
Abstract
Background
Scallops (Bivalvia: Pectinidae) present extraordinary variance in both mitochondrial genome size, structure and content, even when compared to the extreme diversity documented within Mollusca and Bivalvia. In pectinids, mitogenome rearrangements involve protein coding and rRNA genes along with tRNAs, and different genome organization patterns can be observed even at the level of Tribes. Existing pectinid phylogenies fail to resolve some relationships in the family, Chlamydinae being an especially problematic group.
Results
In our study, we sequenced, annotated and characterized the mitochondrial genome of a member of Chlamydinae, Mimachlamys varia—a species of commercial interest and an effective bioindicator—revealing yet another novel gene arrangement in the Pectinidae. The phylogeny based on all mitochondrial protein coding and rRNA genes suggests the paraphyly of the Mimachlamys genus, further commending the taxonomic revision of the classification within the Chlamydinae subfamily. At the scale of the Pectinidae, we found that 15 sequence blocks are involved in mitogenome rearrangements, which behave as separate units.
Conclusions
Our study reveals incongruities between phylogenies based on mitochondrial protein-coding versus rRNA genes within the Pectinidae, suggesting that locus sampling affects phylogenetic inference at the scale of the family. We also conclude that the available taxon sampling does not allow for understanding of the mechanisms responsible for the high variability of mitogenome architecture observed in the Pectinidae, and that unraveling these processes will require denser taxon sampling.
Similar content being viewed by others


Background
Historically, animal mitochondrial genomes were thought to have a conserved size of approximately 16 kbp, as well as a conserved structure, containing 13 protein coding genes (PCGs), two rRNA genes, and 22 tRNA genes and a single non-coding, ‘control region’ that regulates replication and transcription [1, 2]. In contrast, molluscan mitochondrial genomes provide examples of exceptional variation in size, structure and even function. Mitogenome size in Mollusca ranges from ~ 14 kbp up to 56.2 kbp [3,4,5,6,7], gene rearrangements, duplications, losses and inversions are frequent e.g. [8], and there is evidence of the integration of novel genes e.g. [9]. Additionally, a unique, complex mitochondrial inheritance system, called ‘doubly uniparental inheritance’ evolved in gonochoric bivalve mollusks [10,11,12].
Regarding mitogenome structure, scallops (Bivalvia: Pectinidae) constitute one of the most peculiar groups within Mollusca. They include one of the largest mitogenomes known (32–42 kbp; [7], with other species having mitogenomes of different lengths ranging from 16 to 21 kbp [13, 14]. They also present the most variation in mitochondrial gene order, even at relatively shallow phylogenetic depths, i.e. within subfamilies [14]. In Pectinidae, as well as among metazoans in general, gene rearrangements are most common for tRNAs [14,15,16,17]. Several pectinids contain extra tRNAs beyond the essential 22 needed for mitochondrial translation, either duplicated genes, or genes encoding for the same amino acid, but using a different anticodon [18,19,20,21]. However, even more species contain fewer than 22 mitochondrial tRNAs [13, 14, 19, 22, 23], possibly coming from annotation errors, which might be the result of modified tRNA secondary structure, common in other mollusks [24,25,26]. Similarly, atp8 genes were reported earlier as missing from most bivalves, including pectinids [13, 14, 18, 20], although reannotation efforts revealed its presence in several species [15, 21, 27]. The difficulty of annotating the atp8 gene is thought to stem from the poorly conserved sequence of this gene in bivalves [28,29,30]. Unlike what was observed in other animal groups, mitochondrial genome rearrangements in Pectinidae often involve PCGs [13,14,15]. This, however, does not seem to be correlated to sequence similarities among PCGs [14]. Furthermore, like most bivalves, most pectinid mitochondrial genomes contain several inflated non-coding regions, some of which are hypothesized to function as control regions [13, 14, 18, 20], although it remains controversial whether several control regions within a mitogenome could provide normal function [20].
Most existing pectinid phylogenies are based on one, or a handful of gene sequences, of either just mitochondrial, or of both mitochondrial and nuclear origin [31,32,33,34,35,36,37,38,39,40,41]. These phylogenies are often incongruent with morphological classifications of the Pectinidae [40]. Also, while they conclude that the Pectinidae is monophyletic, many lower taxonomic levels are reported as being paraphyletic [31, 40]. Phylogenies based on full mitogenomes have shown their utility in disentangling evolutionary relationships that were controversial when investigated using only a few genes [16, 42,43,44]. There are a few examples of pectinid phylogenies based on complete mitogenomes [14, 15, 18, 22, 27], however, most of these studies rely solely on genetic distance-based methods (except: [15, 22]), without considering the differences in substitution rates among different mitochondrial genes. Despite the diversity of approaches, some phylogenetic relationships remain to be resolved, for example, those within the Chlamydinae subfamily.
