
Abstract
Background
Piglet birth weight variability, a trait also known as the within-litter homogeneity of birth weight, reflects the sow’s prolificacy, because it is positively genetically correlated with preweaning mortality but negatively correlated with the mean growth of piglets during sucking. In addition, the maternal additive genetic variance and heritability has been found exist for this trait, thus, reduction in the variability of piglet birth weight to improve the sow prolificacy is possible by selective breeding.
Results
We performed a genome wide association study (GWAS) in 82 sows with extreme standard deviation of birth weights within the first parity to identify significant SNPs, and finally 266 genome-wide significant SNPs (p < 0.01) were identified. These SNPs were mainly enriched on chromosome 7, 1, 13, 14, 15 and 18. We further scanned genes of the top 50 SNPs with the lowest p values and found some genes involved in plasma glucose homeostasis (GLP1R) and lipid metabolism as well as maternal-fetal lipid transport (AACS, APOB, OSBPL10 and LRP1B) which may contribute to the birth weight variability trait.
Conclusions
Birth weight variability trait has a low heritability. It is not easy to get significant signal by GWAS using small sample size. Herein, we identified some candidate chromosome regions especially chromosome 7 and suggested five genes which may provide some information for the further study.
Similar content being viewed by others

Background
In the past decades, litter size at birth has been considered as the most important index for evaluating sow productivity and great genetic improvement has been successfully obtained for this trait in most of commercial pig breeds [1]–[4]. However, the preweaning mortality is rather high; thus, a more applicable index for evaluating sow productivity is the total number of alive piglets at weaning produced by a sow per year. Preweaning mortality is influenced by a number of factors, and within-litter variation in birth weight (birth weight variability) has been proved to be an essential factor for piglet survival [5]. Several studies have reported that birth weight variability was positively related to preweaning mortality on the phenotypic scale [5]–[7]. Recently, several studies have addressed the genetic effect on birth weight variability within-litter. Damgaard et al. [8] analyzed 22,521 piglets born in 2,003 litters by 1,074 Swedish Yorkshire sows and proved the genetic correlation of birth weight variability with proportion of dead piglets and the mean growth of piglets during suckling was 0.25 and −0.31, respectively. Previous studies have also reported the heritability of birth weight variability ranged from 0.08 to 0.12 [8], [9]. Based on the maternal genetic variance and heritability of piglet birth weight variability trait [8], it is possible to improve the genetic progress of this trait by selective breeding. In addition, selection for sows’ capacity to produce homogeneous litters may reduce the piglets’ mortality, improve the mean growth during sucking and obtain more homogeneous litters at weaning, which makes the pig farm “all-in-all-out” strategy possible to get more economic benefits.
The standard deviation of birth weights within one litter can be used to describe the birth weight variability. So far, the genetic architecture of birth weight variability within-litter is still unknown. Genome wide association study (GWAS) using high-density SNP chip such as the Illumina porcine 60K SNP chip has been proved as an efficient tool to identify and map candidate genes for quantitative traits in pigs [10]-[12]. In this study, we collected 3,305 piglet’s birth weight records from 335 Suzhong sows’ first parities and assigned the sample standard deviation of birth weights within-litter as a phenotypic trait of the sow. Then we performed a GWAS in 82 sows (39 with low variability and 43 with high variability) by using the Illumina porcine 60K SNP chip to identify significant SNPs associated with the birth weight variability at a genome level, then identified the major candidate genes associated with this trait. The filtered SNP loci may be used as a preliminary foundation for further selective breeding.
Results and discussion
Genome-wide significant SNPs from the association studies
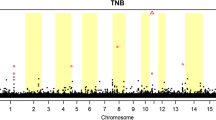
Totally, 53,693 SNPs with genotypes in 82 individuals were used for association analyses after data filtering. The number of genome-wide significant SNPs were 1916 and 266 at α level 0.05 and 0.01, respectively. For the 266 significant SNPs, 17 SNPs have not been mapped to any chromosome, and the other 249 SNPs were mainly enriched on chromosome 1, 7, 13, 14, 15 and 18 (Fig. 1). We further scanned genes of the 249 SNPs located in, and found 71 SNPs were located within 60 annotated genes, 139 SNPs in region of 0.5 Mb away from the nearest genes and no genes had been found in region of 1 Mb for the rest of 39 SNPs (Fig. 2). The detailed information for the top 50 SNPs with the lowest p values is illustrated in Table 1.

