool [43] employed pSI statistics to determine tissue-enriched gene sets using publicly available RNA-seq data across the healthy, adult human body [44]. Within this approach, each cell type profile was compared to all other profiles and transcripts consistently enriched in each cell type were identified. For each transcript, the enrichment score (SI) was calculated and a pSI value was ascribed. Then cell-specific and enriched transcript lists were derived for each cell type at a given pSI threshold. The lower the pSI, the smaller, but more stringently specific, transcript lists were obtained. The TSEA tool accepts an input list of gene symbols and returns the enrichment analysis of their expressions across 25 tissues. As a result, candidate gene lists that overlap cell-specific lists of transcripts expressed in tissue-enriched manner in a particular tissue are identified by Fisher’s exact test with the Benjamini-Hochberg correction. To identify overrepresented lists of tissue-enriched genes and to reveal corresponding human organs or tissues, we set the pSI threshold to 0.05.
Network construction
We employed the GeneMANIA Cytoscape plugin and STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) to identify pairwise relationships among all genes/proteins from the compendium.
STRING [45] contains direct (physical) and indirect (functional) associations derived from different sources, including high-throughput experiments, co-expression, and prior knowledge. The associations of the following types were extracted from STRING in a tab-delimited format: (1) Experimental: protein-protein interactions, (2) Homology: homologous proteins, and (3) Knowledge: functional partners from pathways from the Nature Pathways Interaction Database (NCI) or KEGG. Pairwise relationships of all three types were filtered to include only high-confidence edges with STRING scores greater than 0.4.
We also used the GeneMANIA Cytoscape plugin [46] to identify pairwise physical relationships among genes from the compilation. Data were extracted in a tab-delimited format and filtered to include only edges with GeneMANIA weights greater than 0.01. To obtain a network of protein-protein interactions between objects (Experimental), data on pairwise physical relationships among genes extracted from STRING and GeneMANIA were imported into Cytoscape as two separate networks. Then the networks were merged, and duplicated edges were removed.
The other two networks (Knowledge and Homology) were obtained by importing into Cytoscape the associations of these two types extracted from STRING.
For each gene/protein the following additional data were imported into Cytoscape as attributes of nodes and used to arrange the visualization style: (1) the expression category of the gene (Tissue enriched, Group enriched, etc.); (2) the source of data (Publications, OMIM_allelic_variants, OMIM_all_text, Syndromes, GWAS meta-analysis); (3) knowledge on the biological role of the gene/protein (Rank_1: genes with biological interpretation or Rank_2: genes without biological interpretation).
Module network analysis
The Experimental network was clustered using the ‘Molecular Complex Detection’ (MCODE) algorithm [47] with the default settings. MCODE is a Cytoscape plugin available via the Cytoscape plugin manager [48]. MCODE identifies discrete subnetworks (or clusters) from a larger network (e.g. STRING) and has the advantage over other clustering methods, as it allows direct fine tuning of clusters of interest without relying on the rest of the network.
The top three modules (clusters) of the Experimental network were screened under the conditions of minimum size = 4 and minimum score = 3.3. Then we expanded these three modules by adding the first neighbors of all nodes involved in each module.
To investigate the Gene Ontology (GO) functional enrichment for three expanded lists of genes we employed the DAVID tool. The enriched GO terms from the biological processes vocabulary GOTERM_BP_5 were considered in our study.
Sublist of proteins expressed in brain
The list of genes important for central BW regulation was created by combining two groups of genes from the compendium. The first group included 93 genes revealed by TSEA (see Gene expression analysis section) as brain-specific at pSI threshold = 0.05. The second group included 203 genes whose expression in hypothalamic AGRP-expressing neurons or POMC-expressing neurons differed (with abs[log2(fold-change)] > 1) between mice fed ad libitum and deprived of food for 24 h (Suppl. Table 1 from [49], columns adj.lfc.agrp, adj.lfc.pomc, adj.lfc.AgPo). The two groups of genes were combined, and the resulting sublist of genes (designated below as Brain-specific) comprised 249 genes (Additional file 1: Table S6).
Results
The compendium of genes controlling human body weight and feeding behavior
We collected 578 genes from three sources (Table 1). Among them, 105 genes (Publications) were collected from scientific publications reporting the involvement of genes in FB regulation in humans, mice, or rats. (Additional file 1: Table S1). Association with monogenic non-syndromic obesity was found in 11 genes of the 105 [5]. We extracted 336 genes from OMIM using the query “obesity” OR “hyperphagia” OR “anorexia” (Additional file 1: Table S2). Of them, 73 genes (OMIM_allelic_variants) had OMIM-annotated allelic variants associated with FB abnormalities (hyperphagia, anorexia) or obesity. For the rest 263 genes (OMIM_all_text), the query terms «obesity», «hyperphagia», or «anorexia» were found in textual sections not related to allelic variants. The fourth gene set (Syndromes) was obtained from OMIM and scientific publications. It included 37 genes implicated in syndromes (Bardet-Biedl, Prader–Willi, Alstrom, etc.; 13 syndromes altogether) that included obesity as one of the phenotypic characteristics (Additional file 1: Table S3). The fifth gene set (GWAS meta-analysis) was collected from GWAS meta-analysis papers. It comprised 184 genes listed in articles and located in regions around 164 lead SNPs. The majority of lead SNPs (141 of 164, or 85%) were identified in European ancestry populations. The other 15% were found only in African, East Asian, Australian, and North American populations (Additional file 1: Table S4).
To determine the total number of genes relevant to BW regulation, we merged all gene sets: Publications, OMIM_allelic_variants, OMIM_all_text, Syndromes, and GWAS meta-analysis. With duplicates removed, a list comprising 578 unique genes was obtained (Fig. 1, Additional file 1: Table S5). We found that seven genes (BDNF, MC4R, NTRK2, PCSK1, POMC, SH2B1, TUB) were present in the following three gene sets: Publications, OMIM, GWAS meta-analysis. One gene (BBS4) was found at the intersection of three gene sets: Syndromes, OMIM, and GWAS meta-analysis. Thus, these eight genes found at the intersection of at least three gene sets may be considered the most significant for BW regulation.
Fig. 1
Venn diagram representing the numbers of genes in all gene sets Publications, OMIM, Syndromes, and GWAS meta-analysis used for creating the compendium of human genes controlling BW/FB. The red and blue dashed lines denote groups of genes obtained after ranking genes according to the knowledge of their biological role in body weight control
The ranking of genes according to the knowledge of their biological role in body weight control
According to the ranking procedure described in Extracting genes from diverse data sources. Scoring schemes section, all genes were divided into two groups: (1) Rank_1: genes with biological interpretation; (2) Rank_2: genes without biological interpretation. The first group (Rank_1) included all genes from the following four sets/informational sources: (1) Publications, (2) OMIM_allelic_variants, (3) OMIM_all_text, and (4) Syndromes. Some genes from GWAS meta-analysis data source were also included into Rank_1 if they were characterized in papers as (1) genes notable for their biological relevance to obesity or (2) biological candidates. Thus, 459 genes from the compendium (79% of the total number) were classified to Rank_1: genes with biological interpretation (Fig. 1). The other 21% of genes were classified to Rank_2: genes without biological interpretation. According to the ranking process, the second group of genes was composed entirely of genes from the GWAS meta-analysis data source.
Functional composition of genes from the compendium
First, we determined the fraction of protein-coding genes in the compendium. It was 96.3% (Fig. 2a). The other 21 genes (3.7% of the total number) were non-protein-coding, with ncRNA (microRNA, antisense RNA, etc.) being the largest category. Genes encoding ncRNA comprised 2.5% of the total number of genes in the compendium. The largest portion of non-protein-coding genes were assigned previously to Rank_2: genes without biological interpretation. However, some non-protein-coding genes were categorized into Rank_1: genes with biological interpretation. Among them were MIR148A, H19, LINC00237, MIR103A1, MIR107, MT-TK, NPY6R.
Fig. 2
Distribution of functions of genes from the compendium. Panel a The fractions of protein-coding genes and other genes. Panel b Fractions of major functional groups of genes in the list of protein-coding genes
Next, we divided the list of protein-coding genes into non-overlapping functional groups. We found that the five largest groups were: (1) transmembrane receptors (17% of genes from compendium); (2) signaling molecules (hormones, neuropeptides, cytokines, etc.) (14%); (3) transcription factors (12%); (4) and (5) metabolic and regulatory enzymes (8% and 5%, respectively) (Fig. 2b).
The official symbols of all genes included into the compendium, their data sources, molecular functions of encoded proteins, and Ranks are presented in Additional file 1: Table S5.
The hypothesis that the compendium was enriched in some functional groups of genes used in our classification (Fig. 2b) was tested with the DAVID tool. We performed GO analysis for 578 genes from the compendium and selected GO terms that characterized functional groups presented in Fig. 2b. We found that the compendium was enriched in genes associated with GO terms characterizing most groups: transmembrane receptor activity, hormone activity, neuropeptide hormone activity, cytokine activity, transcription factor activity, signal transducer activity, lipid binding, cilium morphogenesis, BBSome (Table 2). For these GO terms fold enrichments exceeded 1.5, and BH adjusted p-values were less than 0.05. According to GO analysis performed with DAVID, the other three of nine functional groups presented in Fig. 2b (transcriptional coregulators, enzymes (regulatory), and enzymes (metabolic)) were not enriched in the compendium.
Table 2 Overrepresented (BH adjusted p-value < 0.05) GO terms (biological processes, molecular functions, cellular compartments) that characterize functional groups of genes presented in Fig. 2b.