Numerous scallop species are harvested for human consumption and are used as environmental indicators. One example is the variegated scallop (Mimachlamys varia L., 1758), a species that inhabits the subtidal zone of the European Atlantic and the Mediterranean Sea [45]. It was demonstrated that Mim. varia is a potent bioindicator, as it presents significant physiological responses to chemical contaminants, including changes in biomarkers connected to oxidative stress, immune system function and mitochondrial respiration [46,47,48]. Most existing genetic research involving Mim. varia focused on the use of a few genes for phylogenetic or population genetic inference [31, 40, 49]. Viricel et al. [50] published the transcriptome of Mim. varia, providing a valuable resource for further ecophysiological and ecotoxicological research, including analyses of differential gene expression in response to marine pollution and comparative genomic studies within the Pectinidae.
In this study, our goals were to: (I) assemble and characterize the mitochondrial genome of the variegated scallop (Mimachlamys varia); (II) reconstruct the most up to date pectinid phylogeny using all available mitochondrial genomes, including Mim. varia; (III) perform comparative analyses of pectinid mitochondrial gene orders, to investigate the evolution of mitogenome arrangements within this family.
Results
Sequencing of the mitogenome of reference for the variegated scallop
The NOVOPlasty assembly, based on a single cox1 seed, yielded four contigs, 5289, 530, 3458 and 13,528 bp long, recruiting about 5.9 M reads (9.67% of the 61.1 M RNAseq reads submitted to the assembler), with an average depth of coverage of 84,143. NOVOPlasty never retrieved a full, circular mitogenome based on these data; the final assembly is based on the manual assembly of the four, overlapping contigs in Geneious, resulting in a complete, 20,400 bp long circular assembly. In total, 6.9 M reads were remapped onto the assembled genome. After remapping, the breath of coverage is 99.98%, only 5 bases are not covered around the site where the circular genome was split into a linear sequence. Coverage depth values ranged from 11–4,160,626 (Fig. 1). The mean and median coverage depths were 100,437 and 14,687, respectively.

Map of the Mimachlamys varia mitogenome. Protein coding (CDS), rRNA and tRNA genes and the Major Non-coding Region (MNR) are marked with differently colored arrows pointing in the direction of transcription. Innermost scale represents size in kbp. GC content and GC skew are represented by black and purple-orange histograms, respectively. The outermost circle in light gray shows the natural logarithm of the depth of coverage for each base. Photo credit: Thierry Guyot
Genome composition
The full mitogenome contains 13 Protein Coding Genes (PCGs; including an atp8, reported as missing for many bivalves; [13, 14, 18, 20]), 23 tRNAs, and 2 rRNAs (Fig. 1, Table 1). All genes are coded on the “+” strand. Two 2 bp overlaps were detected by MITOS2, between nad1–trnR and cob–trnD. Global A+T content is 56.3%. No heteroplasmy was detected with the two MAF settings used in NOVOPlasty.
Protein-coding genes
All 13 PCGs commonly occurring in metazoan mitochondrial genomes were identified in Mim. varia. The combined length of PCGs was 10,896 bp (45.4% of the complete genome). The total amino acid (AA) length was 3619, excluding stop codons. The most frequently used AAs were Leucine (12.86%), Valine (10.39%), Phenylalanine (9.55%), Glycine (9.45%) and Serine (8.31%). Six genes (cox1, atp8, cob, atp6, cox2, nad4) had the most common start codon ATG, three had ATT (nad6, nad3, nad5), three TTG (nad1, nad4l, cox3) and nad2 had the ATA start codon (Table 1). Eight genes had the TAG and five had the TAA stop codon.
rRNA and tRNA genes
The rrnS and rrnL genes were 964 bp and 1422 bp long, respectively. Out of the total 23 tRNAs identified, most were identified by MITOS2, except trnQ2, which was identified by ARWEN. The length of tRNAs ranges from 63 to 73 nt. Three tRNA genes are present in two copies, trnL with different anticodons (trnL1: UAG, trnL2: UAA), trnM and trnQ with the same anticodon in both copies, CAU and UUG, respectively. All tRNAs were predicted to have the typical cloverleaf structure, except trnQ2 (Additional file 1). While present in two copies in most animal mitogenomes, trnS was not identified in the mitogenome of Mim. varia.