Manhattan-Plot for association of SNP loci with birth weight variability. The X axis indicates in different colors from left to right, SNP locations from chromosomes 1–18 (chromosome location for unmapped SNPs was represented by 0), using Sus scrofa genome build10.2. The Y axis represents the minus log of the P-value for each SNP

The genomic distribution of the 249 significant SNPs (p < 0.05) associated with piglet birth weight variability
Genes associated with glucose and lipid metabolism and transport
Pregnancy is a critical period for both the mother and the fetus, and the maternal factors can affect fetal growth and pregnancy outcomes. In order to sustain appropriate fetal development, the mother must provide nutrients such as glucose, amino acids and lipids to the fetus across the placenta [13]. And therefore, genes affecting the maternal nutrient ingestion, energy metabolism and maternal-fetal nutrient transport may affect the placental development as well as fetal growth and finally result in the neonatal birth weight variation. In this study, we only focused on the 50 most significant SNPs listed in Table 2 for candidate genes scanning. Among the 50 SNPs, 17 SNPs were located within annotated genes. Particularly worth mentioning is that, 19 SNPs were mapped on SSC7 spanning from 37.4 Mb to 39.6 Mb. In this region, we explored 27 annotated genes including one gene GLP1R (p = 0.0018) in regulating plasma glucose levels. From the 50 SNPs we also explored several genes involved in lipid metabolism such as AACS on SSC14 (p = 0.0018), and lipid transport related proteins including APOB on SSC3 (p = 0.0016), OSBPL10 on SSC13 (p = 0.0013), and LRP1B on SSC15 (p = 0.0014).
Glucose is the primary energy substrate essential for the fetal growth and development. However, fetus generates minimal glucose by itself, and most of the glucose is transported from maternal circulation through the glucose transporters [14]. During the transport, the maternal glucose should be taken up by placenta firstly, then entries into the fetal circulation across two layers of cells [15]–[18]. The GLP1R gene encodes glucagon-like peptide 1 (GLP1) receptor, which specifically binds with GLP1 to mediate its biological actions [19]–[21]. In mammals, stimulation of the GLP1R in the pancreatic β cells results in a rise of insulin secretion and lowers plasma glucose levels [22], [23]. The maternal plasma glucose levels dramatically affect the fetal growth, because glucose is the nutrient that crosses the placenta in the greatest quantities by facilitated diffusion along a concentration gradient [24]. A number of studies have demonstrated that abnormal maternal plasma glucose level such as hyperglycemia (hypoglycemia) is associated with fetal overgrowth (restriction) during pregnancy [25]–[30].
Except for glucose, lipids such as triglycerides (TGs) and cholesterol serve many critical roles in fetal growth [31]. The AACS gene encodes acetoacetyl-CoA synthetase, which is an acetoacetate-specific ligase [32]. Acetoacetate is the ketone body substrate for lipid biosynthesis which can be converted into acetoacetyl-CoA by AACS then subsequently used for the synthesis of cholesterol or fatty acid. Knock down of AACS in mouse significantly reduced the total blood cholesterol [33], which suggested AACS may play an important role in plasma cholesterol homeostasis.
Cholesterol is a kind of lipids that plays important roles in fetal development, as it is an essential component of cell membranes, a precursor for steroid hormones and is also essential for activation of various signaling pathways [34], [35]. Although most of the fetus’s cholesterol is synthesized by the fetus itself, more and more evidence suggested that during the first weeks of life, the fetus largely depends on maternal cholesterol as its cholesterol source [36]. The maternal cholesterol is initially taken up by the placenta, and then transported to the fetus by the cholesterol-carrying lipoproteins [37], [38]. The apolipoprotein B (apoB)-containing lipoproteins is an efficient system for delivery of lipids because these lipoproteins contain large amounts of cholesterol, TGs and essential lipids [39]. ApoB (encoded by the APOB gene) is the principal protein component of plasma very low density lipoproteins (VLDL) and low density lipoproteins (LDL), and several genome-wide association studies in pig populations have revealed the APOB gene was associated with the serum total cholesterol (TC) and LDL cholesterol (LDL-C) levels [40]–[42]. ApoB is also an essential component for the assembly and secretion of competent apoB-containing lipoproteins [43], [44]. Human and rat placenta can synthesize and secrete apoB [45]–[47], and a sharp increase in rat placental apoB mRNA during the last 48 h of pregnancy has been reported by Demmer et al. [48]. Mouse yolk sac also secretes apoB, and embryos lacking apoB can not export lipoproteins from yolk sac endoderm cells and die with severe neuro-developmental abnormalities during mid-gestation [49], [50]. All these studies suggest a specific role for the APOB gene in maternal-fetal lipid transport.