To identify specific biological pathways that might be implicated in FB or BW control, we selected pathways overrepresented among genes from the compendium by applying the DAVID tool. The tool allows detection of enriched biological pathways or models for a variety of biological processes presented in the KEGG, REACTOME, BIOCARTA, and PANTHER databases. We suggested that a substantial part of genes from the GWAS meta-analysis set that had no interpretation (Rank_2: genes without biological interpretation) were not involved in the regulation of FB or BMI, so we excluded these genes from the pathway analysis. Just for this reason we analyzed the subset of 459 genes that had biological interpretations (Rank_1: genes with biological interpretation) at the first step. We identified 27 significantly enriched pathways or biological processes (Fig. 3). In all cases, the fold enrichment exceeded 1.5, and BH adjusted p-values were less than 0.05. We applied the hierarchical scheme provided by the KEGG pathway database to classify the enriched pathways into the following categories: (1) signaling molecules and interaction; (2) signal transduction; (3) endocrine system; (4) excretory system; (5) development; (6) endocrine and metabolic diseases; (7) tumors. The next hierarchical level of classification included such processes and entities as: (1) Environmental Information Processing, (2) Organismal Systems, and (3) Human Diseases.
Fig. 3
Association of genes from the compendium with major KEGG, REACTOME and BIOCARTA pathways. Pathways with fold enrichment > 1.5 and BH adjusted p-value < 5*10−2 are presented. Only genes from Rank_1: genes with biological interpretation were involved in analysis
We also performed pathway analyses for five sets of genes (Publication, OMIM_allelic variant, OMIM_all_text, Syndromes, GWAS meta-analysis) (Additional file 2: Figure S1). We found that the Publications set of genes had only six enriched pathways but the fractions of genes involved into two of them were very large (57% for Signaling by GPCR and 39% for Neuroactive ligand-receptor interaction). This observation points to a functional homogeneity of the Publications set and is in accordance with our previous observation that the compendium is enriched with transmembrane receptors and signaling molecules. Four enriched pathways were revealed for the OMIM_allelic variants set. In contrast, the OMIM_all_text set had 32 overrepresented pathways, and 12 of them (marked by plus signs in Additional file 2: Figure S1) were not overrepresented in the group of 459 genes that had biological interpretations (Rank_1: genes with biological interpretation). No enriched pathway was revealed for the very small set Syndromes (37 genes). The GWAS meta-analysis set also had only one enriched pathway (Signalling by NGF), comprising 9 genes (5% of the total number). The last observation points to high functional heterogeneity of genes from the GWAS meta-analysis set and confirms our guess that some genes from the set may have no relevance to FB or BW control.
Gene expression analysis
We used two approaches to characterize the expression patterns of genes from the compendium.
First, we divided genes from the compendium into six categories (Expressed in all, Mixed, Not detected, Tissue enriched, Group enriched, Tissue enhanced) basing on the classification of all human protein-coding genes presented in [42]. We observed differences in the distributions into categories between the whole genome set of protein-coding genes and genes from the compendium. The fraction of genes classified as Expressed in all decreased significantly (p-value < 0.001) in the compendium (Fig. 4a) as compared to the whole genome level (37.1% vs. 43.6%). In a good agreement with this observation, the compendium had an elevated (p-value < 0.001) fraction of genes from the Tissue enhanced category. The fraction of genes classified as Tissue elevated in the compendium was also higher (p-value < 0.001) than in the whole genome set (Fig. 4b). The enrichment in Tissue elevated genes was also found in sets of genes Publications, OMIM_allelic_variants, and OMIM_all_text. On the contrary, the set Syndromes was depleted of genes from the Tissue elevated category (p-value < 0.01). The most pronounced differences in expression patterns were observed between the whole genome set of protein coding genes and the Publications set. This set was depleted of genes classified as Expressed in all tissues at the significance level of 0.001 (Fig. 4a) and enriched (p-value < 0.001) in genes classified as Tissue elevated (Fig. 4b).
Fig. 4
The fractions of genes classified according to tissue expression patterns and calculated for all protein-coding genes (Genome) or for all genes from the compendium (578_all_Compendium) and gene sets Publications, OMIM_allelic_variants, OMIM_all_text, GWAS meta-analysis, Syndromes. Panel a presents the fractions of genes classified into all six expression categories described in [42] and the category Not found. Panel b presents the fractions of genes belonging to three consolidated groups: (1) Expressed in all + Mixed; (2) Not detected + Not found; (3) Tissue elevated. The significances of the Chi-square test comparing the fractions of genes in test groups with the fractions in the whole-genome dataset are indicated with one (p-value < 0.05), two (p-value < 0.01), or three (p-value < 0.001) asterisks. The red and blue dotted lines in panel a and the orange dotted line in panel b denote the levels observed in the whole genome set of protein-coding genes
At the next step, we utilized the TSEA tool to identify overrepresented tissue-specific genes and to reveal corresponding tissues and organs. The same analysis was performed for the list of all 578 genes from the compendium (designated as 578_all_Compendium) and seven subsets of genes (Rank_1: genes with biological interpretation, Rank_2: genes without biological interpretation, Publications, OMIM_allelic_variants, OMIM_all_text, GWAS meta-analysis, and Syndromes).
We found that genes from the 578_all_Compendium list were overrepresented in tissue-enriched (pSI < 0.05) TSEA lists of genes expressed in seven tissues or organs (Fig. 5). The following tissues and organs were found: (1) adipose tissue and breast, two related tissues that store lipids; (2) the adrenal gland, pituitary gland, and pancreas, endocrine glands controlling metabolism via humoral signals; (3) the liver, central organ in lipogenesis, gluconeogenesis and cholesterol metabolism; (4) the whole brain, which performs the central regulation of feeding behavior - processing sensory signals (taste, olfactory, and food texture) and correlates them with other information. The highest number of genes from the set 578_all_Compendium (93 genes) were found for the overlap with the brain list of tissue-enriched (pSI < 0.05) genes (Fig. 6).
Fig. 5
The heat map depicts the results of tissue-specific expression analysis performed with TSEA. Only tissues with overrepresented (p-value <0.05) cell-specific lists of tissue-enriched genes identified at the overlap with genes from the compendium or seven sets (Rank_1: genes with biological interpretation, Rank_2: genes without biological interpretation, Publications, OMIM_allelic_variants, OMIM_all_text, GWAS meta-analysis, and Syndromes) are shown. P-values derived by Fisher’s exact test with the Benjamini-Hochberg correction were obtained from the TSEA tool
The number of genes from the compendium found with TSEA tool at the overlap with the cell-specific lists of transcripts expressed in a tissue-enriched manner. The lists of tissue-enriched transcripts were identified at pSI threshold = 0.05. Organs or tissues with Benjamini-Hochberg corrected p-values < 0.05 are presented
The genes from seven gene sets had different numbers of tissues identified by TSEA (Fig. 5). Six tissues were identified for genes from the Publications set. The highest number of tissues (ten tissues) was identified by TSEA for the group of genes Rank_1: genes with biological interpretation. The group of genes Rank_2: genes without biological interpretation, which, in our opinion, contained a large portion of genes unrelated to BW regulation, had no tissues identified by TSEA. Seven, three, eight, and two tissues were found for sets Publications, OMIM_allelic_variants, OMIM_all_text, and GWAS meta-analysis, respectively. No tissue was identified by TSEA for the set Syndromes at any pSI threshold. Thus, TSEA identified at least two tissues for five out of seven sets examined (Fig. 5). We found that for three of these five sets of genes (Rank_1: genes with biological interpretation, Publications, and GWAS meta-analysis) the highest numbers of genes were found at the overlaps with the brain list of tissue-enriched genes compiled by TSEA tool at the pSI threshold = 0.05 (Fig. 6).
Networks formed by associations between genes/proteins
To obtain more characteristics of functional systems involved in the regulation of body weight, we reconstructed networks presenting pairwise interactions between genes/proteins. Data on pairwise interactions were obtained from STRING [45] and GeneMANIA [46] (see Network construction section) and uploaded into Cytoscape [48]. Thus, three networks comprising interactions of three different types (Experimental, Knowledge, and Homology) were constructed and analyzed with Cytoscape and its plugin MCODE.
The maximal number of edges and the maximal average number of neighbors were found in the Knowledge network (Table 3). The maximal number of nodes was found in the Experimental network. None of the three networks involved all the 578 genes from the compendium (Fig. 7a, Table 3. We found that the PPI network (Experimental) involved the maximal portion of all genes from the compendium (62%). The Knowledge and Homology networks contained 53% and 25% of the total number of genes, respectively. Altogether, all the three networks included 71% of the total number of genes in the compendium (408 out of 578) (Fig. 7a).
Table 3 Characterization of three networks formed by associations between genes/proteins from the compendium
Venn diagrams representing the numbers of genes involved in three networks Experimental, Knowledge, and Homology. Panel a shows the total number of genes in each network. Panel b the lists of 20 genes that had the highest numbers of neighbors in each network
Genes involved in each network were ranked according to the number of neighbors, and the lists of 20 top genes were formed (Additional file 1: Table S7). The first genes were ESR1 in the network Experimental (43 neighbors), AGT (77 neighbors) and NMUR2 (36 neighbors) in the Knowledge and Homology networks respectively. The intersections between three lists containing top 20 genes that had the maximal numbers of links in each network were analyzed (Fig. 7b). The portion of shared genes was very low. We found only five shared genes at the intersection of the Knowledge and Homology lists of top 20 genes.
We found that three lists of top 20 genes were different with respect to their expression patterns, data sources, and functions (Additional file 1: Table S7):
In the network Experimental, most of the top genes (15 genes out of 20) were classified according to data from the Human Protein Atlas [42] as Expressed in all. Sixteen of the twenty genes belonged to the OMIM_all_text gene set.
In the network Knowledge, a considerable portion of genes (13 out of 20) belonged to the tissue-elevated category (see Gene expression analysis section). The genes were included into the compendium mostly on the base of Publications (12 genes) and OMIM_all_text (9 genes) data sources.
In the network Homology, 13 out of 20 genes were from the tissue-elevated category (see Gene expression analysis section) and six genes were classified as Not detected. Eighteen of twenty genes were from the set Publications. All twenty genes encoded G protein–coupled receptors.