Gene order
The Mim. varia mitogenome represents a novel gene order for the Pectinidae. The conserved pectinid gene clusters “nad1-trnR-rrnL-cox1” and “nad4-trnH-trnW” are present, however, the “nad6-trnL2-cob” cluster is split by the insertion of trnF, the MNR and atp8, and the “cob-cox2” cluster is also missing. Notable is the relocation of the “rrnS-nad5” pair, and the location of the MNR between the nad6 and cob genes, a pattern observed in only one other species, Pl. magellanicus.
Structure of the MNR
The major non-coding region (MNR) spans 4206 nt and has an elevated A+T content of 60.1%. There were four repeated regions, with repeat sizes between 10 and 683 bp (Table 2). The largest repeat region contained 4.7 copies of a 683 bp long sequence, with a total length of 3205 bp, making up 76.2% of the MNR. We PCR-amplified the MNR but failed to obtain its complete sequence using Sanger sequencing, possibly because of numerous stem-and loop structures and high %AT in this region (Additional file 2).
Annotation of pectinid genomes
Out of the 29 Pectinidae and 1 outgroup (O. lurida) mitogenomes retrieved from Genbank, only two were incomplete. All protein coding and rRNA genes identified in previous descriptions of these genomes were found and reannotated in this study. The atp8 gene was annotated in all 25 genomes, in which it was not previously annotated, including two paralogous copies in Mizuhopecten yessoensis and Pl. magellanicus, and unusually long versions of the gene in two Ar. irradians GenBank accessions—with alternative start codons—showing only 78.5% sequence similarity with conspecific atp8 genes (Table 3, Additional file 3). Between 20 and 40 tRNA genes were annotated per mitogenome in the 29 pectinid species. Most mitogenomes contained 23 tRNA genes, compared to the 22 commonly found in animals. Gene content was mainly identical within species, but there are slight variations, some of which bears a phylogenetic pattern. For example, the trnS1 genes in Amusium pleuronectes, Pecten albicans and Pecten maximus all have GCU as anticodon, and all species in the Chlamydinae subfamily contain two copies of the trnM gene. Some variations are present in only one species, Pe. albicans missing trnL1 and trnM, Mimachlamys nobilis containing an extra trnT gene, Miz. yessoensis lacking five tRNA genes, while containing two supernumerary ones, and Pl. magellanicus containing 12 trnM, 4 trnF and 3 trnS2 genes (Fig. 2), although most of these were identified as pseudogenes by Smith et al. [20].

Heatmap representing mitochondrial tRNA gene sets observed in the 13 pectinid species included in this study. tRNAs are represented with their one-letter amino-acid code, with the anticodon in parentheses. The deeper blue color corresponds to higher copy number of a tRNA gene
Mito-phylogenomics
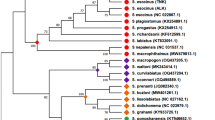
All four subfamilies and 7 out of 9 tribes of the Pectinidae family are represented in this study (Table 4). Two different topologies can be observed among the ML trees reconstructed from the eight datasets (Fig. 3). The two rRNA datasets (G and H) show a distinct topology compared to the other 6 datasets, but the topology based on the rRNA datasets is not well-supported. All PCG ML trees had very similar topology. Mim. varia is placed within the same clade with the other two congeneric species only in the tree based on the E dataset (PCG amino acid sequences). In every other tree Mim. varia is placed in a clade with species in the Crassadomini and Chlamydini tribes. The monophyly of neither of these clades is supported. Similarly, although the topology suggests that Pl. magellanicus is sister to all other pectinids, its placement is never supported as such, rather the topology of the Pectinidae should be considered as a trichotomy represented by the subfamilies Palliolinae, Chlamydinae and a Pectininae + Aequipectini group. The B dataset resulted in the most well-supported tree (Fig. 4A). All eight ML trees presented with proportional branch lengths are shown in Fig. 3.