In mammals, oxysterols are oxygenated forms of cholesterol. Oxysterol-binding protein (OSBP) and its homologs OSBP-related (ORP) or OSBP-like (OSBPL) proteins constitute a conserved family of lipid binding/transfer proteins (LTP), which can accommodate cholesterol, oxysterols and other steroids. The OSBPL10 (also known as ORP10) is a member of the LTP family and has the capacity to bind cholesterol and several acidic phospholipids [51]. Association studies revealed polymorphisms in the OSBPL10 gene displayed linkage and association with the extreme upper end serum triacylglycerol (TAG) and LDL-C levels in dyslipidaemic subjects [52], [53]. Functional studies have also demonstrated the OSBPL10 gene negatively regulates hepatocellular VLDL biosynthesis and suppresses apoB-containing lipoproteins secretion [51].
Finally, LRP1B gene encodes LDL receptor-related protein 1B and mediates cellular cholesterol uptake [54]. Dietrich et al. [55] reported that knockout of Lrp1b in mice results in early embryonic lethality. Association analysis identified LRP1B as a determinant of rat cholesterol concentrations in LDL, and a significant association with child body mass index (BMI) in human [56], [57]. Furthermore, recent studies suggested the LRP1B gene was also involved in glucose homeostasis. Polymorphism of this gene was associated with insulin resistance and in normoglycemic women the maternal glucose levels were associated with DNA methylation changes at LRP1B gene loci in the placenta and cord blood [57], [58]. We summarized the above five candidate genes (GLP1R, AACS, APOB, OSBPL10 and LRP1B) involved in glucose and lipid homeostasis as well as maternal-fetal lipid transport pathways in Fig. 3.

Summary of the five candidate genes involved in glucose and lipid homeostasis as well as maternal-fetal lipid transport pathways
Expression of candidate genes in porcine placenta and endometrium tissues
Genes expressed in placenta or endometrium may play functional roles for fetal development, and the public RNA-seq data give us a good opportunity to check gene expression in specific tissues. We checked the above five candidate genes (GLP1R, AACS, APOB, OSBPL10 and LRP1B) in porcine placenta and endometrium tissues by using the public RNA-seq data or microarray data, and the results were summarized in Table 2. We found the AACS and OSBPL10 gene had relatively high expression both in porcine placenta and endometrium at different gestational stages (day 25, 45, 65, 85, 105 and 113) [59]–[62]. However, the other two genes GLP1R and LRP1B had no expression in these two tissues from the above data. The APOB gene had very low expression in porcine endometrium during early (gestational day 15) and mid-gestation (gestational days 26 and 50) [60], [62]. Based on the Yorkshire endometrium RNA-seq deep sequencing results (Size = 10Gb, unpublished data from our group), the AACS gene was highly expressed in the endometrium during early embryo implantation (RPKM > 250) and the expression of APOB gene was sharply increased at gestational day 15 (RPKM = 16.72) compared with day 12 (RPKM = 1.60).
Interestingly, we found half of the 27 genes on SSC7 listed in Table 1 expressed in porcine placenta (gestational day 113) and endometrium (gestational day 25) including four extremely high expression genes (ZFAND3, FOXQ1, GMDS and MTCH1) [50], [51]. ZFAND3 gene encodes Zing finger AN1-type domain 3 protein which is originally isolated from the mouse testis [63] and expression assay suggested this gene is involved in spermatogenesis [64]. Recently, association studies identified ZFAND3 as a susceptible gene to type 2 diabetes in several human populations [65], [66], which suggested this gene may be involved in plasma glucose homeostasis. The high expression of ZFAND3 gene in porcine placenta and endometrium possibly imply its functional role for embryo (or fetus) development.
Conclusions
Birth weight variability is an economic trait with low heritability. In this study, we performed a GWAS in 82 sows with extreme phenotypic records and identified 266 significant associated SNPs (p < 0.01). For the top 50 significant SNPs, we further scanned the genes within 1 Mb region and finally suggested candidate genes involved in plasma glucose homeostasis (GLP1R) and lipid metabolism as well as maternal-fetal lipid transport (AACS, APOB, OSBPL10 and LRP1B) which may contribute to the current trait we focused on. But, further association analysis in bigger sample size and function studies need be carried out to confirm our present conclusion.