We found that the lists of genes involved in three reconstructed nets (Experimental, Knowledge and Homology) overlapped to a certain extent (Fig. 7a). The merged list of genes involved in all three networks examined contained 408 genes (71% of the total number), which was more than the amounts of genes in each of the three separate networks (Table 3). This fact and the very small fraction of shared genes for three lists of 20 top genes (Fig. 7b) motivated us to consider networks of all three types in our further analysis.
Module network analysis
To explore groups of homologous proteins, a network formed by associations between homologous proteins extracted from STRING database was examined. This network included several unconnected subnetworks (Fig. 8). Two subnetworks (Subnetwork 1, 44 genes, and Subnetwork 4, 6 genes) contained G protein–coupled receptors. Subnetwork 2 (17 genes) was formed by signal transduction proteins (Mitogen-activated protein kinases, other kinases, and some other proteins, such as INSR or IKBKB). Subnetwork 3 (14 genes) included transcription factors from the nuclear receptor superfamily. Each of the next five subnetworks (from fifth to ninth) involved three proteins. Other 24 unconnected subnetworks contained two proteins each (Additional file 1: Table S8).
Fig. 8
Unconnected subnetworks from the network formed by associations between homologous proteins (Homology). The colors of nodes indicate expression categories of genes (see legend) assigned according to data from the Human Protein Atlas [42] (see Gene expression analysis section). Thicker lines represent the stronger associations. Names of genes/proteins from the GWAS meta analysis set are shown in blue. Subnetworks with three or more nodes are outlined by dotted line. TFs – transcription factors
To characterize the functional domains of the network formed by physical interactions between proteins, we explored clusters identified by the MCODE tool (see Module network analysis section). Three clusters with node numbers exceeding 3 and scores exceeding 3.3 were found. As the numbers of nodes in each of these clusters were not large (10, 4, and 4 nodes), we identified the first neighbors for all genes from the clusters and included them into extended groups of genes. By this means, we obtained three extended clusters comprising 15, 33, and 31 nodes (Fig. 9).
Fig. 9
Three extended clusters revealed in the network Experimental formed by physical interactions between proteins from the compendium. Dashed lines denote the initial three clusters comprising 10, 4, and 4 proteins, which were identified with the MCODE tool. For Cluster 1: red check marks denote nine proteins annotated by GO term cilium morphogenesis; four genes marked by blue check marks are localized in primary cilia according to [56]. For Cluster 3: blue lozenges mark proteins associated with GO term response to insulin stimulus. Thicker lines represent stronger associations. The color legend and other designations follow Fig. 8
Taking into account the classification of protein-coding genes according to their tissue-specific expression available from the Human Protein Atlas [42], we observed the expression patterns of genes involved in all three extended clusters. We found that the largest fractions of genes involved in the three extended clusters (100, 79, and 61%, respectively) were classified as expressed in all or mixed.
Then three extended groups/lists of genes were subjected to Gene Ontology (GO) functional enrichment analysis (see Module network analysis section), and it was found that (1) extended group 1 (extended cluster 1) was enriched in genes involved in cilium morphogenesis; (2) 85% of genes from extended group 2 (extended cluster 2) were related to the regulation of transcription; (3) ten proteins from extended group 3 (extended cluster 3), including two of four proteins from initial cluster 3 (IRS2, INSR), were involved in response to insulin stimulus (Fig. 9, and Additional file 1: Table S9).
The ranking of genes from the GWAS meta-analysis set on the base of their weights (numbers of neighbors) in the networks
To obtain additional characteristics for genes from the GWAS meta-analysis set, we considered their links in three networks that were described previously. In the Experimental network we found 74 genes/proteins from the GWAS meta-analysis set that had at least one neighbor. The greatest numbers of the first neighbors were possessed by MAPK3 (22 neighbors), IRS1 (17 neighbors), and NCOA1 (16 neighbors) (Additional file 1: Table S10). In the Knowledge and Homology networks, we found, respectively, 50 and 27 genes/proteins from the GWAS meta-analysis set that had at least one neighbor. The highest numbers of the first neighbors in the Knowledge network were found for POMC (66 neighbors), ADCY3 (65 neighbors) and ADCY9 (64 neighbors). In the Homology network, the three top genes/proteins — HNF4G, MAPK3, RARB — had 12, 11, and 11 first neighbors, respectively. Eighty-five genes/proteins from the GWAS meta-analysis set (~46% of the total number) had at least one first neighbor in at least one of the three networks examined (Fig. 10). Forty of them were from the group Rank_2: genes without biological interpretation. In Fig. 10 (and in all figures below), genes from the group Rank_2: genes without biological interpretation are enclosed in lozenges.
Fig. 10
Genes from the GWAS meta-analysis set that are involved in three networks (Experimental, Knowledge and Homology). Red numerals indicate the numbers of genes that were found in three, two, or one networks. The colors of nodes correspond to RNA expression categories according to data obtained from the Human Protein Atlas [42] (see Gene expression analysis section). An edge width is proportional to the number of neighbors for the corresponding individual gene/protein in each network
Detection of GWAS genes in the Brain-specific sublist
First, we included the annotation of two new characteristics of genes from the compendium into Additional file 1: Table S5: (1) Brain-specific gene according to TSEA for 93 genes that, according to TSEA, belonged to the cell-specific list of transcripts enriched in brain at the pSI threshold = 0.05; (2) differentially expressed gene (DEG) according to [49] for 203 genes found to be differently expressed in hypothalamic AGRP- or POMC-expressing neurons of mice either fed ad libitum or deprived of food. We found that 45 of all 578 genes in the compendium belonged to both categories, and among them 13 genes were from the GWAS meta-analysis set (Fig. 11). Nine of these 13 GWAS genes were from Rank_2: genes without interpretation: CBLN1, PCDH9, DOC2A, STXBP6, LRP1B, LRFN2, RALYL, LINGO2, and HS6ST3. A total of 71 GWAS genes were found to be differentially expressed according to [49] or to be in the TSEA cell-specific list of transcripts enriched in brain at pSI threshold = 0.05. Among these 71 GWAS genes, 34 genes were annotated as Brain-specific gene according to TSEA and 50 genes were annotated as DEGs according to [49] (see Additional file 1: Table S5).
Fig. 11
Venn diagram representing intersections between the GWAS meta-analysis set and two groups of genes Brain-specific gene according to TSEA and DEG according to [49] that gave rise to the Brain-specific sublist of genes from the compendium (see Sublist of proteins expressed in brain section). Callout rectangles show genes that were found at the intersections of the gene set and two gene groups. Genes belonging to the group Rank_2: genes without interpretation are underlined
Subnetwork involving proteins expressed in brain: revealing GWAS genes
At the next step of our study, we reconstructed a network formed by physical interactions between proteins/genes annotated in the Additional file 1: Table S5 as Brain-specific gene according to TSEA or DEG according to [49] (see also Brain-specific sublist, Additional file 1: Table S6). This network was designated Experimental_brain-specific. It included 117 proteins and 172 associations between them (Fig. 12). All nodes in the network were ranked according to their degree (number of neighbors). The highest degree (20 neighbors) was found for estrogen receptor 1 (ESR1). This protein was assigned rank one (Additional file 1: Table S11). Rank two was assigned to PPARG, which had 11 neighbors, and rank three was shared by AR and STAT3, each having 10 neighbors. The Experimental_brain-specific network involved 22 genes from the GWAS meta-analysis set, and six of them were from Rank_2: genes without biological interpretation. Two of these six proteins (NDUFS3, SNRPC) had two neighbors and other four proteins (ERBB4, ETV5, PRKD1, LRP1B) were found to have one neighbor each (Table 4). The numbers of first neighbors ranged from high for AR and STAT3 (both had 10 neighbors) to medium for PARK2 and ADRB2 (8 and 7 neighbors) and low for MAPK9, TOMM40, SERPINE1, and SNRPN (4, 2, 1, and 1 neighbors respectively).
Fig. 12
The Experimental_brain-specific network formed by physical interactions between genes/proteins from the sublist Brain-specific (see Sublist of proteins expressed in brain section). Ellipses denote proteins/genes from the Rank_1: genes with biological interpretation group, lozenges denote proteins/genes from Rank_2: genes without biological interpretation. Names of genes/proteins from the GWAS meta analysis set are shown in blue. Dashed rectangles denote associations that involve genes/proteins from the group Rank_2: genes without biological interpretation. Blue numerals denote the ranks of nodes calculated according to their weight (the number of first neighbors). The color legend and other designations are the same as in Figs. 8, 9, and 10
Table 4 Genes from the GWAS meta-analysis set involved in the Experimental_brain-specific network that were classified into Rank_2: genes without biological interpretation and their first neighbors
We also found that 132 genes from the compendium were present in the Brain-specific sublist (Additional file 1: Table S6), but these genes/proteins were not associated with any other nodes (genes/proteins) in the Experimental_brain-specific network (In Additional file 1: Table S11, these genes are presented in part (B) and are denoted as isolated (single) nodes. We found that 49 genes denoted as isolated nodes were from the GWAS meta-analysis set. Among them, 36 genes were from Rank_2: genes without biological interpretation.
Using the PubMed links presented by STRING and GeneMANIA, we checked pieces of evidence confirming associations between proteins and found that three associations (between ETV5 and AR, LRP1B and SERPINE1, NDUFS3 and ADRB2) were of very good quality. In Table 4, the quality of proofs for these three associations is denoted as Very high. Other associations were confirmed by interactions between homologous proteins in other species (SNRPC and SNRPN) or interactions with paralogous protein in humans or other species (ERBB4 and STAT3, PRKD1 and MAPK9), or even interactions through one intermediate protein (between NDUFS3 and PARK2). In Table 4, the qualities of these proofs are denoted as High or Medium.