Maximum likelihood trees based on all eight datasets (A–H). Branch lengths are scaled by setting the length of the outgroup branch to 1.5 on each tree. Numbers on top of the outgroup branches represent the original length of the branches. Every genus is marked with a different color, which is identical to the color coding on Figs. 4 and 5. Mimachlamys varia is marked with a black diamond shape, the gray line connects the corresponding internal node with the SH-like approximate likelihood ratio test (SH-aLRT) value on the left and ultrafast bootstrap support on the right. Dataset descriptions: A all PCG and rRNA genes treated with Gblocks; B all PCG amino acid sequences and rRNA genes treated with Gblocks; C all PCGs; D all PCGs treated with Gblocks; E all PCG amino acid sequences; F all PCG amino acid sequences treated with Gblocks; G rRNA genes; H rRNA genes treated with Gblocks

Phylogenetic reconstruction of 30 Pectinidae mitogenomes with Ostrea lurida (Ostreidae) as the outgroup, based on the “B” dataset (PCG amino acid and ribosomal DNA sequences). Every genus is marked with a different color, identical to the color coding on Figs. 3 and 5. Mimachlamys varia is marked with a black diamond shape. Fully supported branches are marked with a blue circle on the corresponding node. A Maximum likelihood tree. SH-like approximate likelihood ratio test (SH-aLRT) is presented on the left, ultrafast bootstrap support projected from the extended majority-rule consensus tree is presented on the right. B Bayesian tree. Numbers represent posterior probabilities; the staggered and gray lines connect species names with the corresponding tips. For original branch lengths, see Figs. 3B and 5B
Every Bayesian phylogenetic reconstruction run on every dataset reached convergence (ESS > 200). The results of these analyses slightly differ from the results of the ML method (Fig. 4B). Support values are generally higher than in the ML approach. Pl. magellanicus is placed in the same clade with Chlamydinae in the trees B, E, F, G and H with moderate to high posterior probabilities (0.81–0.98). In the rRNA trees (G and H), there is a trichotomy at the root of the Pectinidae, where the three clades are formed by Palliolinae + Chlamydinae, Pectininae and Aequipectini groups, respectively. The placement of Mim. varia is identical to its placement in the ML trees. All eight Bayesian trees presented with proportional branch lengths are shown in Fig. 5.

Bayesian consensus trees based on all eight datasets (A–H). Branch lengths are scaled by setting the length of the outgroup branch to 1.5 on each tree. Numbers on top of the outgroup branches represent the original length of the branches. Every genus is marked with a different color, that is identical to the color coding of Figs. 3 and 4. Mimachlamys varia is marked with a black diamond shape. Posterior probabilities of the corresponding internal node are presented. Dataset descriptions: A all PCG and rRNA genes masked with Gblocks; B all PCG amino acid sequences and rRNA genes masked with Gblocks; C all PCGs; D all PCGs masked with Gblocks; E all PCG amino acid sequences; F all PCG amino acid sequences masked with Gblocks; G rRNA genes; H rRNA genes masked with Gblocks
Genome rearrangements
Gene collinearity
Fifteen Locally Collinear Blocks were identified among 13 (1 per species) pectinid mitogenomes (Fig. 6). Most of these 15 blocks were present in all mitogenomes, except for LCB2 (missing from Mim. nobilis, Mimachlamys senatoria, Pe. albicans and Pl. magellanicus), LCB9 and LCB14 (both missing from Pl. magellanicus). Ten blocks contained one major gene (PCG or rRNA) and none or some tRNA genes. LCB6 and LCB9 contained one or two PCGs and none or some tRNA genes, LCB7 two to four PCGs and some tRNAs, while LCB2 and LCB4 both contained a set of tRNA genes only. LCBs 4, 7 and 12 were the most variable in terms of length (Fig. 7), due to the fact that these three blocks contained the Major Non-coding Region in 8 out of the 13 species (Fig. 6). The order of LCBs was generally more similar in more closely related species, and identical within Argopecten, between Mim. nobilis and Mim. senatoria and between Chlamys farreri and Crassadoma gigantea. Mim. varia, Miz. yessoensis and Pl. magellanicus presented substantially different genome organization from what could be expected by looking at their phylogenetic position alone. When we look at only a subset of the whole species list, several larger blocks can be observed. Larger blocks present in more than two species are the following: LCBs 5, 1 and 13 are present in the same order in 11 species (different in Mim. yessoensis and Pl. magellanicus); LCBs 5, 1, 13, 7, 8, 9 and 10 form a single block in the Pectininae and the three Argopecten species; LCBs 10, 14, 8, 11, 15, 12 and 4 form a block in the Chlamydinae.

Gene collinearity analysis of 13 Pectinid species performed in Mauve v2.3.1. Color blocks represent homologous regions among different mitogenomes. The level of similarity at each position is shown in the blocks. Below these blocks, the white, red, green and pink boxes represent protein coding, rRNA, tRNA genes, and tandem repeats, respectively

Boxplot showing the length distribution of each LCB (locally colinear block) from the gene collinearity analysis
Common interval analysis
Tandem-duplication/random loss and transposition events occurred seven times along the pectinid phylogeny (Fig. 8). Out of the 7 internal nodes, only one (the common ancestor of chlamydins) was consistent, with a high level of certainty, two were k-consistent, i.e., less reliable, with an intermediate level of certainty (the common ancestor of Chlamydinae + Placopecten and the common ancestor of Pectininae and Argopecten) and the rest were fallback nodes (representing the highest level of uncertainty), including the common ancestor of Pectinidae.