Methods
Pig population and phenotype measurement
The pigs we studied were coming from Suzhong pig seed farm of Jiangsu Academy of Agricultural Institute. We collected the reproductive information from 335 sows, including the total number born (TNB), number born alive (NBA) and the birth weight (BW) records from total of 3,305 first parity’s piglets. Because the farm only had the birth weight of born alive offspring’s records, in this case the sample standard deviation (SD) of born alive birth weights within one litter was described as a phenotype to assess the piglet birth weight variability for each sow.
Genotyping and quality control
A total of 82 sows with extreme SD were genotyped for further association studying, and they were divided into low (group 1, n = 39) and high (group 2, n = 43) SD groups, with the mean SD 0.08 (from 0.04 to 0.12) and 0.21 (from 0.12 to 0.48), respectively. The TNB, NBA and SD information of these sows is summarized in Additional file 1. It is worth mentioning that the TNB is more than four for all the 82 studied sows in order to reduce the effect of litter size. 5 ml blood samples were collected from each sow for genomic DNA isolation using a standard phenol/chloroform method. All DNA samples were qualified with a ratio of A260/280 between 1.80 and 2.0 and standardized into a final concentration of 200 ng/μL. Then, 2 μg DNA sample from each of these sows were genotyped using the Porcine SNP60 Beadchips (Illumina, USA) following the manufacturer’s protocol. Quality control was carried out using PLINK (version 1.07) [67] and executed SNPs with call rate < 80 %, Gentrain score < 40 %, minor allele frequency (MAF) < 0.01, and severely departed from hardy weinberg equilibrium (HWE) (P-value < 0.0001).
Genome-wide association analyses
In this study, compressed mixed linear model (CMLM) from the Genome Association and Prediction Integrated Tool (GAPIT) program package [68] was used for whole genome association analyses. The CMLM statistical model we used was described as following:
y = Xα + Pβ + Kμ + e. Where y is the vector of phenotype, X is a matrix of SNP genotypes, p is a matrix of PC (principle components) for population structure, K is a kinship matrix. Xα and Pβ are regarded as fixed effects, where Pβ is used as a covariate to address the spurious associations that arise from population structure, and Kμ and e are regarded as random effects.
Gene search and functional annotation
Gene searches were carried out in 0.5 Mb sequence upstream and downstream of the significant associated SNPs with the top 50 lowest p value using the Sus scrofa 10.3 genome build. If no genes were identified in the gene-poor regions, then the genes upstream and downstream of the region were considered to possibly represent the locus. Functional annotation clustering was performed for all the identified genes using DAVID software (http://david.abcc.ncifcrf.gov), and the gene enrichment clusters related to reproductive functions and reproductive tissues were taken into consideration.
Declarations
Publication of this article has been funded by the National Natural Science Foundation of China (31272426 and 31372352) and the National Basic Research Program of China (2014CB138500).
This article has been published as part of BMC Genetics Volume 17 Supplement 1, 2016: Selected articles from the 7th International Young Scientists School “Systems Biology and Bioinformatics” (SBB’2015): Genetics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcgenetics/supplements/17/S1.
Additional file
Abbreviations
- GWAS:
-
genome wide association study
- SD:
-
standard deviation
- SNPs:
-
single nucleotide polymorphisms
- MAF:
-
minor allele frequency
- HWE:
-
hardy weinberg equilibrium
- Mb:
-
mega base
- SSC:
-
sus scrofa chromosome
- CMLM:
-
compressed mixed linear model
- GAPIT:
-
Genome Association and Prediction Integrated Tool
- DAVID:
-
The Database for Annotation, Visualization and Integrated Discovery
- TGs:
-
triglycerides
- VLDL:
-
very low density lipoproteins
- LDL:
-
low density lipoproteins
- TC:
-
total cholesterol
- LDL-C:
-
low density lipoprotein cholesterol
- TAG:
-
triacylglycerol
- GLP1R:
-
glucagon-like peptide 1 receptor
- AACS:
-
acetoacetyl-CoA synthetase
- APOB:
-
apolipoprotein B
- OSBPL10:
-
Oxysterol-binding protein like 10
- LRP1B:
-
LDL receptor-related protein 1B
- BMI:
-
body mass index
- RPKM:
-
Reads Per Kilobase of exon model per Million mapped reads
- TNB:
-
total number born
- NBA:
-
number born alive
- BW:
-
birth weight
References
Southwood OI, Kennedy BW: Genetic and environmental trends for litter size in swine. J Anim Sci. 1991, 69 (8): 3177-82.