Discussion
A compendium of human genes regulating FB and BW
To obtain a systematic overview of genes controlling human body weight and feeding behavior, which may serve as therapeutic targets, we created a compendium of genes relevant to the impairment of feeding behavior and elevated body mass index. At present, the compendium contains information about 578 genes, for which there are indications of their direct or indirect relevance to FB or BW regulation (Fig. 1). We did not find any analogs for such a comprehensive gene catalog in publications. A catalog of eleven monogenic obesity genes identified to date and 227 genetic variants associated with polygenic obesity was presented in [5]. Ninety-seven GWS loci associated with BMI obtained from meta-analysis of nearly 340,000 individuals were described in [29]. A list of 38 genes used in commercially available nutrigenomic tests was compiled in [50]. Thirteen of these 38 genes (HMGCR, APOE, APOB, AGT, CRP, ADRB1, TNF, APOA5, LPL, ADRB3, ADRB2, COMT, and UCP2) are present in the compendium described in the current study. The results of nutrigenomics research studies of the 38 genes were subsequently meta-analyzed in [50] to identify possible associations between the genes of interest and dietary intake and/or nutrient-related pathologies. No specific or statistically significant association were identified for any of the 38 genes of interest. The authors made a conclusion that the need for thorough and continuous nutrigenomics research was evident as it would be a highly promising tool in precision medicine. The application of genomic information in the context of nutritional choice requires the continuing education of healthcare professionals [51] and creation of new evidence evaluation by test providers [52]. Therefore, we consider studies presenting compilations of genes and their analysis useful in this context.
Protein-coding genes constituted 96.3%, the overwhelming majority of genes in the compendium (Fig. 2a). Among them, six major overrepresented functional groups were found: (1) transmembrane receptors; (2) transcription factors; (3) signaling molecules (hormones, neuropeptides, cytokines); (4) signal transducers; (5) cilium and BBSome components; (6) lipid binding proteins (Table 2). Thus, we obtained an updated list of molecular functions important for BW regulation. The significance of these molecular functions for FB and BW regulation is obvious. The key role in regulation of FB and energy balance belongs to the central nervous system, being implemented through a complex interplay among neurons. This interplay is supported by neurotransmitters (neuromediators, neuropeptides, and releasing factors), which modulate neuronal activity via interactions with cellular transmembrane receptors [33, 53, 54]. In turn, receptors activate signal transducers, which effect transcription factor activity [55]. Cilia transduce intracellular signaling activated in response to various homeostatic neuropeptides, neurotransmitters, and hormones (such as neuropeptide Y, melanin-concentrating hormone, insulin, leptin, etc.) [56, 57]. Lipid binding proteins are involved in lipid transport and metabolism [58–60].
To evaluate the degree to what specific biochemical/signaling pathways or biological processes might be involved in FB or BW regulation, we attempted to define overrepresented pathways from the KEGG, REACTOME, and BIOCARTA databases with the DAVID tool. The benefits of using the ontological and pathway analyses for functional annotation of group of genes revealed by different criteria have been considered in numerous publications [38, 61, 62]
We identified a broad variety of enriched pathways/processes. Twenty-seven overrepresented pathways were found for the group Rank_1: genes with biological interpretation (Fig. 3), and the total number of overrepresented pathways found after analysis (Additional file 2: Figure S1) performed for additional five gene sets (Publications, OMIM_allelic_variants, OMIM_all_text, Syndromes and GWAS meta-analysis) was 44. This result is in good agreement with the notion that the process of body weight regulation is very complex [29, 63–65]. Pathway analysis provided evidence that elevated BMI might result from abnormalities in a large number of particular cellular or organismal processes (signaling molecules and interaction, signal transduction, or development) and organismal systems (endocrine or excretory system).
It is also important to keep in mind that most pathway models are not exclusive, i.e., they show considerable overlaps, reflecting the frequently observed synergy in signaling [66]. Such overlaps are most frequently observed for (1) signal transduction pathways, which often share a set of protein kinases, and (2) specific types of cancers sharing signal transducers like STAT3, VEGFB, NFKB1, IKBKB, TGFB1, PIK3CA, MAP2K1, MAPK3, MAPK8, MAPK9, etc. We think that it was a redundancy of signal transduction pathways annotated in any of the three database used (KEGG, REACTOME BIOCARTA) and a redundancy of pathways involved in specific types of cancer in KEGG pathway database that predetermined a considerable abundance of enriched pathways related to signal transduction and cancers revealed by DAVID.
Nevertheless, we propose that all enriched pathways be kept in mind as models of biological processes relevant to body weight regulation. We think that in any case their consideration may be useful for designing new pharmacological approaches for the treatment of BW abnormalities.
Gene expression analysis
Tools such as DAVID, PANTER, REVIGO, and others [65, 67, 68] are a rich source of functional data, but they are static resources that rely on manually curated information from GO, KEGG PATHWAY, Reactome Pathway, Biocarta databases, etc. That is why we think that the results of pathway analysis performed with DAVID (or other similar tools) are not completely objective or unbiased. For this reason, in order to obtain additional functional characteristics of genes from the compendium, we utilized another approach, based on a dynamic source of information, such as gene expression across tissues.
We used the classification of 19,709 human protein-coding genes according to their tissue-specific expression presented in the Human Protein Atlas [42]. We found that the compendium was enriched in genes belonging to the tissue-elevated category, which comprised all genes assigned to the tissue enriched, group enriched, and tissue enhanced categories (Fig. 4). To identify tissues or organs important for FB/BW regulation, we utilized the TSEA tool [43]. It evaluates the significance of overlaps between genes from the compendium and cell-specific lists of transcripts enriched in a particular human organ or tissue (see Module network analysis section).
We found a significant (p-value < 0.05) overlap between genes from compendium and the TSEA lists of tissue-enriched transcripts from seven tissues and organs (adipose tissue, adrenal gland, breast, pituitary gland, pancreas, liver, and brain) (Fig. 5). The same analysis was performed for two gene groups (Rank_1: genes with biological interpretation and Rank_2: genes without biological interpretation) and five gene sets (Publications, OMIM_allelic_variants, OMIM_all_text, GWAS meta-analysis, Syndromes). It revealed six more tissues/organs. In the great majority of cases (in the analyses of three gene sets and of the entire compendium), the highest numbers of genes were found in the overlaps with the cell-specific list of genes enriched (pSI < 0.05) in the brain (Fig. 6). This observation confirms the idea that the nervous system plays a critical role in body weight regulation. First, a large portion of genes responsible for body weight regulation are involved in central mechanisms controlling appetite and food intake [22, 29, 32]. Second, a clear enrichment of expression in the brain was found for genes controlling BMI according to GWAS studies [43, 69, 70].
Networks formed by associations between genes/proteins and module network analysis
At the next step, we reconstructed networks presenting pairwise interactions between genes/proteins of three types: (1) Experimental; (2) Knowledge; (3) Homology (see Networks formed by associations between genes/proteins section). Each network involved a unique set of genes (Fig. 7a) and had a unique set of top genes (Fig. 7b, Table 3). That is why we decided to consider networks of all three types in our further analysis.
We recognized clusters (highly interconnected regions) in the networks. Clusters may be of different sorts in different types of networks: (1) Clusters in a protein-protein interaction network (Experimental) are often protein complexes and parts of pathways, whereas (2) clusters in a protein similarity network (Homology) represent protein families. It was found that Homology network included several unconnected subnetworks (Fig. 8) One-third of all proteins involved in this network (50 genes/proteins) were involved into two subnetworks comprising G protein–coupled receptors (Subnetwork 1 and Subnetwork 4) (Additional file 1: Table S8). Thus, module network analysis confirmed our finding obtained from functional annotation (Fig. 2b) that a substantial part of the compendium (nearly one-sixth, or 17%) comprised genes encoding receptors, and G-protein-coupled receptors among them.
One of unconnected subnetworks (Subnetwork 2) in the network Homology consisted of 17 signal transduction molecules (protein kinases, etc.). In addition: (1) Functional analysis (Fig. 2) showed that 9% of genes from the compendium encoded proteins involved in signal transduction. (2) Sixteen overrepresented signal transduction pathways from KEGG, REACTOME and BIOCARTA were revealed by DAVID for the list of genes Rank_1: genes with biological interpretation (Fig. 3). (3) Analysis of PPI network with the MCODE tool revealed a highly scored cluster (and then, extended cluster 3) involving proteins associated with insulin signaling (Fig. 9), a key energy balance signaling pathway [71, 72]. Thus, module network analysis together with functional analysis showed that (1) genes encoding signal transduction molecules constituted a substantial portion of genes in the compendium and (2) signaling molecules formed a dense network of physical and functional associations. According to the pathway analysis performed with DAVID, genes/proteins from overrepresented signaling pathways are involved in BW regulation, supporting the cellular response to well-known regulators of energy homeostasis, such as insulin [72], leptin [73], neurotrophins (BDNF, NGF) [74], adiponectin [75], PPARs [76], POMC [77], NPY [78], ghrelin [79], etc.
A little more than one-fifth of all proteins (22.7%) in the Homology network were involved in the subnetworks comprising transcription factors and coregulatory proteins (Additional file 1: Table S8: Subnetwork 3, Subnetwork 5, Subnetwork 7, Subnetwork 13, Subnetwork 14, Subnetwork 18, Subnetwork 29, and Subnetwork 30). The extended Cluster 2 obtained in the MCODE analysis of the PPI network was also enriched in transcription factors and coregulatory proteins. From the functional analysis (Fig. 2b), we found that transcription factors and coregulatory proteins constituted, respectively, 12% and 2% of all genes in the compendium. Taken together, these observations are indicative of an important role of transcription factors and transcriptional coregulators in BW control. This finding is in a good agreement with our knowledge on the cooperative interactions between transcription factors and other coregulatory proteins that form complicated mechanisms of transcription complex assembly [38, 80–82].