Common interval analysis presented on the most supported Bayesian tree, obtained from dataset “B”, where each tip represents a unique gene order. Internal nodes are named as “An”, where “n” is a number between 0 and 7, and represents ancestral gene orders. C and P indicate consistency and parsimony values, respectively. The colors code consistent (green; highest level of certainty), k-consistent (yellow; intermediate level of certainty) and inconsistent (red; low level of certainty) nodes. Mutation types are indicated on corresponding branches, T: transposition, TDRL: tandem duplication and random loss
Three genes—nad1, rrnL and cox1—were involved in the fewest rearrangement events (Additional file 4).
Maximum likelihood analysis of gene orders
In the maximum likelihood phylogeny reconstructed from gene orders, Argopecten is sister to pectinins, as in the eight ML and eight Bayesian trees, however, within Pectininae, Am. pleuronectes and Pe. maximus form the crown (Fig. 9). The other major differences are the positions of Mim. varia and Miz. yessoensis, the latter becoming the basal species in the Chlamydinae and Mim. varia becoming sister to the crown of the Chlamydinae, formed by the pairs of Mim. nobilis—Mim. senatoria and Cr. gigantea—Ch. farreri (Fig. 9).

Maximum likelihood tree based on gene orders, 42 genes in total, including all 13 species included in this study plus the outgroup, and the complete set of genes in each mitogenome. Numbers represent bootstrap support. Mimachlamys varia is marked with a black diamond shape
Discussion
To the best of our knowledge, our study presents the most complete mitochondrial comparative- and phylogenomic analysis to date in the Pectinidae family, including the newly-assembled mitogenome of Mim. varia. The composition of the latter conforms to most other pectinid mitogenomes published so far. However, it represents a completely novel gene order, previously not described in any other species of Pectinidae [13, 14, 18, 20,21,22,23, 27].
Gene content
In addition to the 12 PCGs present in every bivalve mitogenomes, the atp8 gene was annotated in Mim. varia. This is not surprising, given that atp8 is being annotated in more and more newly published bivalve mitogenomes, as well as identified in mitogenomes where it was originally thought to be missing [15]. In our study, the atp8 gene was annotated in 25 mitogenomes in which it was not previously found, in two species (Miz. yessoensis, Pl. magellanicus) it was even detected in two paralogous copies, and in two accessions of Ar. irradians, only as an unusual version of the gene (alternative start codon and a total length of 222 bp) (Table 3). The atp8 gene is very short (135–222 bp in the Pectinidae), and is among the most variable mitochondrial PCGs, making it very difficult to detect. The discovery of a functional atp8 gene in most pectinids hints at it not being accessory, as previously proposed [51,52,53]. Duplicated genes in mitochondrial genomes are not unheard of [54,55,56], although whether all copies are functional remains unknown.
While it is common for metazoan mitochondrial genomes to contain two trnL and trnS genes, pectinid mitogenomes also commonly contain two trnM genes. In accordance, the mitogenome of Mim. varia contains two trnL and trnM genes, however, it contains no trnS gene, and two trnF and trnQ genes. The two trnF genes show low sequence similarity and trnF (AAA) shows low sequence identity to trnF genes in other species as well as every tRNA gene in Mim. varia. Although with our methodology, trnQ2 was annotated in Mim. varia, this might be an artifact. We base this assumption on the fact that the two trnQ genes in Mim. varia are found next to each other, in the gene block (trnK, trnF, trnQ2, trnQ1, trnE), but the same gene block, containing trnS2 instead of trnQ2, is identified in Mim. senatoria [21] and Cr. gigantea [18], and revealed during reannotation in the current study in Mim. nobilis, Ch. farreri and Miz. yessoensis (Fig. 2, Additional file 3). Also, this trnQ2 gene shows higher sequence similarity to the trnS2 gene than to the trnQ gene in the aforementioned species. While we assume that this difference is the result of an artifact, it is not impossible that in Mim. varia, the trnS gene evolved into trnQ. This is supported by the fact that tRNA genes are often lost in mitochondrial genomes, and that remolding of tRNA anticodons is known to happen in mollusks [9, 21, 57].
Structure of the MNR
High A+T content and stem-loop secondary structures are the common diagnostic traits in identifying the mitochondrial control region [58,59,60]. Both can be observed in the MNR of Mim. varia (Fig. 1, Additional file 2), pointing towards it serving as the control region.