Blasco A, Sorensen D, Bidanel JP: Bayesian inference of genetic parameters and selection response for litter size components in pigs. Genetics. 1998, 149 (1): 301-6.
Noguera JL, Varona L, Babot D, Estany J: Multivariate analysis of litter size for multiple parities with production traits in pigs: I. Bayesian variance component estimation. J Anim Sci. 2002, 80 (10): 2540-7.
Noguera JL, Varona L, Babot D, Estany J: Multivariate analysis of litter size for multiple parities with production traits in pigs: II. Response to selection for litter size and correlated response to production traits. J Anim Sci. 2002, 80 (10): 2548-55.
Roehe R, Kalm E: Estimation of genetic and environmen-tal risk factors associated with pre-weaning mortality in piglets using generalized linear mixed models. J Anim Sci. 2000, 70: 227-40.
English PR, Smith WJ: Some causes of death in neonatal piglets. Vet Annu. 1995, 15: 95-104.
Fahmy MH, Holtmann WB, MacIntyre TM, Moxley JE: Evaluation of piglet mortality in 28 two-breed crosses among eight breeds of pig. Anim Prod. 1978, 26: 277-85. 10.1017/S0003356100040873.
Damgaard LH, Rydhmer L, Løvendahl P, Grandinson K: Genetic parameters for within-litter variation in piglet birth weight and change in within-litter variation during suckling. J Anim Sci. 2003, 81 (3): 604-10.
Wittenburg D, Guiard V, Teuscher F, Reinsch N: Comparison of statistical models to analyse the genetic effect on within-litter variance in pigs. Animal. 2008, 2 (11): 1559-68. 10.1017/S1751731108002851.
Chung HY, Lee KT, Jang GW, Choi JG, Hong JG, Kim TH: A genome-wide analysis of the ultimate pH in swine. Genet Mol Res. 2015, 14 (4): 15668-82. 10.4238/2015.December.1.19.
Bergfelder-Drüing S, Grosse-Brinkhaus C, Lind B, Erbe M, Schellander K, Simianer H, Tholen E: A genome-wide association study in large white and landrace pig populations for number piglets born alive. PLoS One. 2015, 10 (3): e0117468-10.1371/journal.pone.0117468.
Fan B, Onteru SK, Du ZQ, Garrick DJ, Stalder KJ, Rothschild MF: Genome-wide association study identifies Loci for body composition and structural soundness traits in pigs. PLoS One. 2011, 6: e14726-10.1371/journal.pone.0014726.
Brett KE, Ferraro ZM, Yockell-Lelievre J, Gruslin A, Adamo KB: Maternal-fetal nutrient transport in pregnancy pathologies: the role of the placenta. Int J Mol Sci. 2014, 15 (9): 16153-85. 10.3390/ijms150916153.
Kalhan S, Parimi P: Gluconeogenesis in the fetus and neonate. Semin Perinatol. 2000, 24 (2): 94-106. 10.1053/sp.2000.6360.
Jansson T, Illsley NP: Osmotic water permeabilities of human placental microvillous and basal membranes. J Membr Biol. 1993, 132: 147-55. 10.1007/BF00239004.
Johnson LW, Smith CH: Glucose transport across the basal plasma membrane of human placental syncytiotrophoblast. Biochim Biophys Acta. 1985, 815: 44-50. 10.1016/0005-2736(85)90472-9.
Firth JA, Leach L: Not trophoblast alone: a review of the contribution of the fetal microvasculature to transplacental exchange. Placenta. 1996, 17: 89-96. 10.1016/S0143-4004(96)80001-4.
Eaton BM, Leach L, Firth JA: Permeability of the fetal villous microvasculature in the isolated perfused term human placenta. J Physiol. 1993, 463: 141-55. 10.1113/jphysiol.1993.sp019588.
Cardoso JC, Pinto VC, Vieira FA, Clark MS, Power DM: Evolution of secretin family GPCR members in the metazoa. BMC Evol Biol. 2006, 6: 108-10.1186/1471-2148-6-108.
Holst JJ: The physiology of glucagon-like peptide 1. Physiol Rev. 2007, 87: 1409-39. 10.1152/physrev.00034.2006.
Gupta NA, Mells J, Dunham RM, Grakoui A, Handy J, Saxena NK, et al: Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signal-ing pathway. Hepatology. 2010, 51: 1584-92. 10.1002/hep.23569.