Module network analysis performed with the MCODE tool showed that the largest and the highest-scored cluster in the PPI network included proteins associated with cilium morphogenesis. Primary cilia are microtubule-based cellular organelles protruding 1–50 μm from the apical surface of cell membrane; they perceive sensory cues and process extracellular signaling, important for cell functions [83]. Primary cilia have been recognized as the signaling center for processing a large number of homeostasis and developmental signaling pathways (PDGFRalpha, mTOR, Notch, Hedgehog, Wnt, etc.) [84]. Several recent lines of indirect evidence suggest a possible crosstalk between energy balance signaling and ciliary signaling [56]. Receptors for energy homeostatic neuropeptides, neurotransmitters, and growth factors, for example, NPYR (neuropeptide Y receptor), MCHR (melanin-concentrating hormone receptor), and IGFR (insulin growth factor receptor), are located in the cilium, and they employ the ciliogenesis machinery to transduce signals properly [57]. During adipocyte differentiation of human adipose stem cells, the primary cilium goes through dynamic size modifications. This suggests that the cilium has various functions during adipogenesis [85]. A recent study by Seo S. et al. [86] has shown that proteins distorted in the human ciliary disorder Bardet-Biedl syndrome (BBS proteins) are required for leptin receptor (LepR) signaling in the hypothalamus: (1) Bbs2(−/−), Bbs4(−/−) and Bbs6(−/−) mice are resistant to the action of leptin to reduce food intake and body weight; (2) BBS1 protein physically interacts with the leptin receptor (LepR). In our study, LepR was also found to be involved into the network formed by PPIs between proteins responsible for cilium morphogenesis (Fig. 9, extended cluster 1). These observations prove the key role of primary cilia in energy balance and indicate that they may be involved in energy balance as a signaling center for processing numerous homeostasis and developmental signaling pathways, including the LepR signaling pathway.
We found that genes involved in clusters identified with the MCODE tool and their first neighbors (three extended clusters, Fig. 9) were enriched in genes that were classified to the Expressed in all and Mixed categories. This observation is in a good agreement with the fact that 17 of 20 top genes that have maximal numbers of neighbors in the Experimental network (Additional file 1: Table S7) also belong to the Expressed in all and Mixed categories. It also indicates that physical interactions between proteins from all three extended clusters revealed in our study (Fig. 9) may be functionally significant and important in a broad range of human tissues and organs.
The ranking of genes from the GWAS meta-analysis set according to the number of neighbors
Meta-analysis of genome-wide association studies (GWAS) resulted in the identification of hundreds of genetic variants associated with elevated body weight. The compendium presented in this report incorporates 164 lead SNPs and 184 genes mentioned in the GWAS meta-analysis papers (Fig. 1, Additional file 1: Tables S4 and S5). However, in most cases it is not easy to find out how these genetic variants influence body weight. In the current study, 119 genes from the GWAS meta-analysis set (or ~65% of all GWAS genes) were classified to the group Rank_2: genes without biological interpretation. That is why we concentrated on the exploration of putative mechanisms involving GWAS genes into BW regulation.
With this task in mind, we examined three networks (Experimental, Knowledge, Homology) and ranked GWAS genes according to the number of neighbors in these three networks (Fig. 10, and Additional file 1: Table S10). In our opinion, associations revealed for some GWAS genes from the group Rank_2: genes without biological interpretation may serve as an additional proof of their relevance to the system of BW regulation.
Identification of GWAS genes in the brain-specific sublist
Expression analysis confirmed the idea that the central nervous system played a critical role in BW regulation: Employment of the TSEA tool showed that the highest number of genes from the compendium overlapped the tissue-enriched list of genes compiled for the brain (Fig. 6). Therefore, at the next step we created a brain-specific sublist comprising 249 genes (see Sublist of proteins expressed in brain section, and Additional file 1: Table S6). We used this sublist for two tasks: (1) analysis of the intersection between the brain-specific sublist and genes from the GWAS meta-analysis set and (2) construction of the network Experimental_brain-specific and its further analysis.
We revealed 71 genes from the GWAS meta-analysis set that were found among 249 genes from the sublist brain-specific (Fig. 11). That meant that these 71 GWAS genes were either differently expressed in hypothalamic POMC- or AGRP-expressing neurons of mice fed ad libitum and deprived of food [49] (21 genes), or were determined by TSEA as specific to brain at the pSI threshold = 0.05 (see Gene expression analysis section) (37 genes), or were found within both categories (13 genes). As shown in Fig. 11, more than a half of GWAS genes found in the intersection between GWAS genes and the brain-specific sublist belonged to the group Rank_2: genes without interpretation. Moreover, nine genes (CBLN1, PCDH9, DOC2A, STXBP6, LRP1B, LRFN2, RALYL, LINGO2, and HS6ST3) that belonged to Rank_2: genes without interpretation were found in the intersection of groups Brain-specific gene according to TSEA and DEG according to [49]. Thus, we conclude that these nine GWAS genes are good candidates for further functional analysis. We found that one of these nine genes (LRP1B) belonged to the network Experimental_Brain-specific, (Fig. 12), and its presumed functional relevance to BW regulation will be discussed below.
The subnetwork involving proteins expressed in brain: search for GWAS genes
At the final step, we examined the brain-specific network of genes/proteins. This network encompassed physical interactions between proteins from the brain-specific sublist (Additional file 1: Table S6). The Experimental_brain-specific network involved 117 genes from the compendium. Among them, 22 genes were from the GWAS meta-analysis set (Additional file 1: Table S11). Six GWAS genes (ETV5, LRP1B, NDUFS3, SNRPC, ERBB4, and PRKD1) of these 22 belonged to the group Rank_2: genes without biological interpretation. For three PPIs (between ETV5 and AR, LRP1B and SERPINE1, NDUFS3 and ADRB2), the quality of evidence for direct physical interactions was very high (Table 4). Therefore, it is highly likely that ETV5, LRP1B, NDUFS3 are involved in the brain-specific network. Associations involving three other genes (SNRPC, ERBB4, PRKD1) may also take place, as follows from the evidence for PPIs found in other species. In addition, SNRPC had two PPIs (with TOMM40 and SNRPN), confirming the idea that this gene/protein was involved in the Experimental brain-specific network.
Thus, we suppose that these six GWAS genes (NDUFS3 SNRPC ERBB4, ETV5, PRKD1, and LRP1B) participate in BW regulation via PPIs with proteins whose biological roles in BW regulation are already known. On the base of the most confident PPIs, we propose three putative regulatory schemes:
Putative regulatory pathways involving physical interactions revealed within the network Experimental_brain-specific: Panel a. Interactions between ETV5 and AR. Panel b. Interactions between LRP1B and SERPINE1. Panel c. The regulatory scheme involving NDUFS3, PARK2, and ADRB2. Ellipses indicate proteins/genes from the group Rank_1: genes with biological interpretation. Lozenges indicate proteins/genes from the group Rank_2: genes without biological interpretation. Genes/proteins from the GWAS meta-analysis set are shown in blue
The androgen receptor (AR) belongs to the superclass of nuclear receptors. It serves as a negative regulator of adipose tissue development in adult males: an increase in the wet weights of white adipose tissues was found in 30-week-old male androgen receptor knockout mice [87]. In the brain, particularly, in the hypothalamus, ARs appear to participate in the regulation of insulin sensitivity and glucose homeostasis [88]. Experiments with AR-deficient mice demonstrated that the loss of neuronal AR led to increased activation of hypothalamic nuclear factor κB (NF-κB), which induces the expression of protein-tyrosine phosphatase 1B (PTP1B) [89]. In turn, PTP1B interferes with hypothalamic insulin signaling via insulin receptor (INSR) dephosphorylation [90].
ETV5 was found to be one of the top 30 genes differentially expressed in POMC-expressing neurons of mice fed ad libitum and deprived of food [49]. According to [91], ARs may negatively regulate the transcriptional activity of target genes through protein-protein interactions with the other transcription factor, ETV5, rather than through direct specific interaction with promoter DNA. Thus, ETV5 may participate in the regulatory mechanism involving NF-κB downregulation and subsequent decline in PTP1B activity. In turn, decreased PTP1B activity may affect the rate of insulin receptor dephosphorylation and thereby impair insulin signaling in hypothalamic neurons (Fig. 13a).
The SERPINE1 gene encodes serpin family E member 1, also known as endothelial plasminogen activator inhibitor-1 (PAI1). PAI1/SERPINE1 is an adipokine, a member of the serine protease inhibitor family. Adipocytes are the main contributor to the elevated levels of PAI-1/SERPINE1 seen in obesity. However, PAI1/SERPINE1 is also produced within the brain by neurons, astrocytes and microglia, and it may regulate migration of microglia, survival of neurites, and apoptosis [92]. PAI1/SERPINE1 was found to be differentially expressed in mouse hypothalamic neurons according to [41] (Sublist Brain-specific,Sublist of proteins expressed in brain section, and Additional file 1: Table S6). With regard to the function of PAI1/SERPINE1 in a non-obese hypothalamus, it is pertinent to mention that transgenic mice overexpressing urokinase-type plasminogen activator (uPA) in the paraventricular nucleus of the hypothalamus and some other brain regions also exhibit reduced body weight and energy intake [93]. PAI1/SERPINE1 is considered the primary inhibitor of uPA. Thus, overexpression of PAI1/SERPINE1 within the hypothalamus can attenuate expression of uPA and, as a result of thereof, increase energy intake and body weight, which are the main parameters in obesity [92]. PAI1/SERPINE1 prevents the disintegration of formed neuronal networks by promoting or maintaining neuroprotective signaling through the MAPK(ERK) pathway [94]. PAI1/SERPINE1 activates microglial cells through the LRP1/JAK/STAT1 axis and promotes migration of microglial cells in culture [95].
LDL receptor related protein 1B (LRP1B) is a member of the low-density lipoprotein receptor gene family. It has been shown that PAI1/SERPINE1 is a ligand for LRP1B [96]. On the cell surface, LRP1B binds and internalizes PAI1/SERPINE1, mediating the function of the urokinase plasminogen activator/receptor [96]. LRP1B is expressed in the whole brain specifically according to TSEA and is a DEG in murine POMC- or AGRP-expressing hypothalamic neurons [49] as well (Fig. 11, and Additional file 1: Table S6). Therefore, we hypothesize that LRP1B mediates the effect of PAI1/SERPINE1 on hypothalamic neurons, promoting cell survival and leading to increased energy intake (Fig. 13b). This regulatory loop may involve: (1) PAI1/SERPINE1 produced by adipocytes, and (2) PAI1/SERPINE1 produced within the brain by neurons, astrocytes and microglia as well, where it mediates paracrine and autocrine signaling between hypothalamic cells. In such a way, the interaction between LRP1B and PAI1/SERPINE1 may contribute to the crosstalk between central and peripheral signals produced within the brain and by adipocytes respectively.