Annotation of pectinid genomes
The fact that we were able the annotate both the atp8 gene and some new tRNA genes in most pectinid accessions downloaded for this study show that the lack of these genes were most likely artifacts emerging from the difficulty of annotating these features in bivalves. With the expansion of annotation databases and the advancement of annotation tools, these difficulties are getting easier and easier to overcome, leading to progressively more precise annotations.
Variations in tRNA gene content presented by singular species (especially those with only one mitogenome annotated: Pe. albicans and Mim. varia) might well be resulting from sequencing errors, incomplete genomes, or simply misannotations. While the possibility of these cannot be completely excluded, even in the case of species that have multiple genomes published (Mim. nobilis, Miz. yessoensis, Pl. magellanicus), those presenting a phylogenetic pattern are most probably real. A peculiar case is that of trnM, as it is present in more than half of all studied species (Fig. 2). This gene is present in two copies in several invertebrate mitogenomes [61, 62], including some bivalves [25]. The invertebrate mitochondrial genetic code contains five start codons, most of which code for amino acids other than methionine in vertebrate mitochondrial genomes. This means that trnM must do ‘double duty’ as the tRNA for both methionine and formyl-methionine, matching five instead of two start codons [58], which could explain why some invertebrates, including some pectinids evolved to have more copies of it in their mitochondrial genomes.
Numerous pectinid mitogenomes are longer than the usual metazoan mitochondrial genome (~ 16 kbp), for example Aequipecten opercularis 21–28.2 kbp, Chlamys hastata 23.9–27.2 kbp, Chlamys islandica 22.5–25 kbp, Cr. gigantea 22.8–24.8 kbp [63], Mim. varia 20.4 kbp (the current study), Miz. yessoensis 20.5–21 kbp [14], Pl. magellanicus 30.7–40.7 kbp [20], and Pe. maximus [22, 63, 64], although the available mitogenome for the latter species is only 17.2 kbp long. This is mostly due to inflated non-coding regions, as demonstrated by Ghiselli et al. [58] in the whole Mollusca phylum. We identified tandem repeats of total length larger than 500 bp in the non-coding regions of Pl. magellanicus, Ch. farreri (compare with [13]), Mim. varia and Am. pleuronectes (Additional file 5) and repeats of around 500 bp length scattered throughout the mitogenome of Miz. yessoensis (Additional file 3). Gjetvaj et al. [63] also found that most of the pectinid mitogenomes they studied, contain tandem repeats of various sizes and repeat numbers in their non-coding region, and they found no significant sequence similarity among these repeats. This supports the assumption that the repeats arose independently in every lineage. There is also apparent intraspecific variation in mitochondrial genome size within the Pectinidae [63,64,65].
While it is likely that correlation exist between the number and length of tandem repeats and genome size, the available number of pectinid mitogenomes does not allow to establish correlations between these traits.
The presence of multiple repeats make it seem very likely that tandem duplication is frequent in pectinid mitogenomes, perhaps enabled by the fact that all genes are coded on the “+” strand. However, this does not seem to influence the mitogenome architecture of other marine bivalves, most of which also code all genes on the “+” strand [13]. In plants, which usually have significantly larger mitogenomes than animals and contain numerous repeated sequences, it is suggested that these repeats promote recombination [66,67,68]. This might play a role in the evolution of the mitochondrial genome in the Pectinidae as well.
The prevalence of non-coding regions and, in some instances, extra tRNAs in Pectinidae make this group an interesting candidate to study the taxonomic distribution of small mitochondrial highly-transcribed RNAs (smithRNAs, [69]). This novel class of sncRNAs (short non-coding RNAs), initially described from the Manila clam Ruditapes philippinarum (class Venerida) and detected in zebrafish, fruitflies and mice [70], could influence the expression of nuclear-encoded genes and be involved in gonad formation. They were quasi-exclusively found in non-coding regions and tRNAs [69].