Doyle ME, Egan JM: Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther. 2007, 113: 546-93. 10.1016/j.pharmthera.2006.11.007.
Drucker DJ: The biology of incretin hormones. Cell Metab. 2006, 3: 153-65. 10.1016/j.cmet.2006.01.004.
Baumann MU, Deborde S, Illsley NP: Placental glucose transfer and fetal growth. Endocrine. 2002, 19: 13-22. 10.1385/ENDO:19:1:13.
Metzger BE, Lowe LP, Dyer AR, Trimble ER, Chaovarindr U, HAPO Study Cooperative Research Group, et al: Hyperglycemia and adverse pregnancy outcomes. N Engl J Med. 2008, 358 (19): 1991-2002. 10.1056/NEJMoa0707943.
Illsley NP, Sellers MC, Wright RL: Glycaemic regulation of glucose transporter expression and activity in the human placenta. Placenta. 1998, 19: 517-24. 10.1016/S0143-4004(98)91045-1.
Jarmuzek P, Wielgos M, Bomba-Opon D: Placental pathologic changes in gestational diabetes mellitus. Neuro Endocrinol Lett. 2015, 36 (2): 101-5.
Ouhilal S, Vuguin P, Cui L, Du XQ, Gelling RW, Reznik SE, et al: Hypoglycemia, hyperglucagonemia, and fetoplacental defects in glucagon receptor knockout mice: a role for glucagon action in pregnancy maintenance. Am J Physiol Endocrinol Metab. 2012, 302 (5): E522-31. 10.1152/ajpendo.00420.2011.
Kc K, Shakya S, Zhang H: Gestational diabetes mellitus and macrosomia: a literature review. Ann Nutr Metab. 2015, 66 (Suppl 2): 14-20. 10.1159/000371628.
Ericsson A, Säljö K, Sjöstrand E, Jansson N, Prasad PD, Powell TL, et al: Brief hyperglycaemia in the early pregnant rat increases fetal weight at term by stimulating placental growth and affecting placental nutrient transport. J Physiol. 2007, 581 (Pt 3): 1323-32. 10.1113/jphysiol.2007.131185.
Polin RA, Fox WW, Abman SH: Fetal and Neonatal Physiology. 1998, 2
Yamasaki M, Hasegawa S, Suzuki H, Hidai K, Saitoh Y, Fukui T: Acetoacetyl-CoA synthetase gene is abundant in rat adipose, and related with fatty acid synthesis in mature adipocytes. Biochem Biophys Res Commun. 2005, 335 (1): 215-9. 10.1016/j.bbrc.2005.07.053.
Hasegawa S, Noda K, Maeda A, Matsuoka M, Yamasaki M, Fukui T: Acetoacetyl-CoA synthetase, a ketone body-utilizing enzyme, is controlled by SREBP-2 and affects serum cholesterol levels. Mol Genet Metab. 2012, 107 (3): 553-60. 10.1016/j.ymgme.2012.08.017.
Baardman ME, Kerstjens-Frederikse WS, Berger RM, Bakker MK, Hofstra RM, Plösch T: The role of maternal-fetal cholesterol transport in early fetal life: current insights. Biol Reprod. 2013, 88 (1): 24-10.1095/biolreprod.112.102442.
Christenson LK, Devoto L: Cholesterol transport and steroidogenesis by the corpus luteum. Reprod Biol Endocrinol. 2003, 1: 90-10.1186/1477-7827-1-90.
Woollett LA: Review: transport of maternal cholesterol to the fetal circulation. Placenta. 2011, 32 (Suppl 2): S218-21. 10.1016/j.placenta.2011.01.011.
Fujimoto VY, Kane JP, Ishida BY, Bloom MS, Browne RW: High-density lipoprotein metabolism and the human embryo. Hum Reprod Update. 2010, 16 (1): 20-38. 10.1093/humupd/dmp029.
Roy R, Bélanger A: Elevated levels of endogenous pregnenolone fatty acid esters in follicular fluid high density lipoproteins support progesterone synthesis in porcine granulosa cells. Endocrinology. 1992, 131 (3): 1390-6.
Herrera E, Amusquivar E: Lipid metabolism in the fetus and the newborn. Diabetes Metab Res Rev. 2000, 16 (3): 202-10. 10.1002/1520-7560(200005/06)16:3<202::AID-DMRR116>3.0.CO;2-#.