ADRB2 encodes beta-2 adrenoceptor, which is a major lipolytic receptor in human fat cells [97]. ADRB2 is involved in the regulation of the catecholamine function, and it may be of particular importance for human obesity because of the central role of catecholamines in energy expenditure. Loss of noradrenergic terminals in ventral bundle termination areas, e.g., the hypothalamus, may lead to hyperphagia in mice [98]. OMIM contains convincing proofs of associations between ADRB2 allelic variants and obesity. In mice, ADRB2 is differentially expressed in hypothalamic neurons, which confirms its involvement in the central regulation of feeding behavior. Also in mice, ADRB2 demonstrates more than tenfold difference in expression level between AGRP- and POMC-expressing neurons in the fed group [49].
NADH:ubiquinone oxidoreductase core subunit S3 (NDUFS3) encodes one of the iron-sulfur protein components of mitochondrial NADH:ubiquinone oxidoreductase (complex I). At present, we have no idea concerning potential molecular mechanisms effecting the central regulation of energy homeostasis (or BW) and involving physical interactions between ADRB2 and NDUFS3. A small piece of evidence indirectly confirming the idea that NDUFS3 affects FB was found in the KEGG pathways database. According to KEGG, NDUFS3 is involved in molecular events associated with Parkinson disease (KEGG Pathway ID = hsa05012 - Parkinson’s disease - Homo sapiens). NDUFS3 is a component of the first enzyme complex in the electron transport chain of mitochondria. Cleavage of NDUFS3 disrupts mitochondrial metabolism and generates reactive oxygen species (ROS), which trigger the programmed cell death pathway [99]. On the other hand, weight loss has also been reported in advanced stages of Parkinson’s disease. These weight changes are multifactorial. They involve changes in energy expenditure, perturbation of homeostatic control, and feeding behavior. [100]. Thus, we assume that interactions between NDUFS3 and ADRB2 may participate in the crosstalk between catecholamine signaling and antioxidative defense mechanisms that provide neuronal longevity and activity (Fig. 13c).
Parkin RBR E3 ubiquitin protein ligase, also known as PARK2, was the second protein found to be the first neighbor for NDUFS3 in the Experimental_brain-specific network (Fig. 12). The enzyme is a component of the multiprotein E3 ubiquitin ligase complex that mediates the targeting of substrate proteins for proteasomal degradation. PARK2 regulates cell proliferation or cell survival [101]. In mice, E3 ligase Park2 is upregulated in AGRP-expressing neurons during food deprivation [49]. The quality of evidence confirming interactions between NDUFS3 and PARK2 was not high (Table 4). Nevertheless, PARK2 involvement in autosomal recessive juvenile Parkinson disease [102] confirms the regulatory scheme presented in Fig. 13c.
Conclusions
Obesity is a complex disorder, involving multiple genes and multiple biological processes and physiological systems in the human body. The main objectives of the present study were: (1) to collect as full as possible list of genes involved in FB and BW regulation and to formalize it as a compendium; (2) to obtain functional characteristics of genes by using different theoretical approaches and to create a catalog of biological processes, biochemical/signaling pathways, and organs/tissues important for the regulation of BW and FB; and (3) to rank GWAS genes.
At present, the compendium contains 578 human genes for which there are indications of their direct or indirect relevance to the regulation of feeding behavior or body weight.
Here we present functional characteristics of genes regulating FB and BW that were revealed based on the compendium by using several complementary theoretical approaches. We admit that none of them can objectively define the most important feature of genes. Nevertheless, we found that the integration of approaches was useful because the combined result demonstrated the complexity and hierarchy of FB and BW regulation.
On the results of our complex analysis, we revealed and catalogued molecular functions of encoded proteins, biological processes, and biochemical or signaling pathways enriched in genes from the compendium. We outlined a group of tissues and organs important for FB and BW control (Fig. 14). We analyzed networks formed by associations between genes/proteins from the compendium and revealed notable clusters formed by G protein-coupled receptors and nuclear receptors, as well as extended clusters of genes involved in the following basic intracellular processes: cilium morphogenesis, transcription regulation, and insulin signaling. We analyzed expression data from the Human Protein Atlas [42] and concluded that physical interactions between proteins involved in extended clusters associated with basic intracellular processes may be functionally significant in a broad range of human tissues and organs. Thus, on the base of module network analysis we organized data on functional groups of genes from compendium that orchestrate the biological activities of different cell types from different tissues and organs according to the demands of the whole human body (Additional file 1: Tables S5, S8, S9).
Fig. 14
A catalog of functional characteristics of genes regulating BW and FB revealed in the current study. Here we present only the most important molecular functions, biochemical/signaling pathways, organs/tissues, and biological processes: (1) non-overlapping functional groups of genes that were overrepresented in the compendium; (2) the classification of enriched pathways performed using the hierarchical scheme provided by the KEGG pathway database (the full list of pathways is presented in Fig. 3 and Additional file 2: Figure S1); (3) TSEA organs and tissues with the cell-specific lists of tissue-enriched transcripts/genes that were overrepresented with genes from the group Rank_1: genes with biological interpretation; (4) homologous groups of proteins including more than three proteins; (5) extended clusters with scores of initial clusters exceeding 3.3
The expression analysis demonstrated that the highest number of genes from the compendium belonged to the brain list of tissue-enriched genes. Thus, our result confirms the widespread opinion that the central nervous system plays a critical role in body weight regulation. In view of this fact, we reconstructed the brain-specific network formed by physical interactions among genes/proteins from the compendium. Resting on the analysis of the brain-specific network, we proposed potential mechanisms involving three GWAS genes (ETV5, LRP1B, and NDUFS3) in body weight regulation.
The assortment of genes in the compendium provided us with the possibility to prioritize genes from the GWAS meta-analysis set. Prioritization of the GWAS genes is a distinct and important problem due to the lack of information on the biological roles of many GWAS genes in BW control. We performed five ranking procedures (Fig. 15) taking into account: (1) knowledge on the biological role in FB/BW regulation; (2) the numbers of neighbors in three networks; (3) the involvement in the brain-specific sublist of genes; (4) the numbers of physical interactions within the brain-specific network; and (5) the confidence of PPIs in the brain-specific network (this procedure was performed for GWAS genes from the group Rank_2: genes without interpretation).
Fig. 15
Ranking and classification of GWAS genes from the compendium according to various criteria. For ranking based on the number of neighbors in three networks, we present three top genes for each network. For GWAS genes involved in the brain-specific network, three top genes (selected according to the number of neighbors) are presented
Here we present the most comprehensive catalog of functional characteristics of genes controlling body weight than those published before in the most recent studies [5, 29]. Use of different sources of data (Publications, OMIM, and GWAS) allowed us to select and analyze more than five hundred genes. As a result, we (1) formed a more complete list of tissues and organs important in body weight control (aside from the brain or parts of the central nervous system) and (2) distinguished a unique combination of functional characteristics of proteins notable for body weight regulation. The framework formed by these proteins (G protein-coupled receptors, cilium and BBSome components, transcription factors, and proteins involved in insulin signaling) regulates the biological activities of particular cells in accordance with the energy state at the whole-body level.
Our analyses can be regarded as a new step towards a comprehensive list of genes, tissues/organs, biological processes, and pathways involved in FB and BW regulation. It may provide grounds for the development of more holistic disease models and new therapeutics, which is one of the major concerns of obesity research.
Abbreviations
BMI:
Body mass index
BW:
Body weight
DEG:
Differentially expressed gene
eQTL:
Cis-expression quantitative trait locus
FB:
Feeding behavior
GWAS:
Genome-wide association studies
LD:
Linkage disequilibrium
PPI:
Protein-protein interaction
SNP:
Single nucleotide polymorphism
TF:
Transcription factor
References
Stunkard AJ, Foch TT, Hrubec Z. A twin study of human obesity. JAMA. 1986;256(1):51–4.
Wardle J, Carnell S, Haworth CM, Plomin R. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr. 2008;87(2):398–404.
Blakemore AI, Froguel P. Investigation of Mendelian forms of obesity holds out the prospect of personalized medicine. Ann N Y Acad Sci. 2010;1214:180–9. doi:10.1111/j.1749-6632.2010.05880.x.
Pigeyre M, Yazdi FT, Kaur Y, Meyre D. Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin Sci (Lond). 2016;130(12):943–86. doi:10.1042/CS20160136.
Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, Cheetham CH, Earley AR, Barnett AH, Prins JB, O’Rahilly S. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387(6636):903–8.
Clément K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM, Basdevant A, Bougnères P, Lebouc Y, Froguel P, Guy-Grand B. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392(6674):398–401.
Krude H, Biebermann H, Luck W, Horn R, Brabant G, Grüters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. 1998;19(2):155–7.
Vaisse C, Clement K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20(2):113–4.
Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, Hutton JC, O’Rahilly S. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16(3):303–6.
Holder Jr JL, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9(1):101–8.
Faivre L, Cormier-Daire V, Lapierre JM, Colleaux L, Jacquemont S, Geneviéve D, Saunier P, Munnich A, Turleau C, Romana S, Prieur M, De Blois MC, Vekemans M. Deletion of the SIM1 gene (6q16.2) in a patient with a Prader-Willi-like phenotype. J Med Genet. 2002;39(8):594–6.
Gray J, Yeo GS, Cox JJ, Morton J, Adlam AL, Keogh JM, Yanovski JA, El Gharbawy A, Han JC, Tung YC, Hodges JR, Raymond FL, O’rahilly S, Farooqi IS. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006;55(12):3366–71.
Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, O’Rahilly S, Farooqi IS. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7(11):1187–9. Epub 2004 Oct 24.