Mito-phylogenomics
The phylogenies presented in our study are in accordance with previous results, both with those using a few genes, and those using the complete set of mitochondrial PCGs. One novel result is that in this group, the two rRNA genes seem to have little power to resolve deep divergences, as seen in the different placement of the Pectininae in both the ML and Bayesian approaches. In accordance with Puslednik et al. [40] and Alejandrino et al. [31], we found that Mim. varia is not placed into one monophyletic clade with the other two Mimachlamys species. Generally, although Chlamydinae itself is monophyletic, its lower taxonomic levels are not well-resolved, and several genera are paraphyletic, as it was presented in previous studies [22, 27, 31, 40, 71]. In contrast to Puslednik et al. [40], who concluded Aequipectini to be the basal clade in Pectinidae (similarly as shown in our rRNA datasets, G and H), we have found that Aequipectini always form a monophyletic group with Pectininae in both of our ML and Bayesian approaches, when we included PCGs in the analysis. It appears that while mitochondrial rRNA genes and the nuclear H3 gene are in accordance [40], mitochondrial PCGs paint a slightly different picture, as can also be seen in, for example, Marín et al. [22] and Yao et al. [27]. However, this could at least be partly caused by the small number of taxa involved in our study, and the different choice of outgroups, as both taxon sampling and outgroup selection are known to influence topologies [40]. Despite the Pectinidae being one of the largest families in Bivalvia, containing around 350 species [72], existing phylogenies usually contain only a handful of species, with the most making up only 31% of all species [31]. Chlamydinae is an especially problematic group within the Pectinidae, perhaps partly because of the low sampling relative to the high number of species in the group. While Puslednik et al. [40] conclude that Chlamydinae—in contrast to Waller’s hypothesis (i.e., that Chlamydinae is paraphyletic, and have provided the ancestral stock for Palliolinae and Pectininae)—is in fact the crown group of the Pectinidae. However, their more recent study [31], doubled taxon sampling and used one more nuclear gene (28S), and subsequently came to the opposite conclusion, confirming Waller’s original hypothesis. Another, recent study by Smedley et al. [71] included yet another nuclear gene (18S), and—as they were studying the Pectinoidea—numerous outgroup species to Pectinidae. They revealed a similar topology to Alejandrino et al. [31], but with substantially lower support. This shows the importance of marker selection and appropriate taxon sampling on pectinid phylogenetics.
The phylogeny reconstructed from gene order data shows a similar, although not identical topology to the one reconstructed from sequence data. The difference between the two trees within the Pectininae can be explained with the likely incompleteness of the Pe. albicans mitogenome, missing a few tRNA genes. In accordance with our phylogenomic results, the gene order phylogeny fails to properly resolve Chlamydinae, which, again, can be attributed to low taxon sampling within this group.
The lack of sampling is even more prominent if we look at phylogenomic studies, as the number of published pectinid mitogenomes is currently low and only slowly growing. Although our study does not resolve this problem, it extends the list of published pectinid mitogenomes, shows the utility of mito-phylogenomics within the Pectinidae and expands our knowledge of the evolutionary history of mitochondria within this remarkable group of bivalves.
Genome rearrangements
Gene collinearity
According to the ‘punctuation model’ [73], mitochondrial genomes are generally transcribed as a single polycistronic RNA from each strand, followed by the enzymatic removal of tRNAs, leading to gene-specific mRNAs [74]. In pectinids, major genes are not always separated by tRNAs (Fig. 6). It is possible that these cistrons are punctuated by secondary structures instead of tRNAs, or that they remain bicistrons. However, given that LCBs are the units of genome rearrangement, and most of them contain one major gene and some tRNA genes, it is probable that these are all separated during mRNA maturation. The one exception from this rule is LCB7, that contains the nad6 and cob genes in every studied pectinid species but contains the atp8 gene in addition to these in Mim. varia and Pectininae + Aequipectini, although in different position, and atp8 together with atp6 in Pl. magellanicus (Fig. 6, Additional file 3). Nevertheless, the variance in the composition of this block points towards it not being a single unit in rearrangements. The order of LCBs also shows a clear phylogenetic pattern. The divergence from this pattern in the case of the three outlier species, Mim. varia, Miz. yessoensis and Pl. magellanicus is most likely stemming from limited taxon sampling, i.e. they are relatively divergent from their closest relatives in our sampling, and their true close relatives would most likely show a similar gene order to theirs, as evidenced in other, better sampled groups in our study (e.g. Argopecten).
Common interval analysis
To the best of our knowledge, our study is the first attempt to reconstruct, at least partially, the ancestral pectinid mitogenome. The CREx method is widely used in other animal groups, for example insects, e.g. [75, 76]. While the ancestral gene order could be inferred for Insecta, shared by most groups, and some lineages showing some rearrangement, this is not the case for Mollusca. Katharina tunicata [17, 77] and Solemya velum [30] are hypothesized to carry a mitochondrial genome similar in organization to the ancestral Molluscan and Conchiferan mitogenomes, respectively. However, pectinid mitogenomes are very divergent from these two species, and from closely related groups with available mitogenomes, which severely limits the effectiveness of using these as outgroups, given the large chance for homoplasy. Our method tries to overcome this difficulty, with first reconstructing the putative gene order of internal nodes, including the common ancestor of the Pectinidae, followed by the inference of rearrangement events. Although some internal nodes were not consistent, we recovered the putative gene order of the common ancestor of the Chlamydinae, a monophyletic group in most phylogenetic analyses, including ours. While most previous studies focusing on mitochondrial genome rearrangements in the Pectinidae involved only a few species, we compare genome rearrangements among all available pectinid mitogenomes, with methods not previously used in this group. Confirming the assumption of Marín et al. [22], we have found that TDRL (Tandem Duplication/Random Loss) events are equally important as transpositions in pectinid mitogenome rearrangements.