Chen C, Yang B, Zeng Z, Yang H, Liu C, Ren J, et al: Genetic dissection of blood lipid traits by integrating genome-wide association study and gene expression profiling in a porcine model. BMC Genomics. 2013, 14: 848-10.1186/1471-2164-14-848.
Yang H, Huang X, Zeng Z, Zhang W, Liu C, Fang S, et al: Genome-wide association analysis for blood lipid traits measured in three pig populations reveals a substantial level of genetic heterogeneity. PLoS One. 2015, 10 (6): e0131667-10.1371/journal.pone.0131667.
Pena RN, Cánovas A, Varona L, Díaz I, Gallardo D, Ramírez O, et al: Nucleotide sequence and association analysis of pig apolipoprotein-B and LDL-receptor genes. Anim Biotechnol. 2009, 20 (3): 110-23. 10.1080/10495390902892518.
Kamper M, Manns CC, Plieschnig JA, Schneider WJ, Ivessa NE, Hermann M: Estrogen enhances secretion of apolipoprotein B-100 containing lipoproteins by BeWo cells. Biochimie. 2015, 112: 121-8. 10.1016/j.biochi.2015.03.002.
Oikawa S, Katohm N: Reduced concentrations of apolipoproteins B-100 and A-I in serum from cows with retained placenta. Can J Vet Res. 1997, 61 (4): 312-4.
Madsen EM, Lindegaard ML, Andersen CB, Damm P, Nielsen LB: Human placenta secretes apolipoprotein B-100-containing lipoproteins. J Biol Chem. 2004, 279 (53): 55271-6. 10.1074/jbc.M411404200.
Farese RV, Cases S, Ruland SL, Kayden HJ, Wong JS, Young SG, et al: A novel function for apolipoprotein B: lipoprotein synthesis in the yolk sac is critical for maternal-fetal lipid transport in mice. J Lipid Res. 1996, 37 (2): 347-60.
Terasawa Y, Cases SJ, Wong JS, Jamil H, Jothi S, Traber MG, et al: Apolipoprotein B-related gene expression and ultrastructural characteristics of lipoprotein secretion in mouse yolk sac during embryonic development. J Lipid Res. 1999, 40 (11): 1967-77.
Demmer LA, Levin MS, Elovson J, Reuben MA, Lusis AJ, Gordon JI: Tissue-specific expression and developmental regulation of the rat apolipoprotein B gene. Proc Natl Acad Sci U S A. 1986, 83 (21): 8102-6. 10.1073/pnas.83.21.8102.
Raabe M, Flynn LM, Zlot CH, Wong JS, Véniant MM, Hamilton RL, et al: Knockout of the abetalipoproteinemia gene in mice: reduced lipoprotein secretion in heterozygotes and embryonic lethality in homozygotes. Proc Natl Acad Sci U S A. 1998, 95 (15): 8686-91. 10.1073/pnas.95.15.8686.
Farese RV, Ruland SL, Flynn LM, Stokowski RP, Young SG: Knockout of the mouse apolipoprotein B gene results in embryonic lethality in homozygotes and protection against diet-induced hypercholesterolemia in heterozygotes. Proc Natl Acad Sci U S A. 1995, 92 (5): 1774-8. 10.1073/pnas.92.5.1774.
Nissilä E, Ohsaki Y, Weber-Boyvat M, Perttilä J, Ikonen E, Olkkonen VM: ORP10, a cholesterol binding protein associated with microtubules, regulates apolipoprotein B-100 secretion. Biochim Biophys Acta. 2012, 1821 (12): 1472-84. 10.1016/j.bbalip.2012.08.004.
Perttilä J, Merikanto K, Naukkarinen J, Surakka I, Martin NW, Tanhuanpää K, et al: OSBPL10, a novel candidate gene for high triglyceride trait in dyslipidemic Finnish subjects, regulates cellular lipid metabolism. J Mol Med (Berl). 2009, 87 (8): 825-35. 10.1007/s00109-009-0490-z.
Koriyama H, Nakagami H, Katsuya T, Akasaka H, Saitoh S, Shimamoto K, et al: Variation in OSBPL10 is associated with dyslipidemia. Hypertens Res. 2010, 33 (5): 511-4. 10.1038/hr.2010.28.
Haas J, Beer AG, Widschwendter P, Oberdanner J, Salzmann K, Sarg B, et al: LRP1b shows restricted expression in human tissues and binds to several extracellular ligands, including fibrinogen and apoE-carrying lipoproteins. Atherosclerosis. 2011, 216 (2): 342-7. 10.1016/j.atherosclerosis.2011.02.030.