Pearce LR, Joe R, Doche ME, Su HW, Keogh JM, Henning E, Argetsinger LS, Bochukova EG, Cline JM, Garg S, Saeed S, Shoelson S, O’Rahilly S, Barroso I, Rui L, Farooqi IS, Carter-Su C. Functional characterization of obesity-associated variants involving the α and β isoforms of human SH2B1. Endocrinology. 2014;155(9):3219–26. doi:10.1210/en.2014-1264.
Borman AD, Pearce LR, Mackay DS, Nagel-Wolfrum K, Davidson AE, Henderson R, Garg S, Waseem NH, Webster AR, Plagnol V, Wolfrum U, Farooqi IS, Moore AT. A homozygous mutation in the TUB gene associated with retinal dystrophy and obesity. Hum Mutat. 2014;35(3):289–93. doi:10.1002/humu.22482.
Fall T, Ingelsson E. Genome-wide association studies of obesity and metabolic syndrome. Mol Cell Endocrinol. 2014;382(1):740–57. doi:10.1016/j.mce.2012.08.018.
Beck T, Hastings RK, Gollapudi S, Free RC, Brookes AJ. GWAS Central: a comprehensive resource for the comparison and interrogation of genome-wide association studies. Eur J Hum Genet. 2014;22(7):949–52. doi:10.1038/ejhg.2013.274. Epub 2013 Dec 4.
Lyon HN, Emilsson V, Hinney A, Heid IM, Lasky-Su J, Zhu X, Thorleifsson G, Gunnarsdottir S, Walters GB, Thorsteinsdottir U, Kong A, Gulcher J, Nguyen TT, Scherag A, Pfeufer A, Meitinger T, Brönner G, Rief W, Soto-Quiros ME, Avila L, Klanderman B, Raby BA, Silverman EK, Weiss ST, Laird N, Ding X, Groop L, Tuomi T, Isomaa B, Bengtsson K, Butler JL, Cooper RS, Fox CS, O’Donnell CJ, Vollmert C, Celedón JC, Wichmann HE, Hebebrand J, Stefansson K, Lange C, Hirschhorn JN. The association of a SNP upstream of INSIG2 with body mass index is reproduced in several but not all cohorts. PLoS Genet. 2007;3(4):e61.
Monda KL, et al. A meta-analysis identifies new loci associated with body mass index in individuals of African ancestry. Nat Genet. 2013;45(6):690–6. doi:10.1038/ng.2608.
Guo Y, Lanktree MB, Taylor KC, Hakonarson H, Lange LA, Keating BJ, IBC 50K SNP array BMI Consortium. Gene-centric meta-analyses of 108 912 individuals confirm known body mass index loci and reveal three novel signals. Hum Mol Genet. 2013;22(1):184–201. doi:10.1093/hmg/dds396.
Berndt SI, et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet. 2013;45(5):501–12. doi:10.1038/ng.2606.
Olszewski PK, Cedernaes J, Olsson F, Levine AS, Schiöth HB. Analysis of the network of feeding neuroregulators using the Allen Brain Atlas. Neurosci Biobehav Rev. 2008;32(5):945–56. doi:10.1016/j.neubiorev.2008.01.007.
Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43(Database issue):D789–98. doi:10.1093/nar/gku1205.
Wingender E, Schoeps T, Haubrock M, Dönitz J. TFClass: a classification of human transcription factors and their rodent orthologs. Nucleic Acids Res. 2015;43(Database issue):D97–D102. doi:10.1093/nar/gku1064.
Ignatieva EV, Levitsky VG, Kolchanov NA. Human Genes Encoding Transcription Factors and Chromatin-Modifying Proteins Have Low Levels of Promoter Polymorphism: A Study of 1000 Genomes Project Data. Int J Genomics. 2015a;2015:260159. doi: 10.1155/2015/260159.
Shipra A, Chetan K, Rao MR. CREMOFAC--a database of chromatin remodeling factors. Bioinformatics. 2006;22(23):2940–4.
Wang Q, Huang J, Sun H, Liu J, Wang J, Wang Q, Qin Q, Mei S, Zhao C, Yang X, Liu XS, Zhang Y. CR Cistrome: a ChIP-Seq database for chromatin regulators and histone modification linkages in human and mouse. Nucleic Acids Res. 2014;42(Database issue):D450–8. doi:10.1093/nar/gkt1151.
Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8(9):R183.
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Pontén F. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi:10.1126/science.1260419.
Wells A, Kopp N, Xu X, O’Brien DR, Yang W, Nehorai A, Adair-Kirk TL, Kopan R, Dougherty JD. The anatomical distribution of genetic associations. Nucleic Acids Res. 2015;43(22):10804–20. doi:10.1093/nar/gkv1262.
Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–8. doi:10.1093/nar/gkq973.
Montojo J, Zuberi K, Rodriguez H, Bader GD, Morris Q. GeneMANIA: Fast gene network construction and function prediction for Cytoscape. F1000Res. 2014;3:153. doi:10.12688/f1000research.4572.1. eCollection 2014.
Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–2. doi:10.1093/bioinformatics/btq675.
Henry FE, Sugino K, Tozer A, Branco T, Sternson SM. Cell type-specific transcriptomics of hypothalamic energy-sensing neuron responses to weight-loss. Elife. 2015;4. doi: 10.7554/eLife.09800
Pavlidis C, Lanara Z, Balasopoulou A, Nebel JC, Katsila T, Patrinos GP. Meta-Analysis of Genes in Commercially Available Nutrigenomic Tests Denotes Lack of Association with Dietary Intake and Nutrient-Related Pathologies. OMICS. 2015;19(9):512–20. doi:10.1089/omi.2015.0109.
Pavlidis C, Karamatri A, Barakou A, et al. Ascertainment and critical assessment of the views of the general public and healthcare professionals on nutrigenomics in Greece. Personalized Medicine. 2012;9(2):201–10.
Pavlidis C, Nebel JC, Katsila T, Patrinos GP. Nutrigenomics 2.0: The Need for Ongoing and Independent Evaluation and Synthesis of Commercial Nutrigenomics Tests’ Scientific Knowledge Base for Responsible Innovation. OMICS. 2016;20(2):65–8. doi:10.1089/omi.2015.0170.
Rediger A, Piechowski CL, Habegger K, Grüters A, Krude H, Tschöp MH, Kleinau G, Biebermann H. MC4R dimerization in the paraventricular nucleus and GHSR/MC3R heterodimerization in the arcuate nucleus: is there relevance for body weight regulation? Neuroendocrinology. 2012;95(4):277–88. doi:10.1159/000334903.
Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci U S A. 2008;105(11):4242–6. doi:10.1073/pnas.0711027105.
Ignatieva EV, Levitsky VG, Yudin NS, Moshkin MP, Kolchanov NA. Genetic basis of olfactory cognition: extremely high level of DNA sequence polymorphism in promoter regions of the human olfactory receptor genes revealed using the 1000 Genomes Project dataset. Front Psychol. 2014;5:247. doi:10.3389/fpsyg.2014.00247.
Lempradl A, Pospisilik JA, Penninger JM. Exploring the emerging complexity in transcriptional regulation of energy homeostasis. Nat Rev Genet. 2015;16(11):665–81. doi:10.1038/nrg3941.
Oishi Y, Manabe I. Integrated regulation of the cellular metabolism and function of immune cells in adipose tissue. Clin Exp Pharmacol Physiol. 2016;43(3):294–303. doi:10.1111/1440-1681.12539.
Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8(8):1551–66. doi:10.1038/nprot.2013.092.
Godard P, van Eyll J. Pathway analysis from lists of microRNAs: common pitfalls and alternative strategy. Nucleic Acids Res. 2015;43(7):3490–7. doi:10.1093/nar/gkv249.
Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6(7):e21800. doi:10.1371/journal.pone.0021800.
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(Database issue):D109–14. doi:10.1093/nar/gkr988.
Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, Rybin D, Liu CT, Bielak LF, Prokopenko I, Amin N, Barnes D, Cadby G, Hottenga JJ, Ingelsson E, Jackson AU, Johnson T, Kanoni S, Ladenvall C, Lagou V, Lahti J, Lecoeur C, Liu Y, Martinez-Larrad MT, Montasser ME, Navarro P, Perry JR, Rasmussen-Torvik LJ, Salo P, Sattar N, Shungin D, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44(6):659–69. doi:10.1038/ng.2274.
Fox CS, Liu Y, White CC, Feitosa M, Smith AV, Heard-Costa N, Lohman K, Consortium GIANT, Consortium MAGIC, Consortium GLGC, Johnson AD, Foster MC, Greenawalt DM, Griffin P, Ding J, Newman AB, Tylavsky F, Miljkovic I, Kritchevsky SB, Launer L, Garcia M, Eiriksdottir G, Carr JJ, Gudnason V, Harris TB, Cupples LA, Borecki IB. Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet. 2012;8(5):e1002695. doi:10.1371/journal.pgen.1002695.
Varela L, Horvath TL. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep. 2012;13(12):1079–86. doi:10.1038/embor.2012.174.
Vogt MC, Brüning JC. CNS insulin signaling in the control of energy homeostasis and glucose metabolism - from embryo to old age. Trends Endocrinol Metab. 2013;24(2):76–84. doi:10.1016/j.tem.2012.11.004.
Zabeau L, Peelman F, Tavernier J. Leptin: From structural insights to the design of antagonists. Life Sci. 2015;140:49–56. doi:10.1016/j.lfs.2015.04.015.
Fargali S, Sadahiro M, Jiang C, Frick AL, Indall T, Cogliani V, Welagen J, Lin WJ, Salton SR. Role of neurotrophins in the development and function of neural circuits that regulate energy homeostasis. J Mol Neurosci. 2012;48(3):654–9.
Bugge A, Mandrup S. Molecular Mechanisms and Genome-Wide Aspects of PPAR Subtype Specific Transactivation. PPAR Res. 2010;2010. doi: 10.1155/2010/169506.
Lowry P. 60 YEARS OF POMC: Purification and biological characterisation of melanotrophins and corticotrophins. J Mol Endocrinol. 2016;56(4):T1-12. doi:10.1530/JME-15-0260.
Loh K, Herzog H, Shi YC. Regulation of energy homeostasis by the NPY system. Trends Endocrinol Metab. 2015;26(3):125–35. doi:10.1016/j.tem.2015.01.003.
Edwards A, Abizaid A. Driving the need to feed: Insight into the collaborative interaction between ghrelin and endocannabinoid systems in modulating brain reward systems. Neurosci Biobehav Rev. 2016;66:33–53. doi:10.1016/j.neubiorev.2016.03.032.
Cheng C, Alexander R, Min R, Leng J, Yip KY, Rozowsky J, Yan KK, Dong X, Djebali S, Ruan Y, Davis CA, Carninci P, Lassman T, Gingeras TR, Guigó R, Birney E, Weng Z, Snyder M, Gerstein M. Understanding transcriptional regulation by integrative analysis of transcription factor binding data. Genome Res. 2012;22(9):1658–67. doi:10.1101/gr.136838.111.
Ko HW. The primary cilium as a multiple cellular signaling scaffold in development and disease. BMB Rep. 2012;45(8):427–32. doi:10.5483/BMBRep.2012.45.8.167.
Forcioli-Conti N, Lacas-Gervais S, Dani C, Peraldi P. The primary cilium undergoes dynamic size modifications during adipocyte differentiation of human adipose stem cells. Biochem Biophys Res Commun. 2015;458(1):117–22. doi:10.1016/j.bbrc.2015.01.078.
Sato T, Matsumoto T, Yamada T, Watanabe T, Kawano H, Kato S. Late onset of obesity in male androgen receptor-deficient (AR KO) mice. Biochem Biophys Res Commun. 2003;300(1):167–71.
Chang C, Yeh S, Lee SO, Chang TM. Androgen receptor (AR) pathophysiological roles in androgen-related diseases in skin, bone/muscle, metabolic syndrome and neuron/immune systems: lessons learned from mice lacking AR in specific cells. Nucl Recept Signal. 2013;11:e001. doi:10.1621/nrs.11001. eCollection 2013.
Zhang YL, Tam M, Kirincich S, Wan ZK, Wilson D, Wu JJ, Lee J, Tobin JF, Erbe DV. An enzyme-linked immunosorbent assay to measure insulin receptor dephosphorylation by PTP1B. Anal Biochem. 2007;365(2):174–84. Epub 2007 Mar 31.
Schneikert J, Peterziel H, Defossez PA, Klocker H, de Launoit Y, Cato AC. Androgen receptor-Ets protein interaction is a novel mechanism for steroid hormone-mediated down-modulation of matrix metalloproteinase expression. J Biol Chem. 1996;271(39):23907–13.
Arnoldussen IA, Kiliaan AJ, Gustafson DR. Obesity and dementia: adipokines interact with the brain. Eur Neuropsychopharmacol. 2014;24(12):1982–99. doi:10.1016/j.euroneuro.2014.03.002.
Miskin R, Masos T. Transgenic mice overexpressing urokinase-type plasminogen activator in the brain exhibit reduced food consumption, body weight and size, and increased longevity. J Gerontol A Biol Sci Med Sci. 1997;52(2):B118–24.
Soeda S, Koyanagi S, Kuramoto Y, Kimura M, Oda M, Kozako T, Hayashida S, Shimeno H. Anti-apoptotic roles of plasminogen activator inhibitor-1 as a neurotrophic factor in the central nervous system. Thromb Haemost. 2008;100(6):1014–20.
Jeon H, Kim JH, Kim JH, Lee WH, Lee MS, Suk K. Plasminogen activator inhibitor type 1 regulates microglial motility and phagocytic activity. J Neuroinflammation. 2012;9:149. doi:10.1186/1742-2094-9-149.
Liu CX, Li Y, Obermoeller-McCormick LM, Schwartz AL, Bu G. The putative tumor suppressor LRP1B, a novel member of the low density lipoprotein (LDL) receptor family, exhibits both overlapping and distinct properties with the LDL receptor-related protein. J Biol Chem. 2001;276(31):28889–96.
Kistner A, Lhommée E, Krack P. Mechanisms of body weight fluctuations in Parkinson’s disease. Front Neurol. 2014;5:84. doi:10.3389/fneur.2014.00084. eCollection 2014.
Bouman L, Schlierf A, Lutz AK, Shan J, Deinlein A, Kast J, Galehdar Z, Palmisano V, Patenge N, Berg D, Gasser T, Augustin R, Trümbach D, Irrcher I, Park DS, Wurst W, Kilberg MS, Tatzelt J, Winklhofer KF. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011;18(5):769–82. doi:10.1038/cdd.2010.142.
Poorkaj P, Nutt JG, James D, Gancher S, Bird TD, Steinbart E, Schellenberg GD, Payami H. Parkin mutation analysis in clinic patients with early-onset Parkinson disease. Am J Med Genet A. 2004;129A(1):44–50.
Zanon A, Rakovic A, Blankenburg H, Doncheva NT, Schwienbacher C, Serafin A, Alexa A, Weichenberger CX, Albrecht M, Klein C, Hicks AA, Pramstaller PP, Domingues FS, Pichler I. Profiling of Parkin-binding partners using tandem affinity purification. PLoS One. 2013;8(11):e78648. doi:10.1371/journal.pone.0078648. eCollection 2013.
Wu GC, Wodak SJ, Tillier ER, Paccanaro A, Marcotte EM, Emili A. A census of human soluble protein complexes. Cell. 2012;150(5):1068–81. doi:10.1016/j.cell.2012.08.011.
Hurd C, Waldron RT, Rozengurt E. Protein kinase D complexes with C-Jun N-terminal kinase via activation loop phosphorylation and phosphorylates the C-Jun N-terminus. Oncogene. 2002;21(14):2154–60.
The authors are grateful to Victor V. Gulevich for checking the English translation. We thank anonymous reviewers for their valuable comments on our manuscript.
The present study was supported in part by the Government of the Russian Federation, grant no. 14.B25.31.0033 to E. I. Rogaev, Resolution No. 220 (compiling the compendium, functional, pathway and gene expression analysis, construction and analysis of the brain-specific network). Elena V. Ignatieva acknowledges the support of the module network analysis by the Federal Agency of Scientific Organizations, project 0324-2015-0003. The publication of this work was supported by the Government of the Russian Federation, grant no. 14.B25.31.0033 to E. I. Rogaev, Resolution No. 220.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its Additional files 1 and 2.
Authors’ contributions
NAK, EIR and DAA participated in project conception and in the study design. EVI extracted data from literature and databases, performed data analysis, and drafted the manuscript. OVS was responsible for the extraction of data from STRING and correction of the manuscript. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Center for Brain Neurobiology and Neurogenetics, The Federal Research Center Institute of Cytology and Genetics of Siberian Branch of the Russian Academy of Sciences, Novosibirsk, 630090, Russia
Elena V. Ignatieva, Dmitry A. Afonnikov, Olga V. Saik & Evgeny I. Rogaev
Novosibirsk State University, Novosibirsk, 630090, Russia
Elena V. Ignatieva, Dmitry A. Afonnikov & Nikolay A. Kolchanov
Laboratory of Evolutionary Bioinformatics and Theoretical Genetics, The Federal Research Center Institute of Cytology and Genetics of Siberian Branch of the Russian Academy of Sciences, Novosibirsk, 630090, Russia
Elena V. Ignatieva & Dmitry A. Afonnikov
Department of Systems Biology, The Federal Research Center Institute of Cytology and Genetics of Siberian Branch of the Russian Academy of Sciences, Novosibirsk, 630090, Russia
Nikolay A. Kolchanov
BNRI, Department of Psychiatry, University of Massachusetts Medical School, Worcester, MA, 15604, USA
Human genes (105) controlling feeding behavior that were collected from scientific publications (research papers and review articles); Table S2. Human genes (336) controlling feeding behavior or body weight that were collected from OMIM; Table S3. Human genes (37) associated with syndromes that include obesity as one of the phenotypic characteristics (Bardet-Biedl, Prader-Willi, Alstrom, etc); Table S4. Data from the GWAS meta-analysis papers. Lead SNPs with genome-wide significant (p-value < 5 × 10−8) associations with BMI (164); and genes listed in GWAS meta-analysis articles and located in the regions around the 164 lead SNPs (184). Table S5. All genes from the compendium (578 genes); Table S6. The brain-specific sublist of genes (column C) that was obtained by merging two lists (presented in columns A and B); Table S7. Top twenty genes that have the maximal numbers of the first neighbors in each network (Experimental, Knowledge, Homology) and their main characteristics; Table S8. Unconnected subnetworks found within the network Homology; Table S9. Top-scoring GO terms from the chart GOTERM_BP_5 obtained with the DAVID tool for three lists of genes. The lists of genes analyzed here were obtained by extending three top-scoring clusters revealed in the protein interaction network Experimental with the MCODE tool (see Module network analysis section). Only GO terms with FDR below E-07 are presented; Table S10. Genes from the GWAS meta-analysis set that are involved in three networks (Experimental, Knowledge, Homology) and have at least one neighbor; Table S11. Genes involved in the brain-specific network of PPIs (Section A) and single (isolated) genes (Section B) that belong to the sublist brain-specific. (XLSX 188 kb)
Association of genes from the compendium with major KEGG, REACTOME and BIOCARTA pathways. Pathways with fold enrichment > 1.5 and BH adjusted p-value < 0.05 are presented. This figure presents the results for the sets of genes Publications, OMIM_allelic_variants, OMIM_all_text, Syndromes and GWAS meta-analysis. (DOCX 600 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Ignatieva, E.V., Afonnikov, D.A., Saik, O.V. et al. A compendium of human genes regulating feeding behavior and body weight, its functional characterization and identification of GWAS genes involved in brain-specific PPI network.
BMC Genet17
(Suppl 3), 158 (2016). https://doi.org/10.1186/s12863-016-0466-2