Dietrich MF, van der Weyden L, Prosser HM, Bradley A, Herz J, Adams DJ: Ectodomains of the LDL receptor-related proteins LRP1b and LRP4 have anchorage independent functions in vivo. PLoS One. 2010, 5 (4): e9960-10.1371/journal.pone.0009960.
Hodúlová M, Šedová L, Křenová D, Liška F, Krupková M, Kazdová L, et al: Genomic determinants of triglyceride and cholesterol distribution into lipoprotein fractions in the rat. PLoS One. 2014, 9 (10): e109983-10.1371/journal.pone.0109983.
Burgdorf KS, Gjesing AP, Grarup N, Justesen JM, Sandholt CH, Witte DR, et al: Association studies of novel obesity-related gene variants with quantitative metabolic phenotypes in a population-based sample of 6,039 Danish individuals. Diabetologia. 2012, 55 (1): 105-13. 10.1007/s00125-011-2320-4.
Houde AA, Ruchat SM, Allard C, Baillargeon JP, St-Pierre J, Perron P, et al: LRP1B, BRD2 and CACNA1D: new candidate genes in fetal metabolic programming of newborns exposed to maternal hyperglycemia. Epigenomics. 2015, 7 (7): 1111-22. 10.2217/epi.15.72.
Bischoff SR, Tsai SQ, Hardison NE, Motsinger-Reif AA, Freking BA, Nonneman DJ, et al: Differences in X-chromosome transcriptional activity and cholesterol metabolism between placentae from swine breeds from Asian and Western origins. PLoS One. 2013, 8 (1): e55345-10.1371/journal.pone.0055345.
Huang J, Liu R, Su L, Xiao Q, Yu M: Transcriptome analysis revealed the embryo-induced gene expression patterns in the endometrium from Meishan and Yorkshire pigs. Int J Mol Sci. 2015, 16 (9): 22692-710. 10.3390/ijms160922692.
SRX178700: Pig placenta day 113 of pregnancy RNA-seq. 2015. http://www.ncbi.nlm.nih.gov/sra/?term=SRR543893. Accessed October 9, 2015.
SRX1301560: Next Generation Sequencing Facilitates Quantitative Analysis of Endometrium Transcriptomes During Porcine Embryo Implantation. 2015. http://www.ncbi.nlm.nih.gov/sra/SRX1301560. Accessed October 9, 2015.
López-Fernández LA, del Mazo J: Characterization of genes expressed early in mouse spermatogenesis, isolated from a subtractive cDNA library. Mamm Genome. 1996, 7 (9): 698-700. 10.1007/s003359900210.
de Luis O, López-Fernández LA, del Mazo J: Tex27, a gene containing a zinc-finger domain, is up-regulated during the haploid stages of spermatogenesis. Exp Cell Res. 1999, 249 (2): 320-6. 10.1006/excr.1999.4482.
Keaton JM, Cooke Bailey JN, Palmer ND, Freedman BI, Langefeld CD, Ng MC, et al: A comparison of type 2 diabetes risk allele load between African Americans and European Americans. Hum Genet. 2014, 133 (12): 1487-95. 10.1007/s00439-014-1486-5.
Sakai K, Imamura M, Tanaka Y, Iwata M, Hirose H, Kaku K, et al: Replication study for the association of 9 East Asian GWAS-derived loci with susceptibility to type 2 diabetes in a Japanese population. PLoS One. 2013, 8 (9): e76317-10.1371/journal.pone.0076317.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007, 81 (3): 559-75. 10.1086/519795.
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, et al: GAPIT: genome association and prediction integrated tool. Bioinformatics. 2012, 28 (18): 2397-9. 10.1093/bioinformatics/bts444.
Acknowledgements
We thank Yunbin Ren for blood sample collection and DNA isolation. This work was financially supported by the National Natural Science Foundation of China (31272426 and 31372352) and the National Basic Research Program of China (2014CB138500).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no conflict of interest.
Authors’ contributions
XW, MY and XL designed the experiments. XW and DD performed the experiments. XL and XL analyzed the data. XL and XW wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Wang, X., Liu, X., Deng, D. et al. Genetic determinants of pig birth weight variability. BMC Genet 17 (Suppl 1), S15 (2016). https://doi.org/10.1186/s12863-015-0309-6
Published:
DOI: https://doi.org/10.1186/s12863-015-0309-6