Conclusions
Scallop fisheries are commercially important and steadily growing worldwide [78, 79]. Considering that mitochondrial function is implicated in growth and environmental resilience [80, 81], it is important to understand the evolution of the unusual mitochondrial genomes of pectinids, as it would aid fisheries management in our changing climate.
In our study, we annotate and characterize the mitochondrial genome of Mimachlamys varia, we demonstrate that complete mitochondrial genomes are powerful resources in reconstructing pectinid phylogenies, and we also show the utility of several tools, not previously used in this group, to investigate mitochondrial genome rearrangements within the Pectinidae. We present annotations of the atp8 gene in several pectinid species, where it was previously thought to be missing.
We show incongruities between phylogenies based on mitochondrial PCGs and rRNA genes, suggesting that locus sampling affects phylogenetic inference at the scale of the Pectinidae family. Both the lack of suitable outgroups—more closely related to the Pectinidae (e.g. Limidae, Entoliidae, Spondylidae and Propeamussiidae) than the currently available species—and the lack of sufficient taxon sampling, especially within Chlamydinae and Palliolinae, are limiting factors for current research. We therefore believe that sequencing and assembly of more mitogenomes, particularly from the aforementioned groups, would greatly improve the power of further research of both mito-phylogenomics and the evolution of mitogenome rearrangements in the Pectinidae.
Methods
Sequencing of the mitogenome of reference for the variegated scallop
One individual of Mim. varia (adult male, shell length: 46 mm, shell height: 40 mm) was collected in Angoulins, France, in order to sequence and annotate a reference transcriptome (nuclear and mitochondrial) for the species. RNA was purified from five tissues (digestive gland, mantle, gills, adductor muscle and gonads) using the Nucleospin RNA Set for Nucleozol kit (Macherey–Nagel). After quality control, extractions were pooled in equal amounts (4 μg of RNA per tissue type). The library was sequenced on Illumina HiSeq 2500 (2 × 300 bp). Full sample preparation and sequencing information is provided in Viricel et al. [50]; raw reads can be accessed on the NBCI Sequence Read Archive (Acc. SRP127478), and transcripts can be accessed at the Transcriptome Shotgun Assembly database (GGGO01000000).
Paired reads, minimally cleaned with Trimmomatic v. 0.36 ([82]; only adapters were removed as recommended in the NOVOPlasty manual), were used as input data for assembly with NOVOPlasty v. 4.2 [83] to assemble the mitochondrial genome. Quality-control before and after read filtering was done using FastQC v0.11.5 [84]. The size of the mitogenome being highly variable among pectinids (from 16,079 bp in Argopecten ventricosus, KT161261.1, to 32,115 bp in Placopecten magellanicus, NC_007234.1, [85]), we set a wide range for possible mitogenome sizes (15–35 kbp) as a parameter in NOVOPlasty. K-mer size was set to 39. We used a portion of the mitochondrial gene cox1 as an initial seed for mitogenome assembly (we used the most common haplotype found along the coast of France, KU680872.1; [49]). When NOVOPlasty runs resulted in multiple contigs (mitogenome fragments), they were assembled in Geneious v2021.0.3 (https://www.geneious.com). Reads were mapped onto the assembly using bowtie2 v2.3.5.1 [86] and coverage statistics were generated with samtools v1.10 [87]. Once the reference mitogenome was assembled, we used NOVOPlasty to detect intra-individual heteroplasmy [88] using a minimum Minor Allele Frequency (MAF) of 0.1 and 0.01 in two separate runs. Finally, the MITOS2 web server (http://mitos2.bioinf.uni-leipzig.de/index.py) [89] was used to annotate the reference with default parameters, using the RefSeq 63 Metazoa database as reference and the protein-coding gene prediction method of Al Arab et al. [90]. The mitogenome was loaded into Geneious v2021.0.3. and checked manually for errors in annotations. ARWEN v1.2.3 [91], was used to look for additional tRNA genes and secondary structures of tRNAs were predicted using MITOS2 and the RNAfold web server, with default options. The Major Non-coding Region (MNR) was scanned for repeated regions using Tandem Repeats Finder v 4.09 (https://tandem.bu.edu/trf/trf.html) [92] and secondary structures were predicted using the mfold Web Server (http://www.unafold.org/) [93]. The circular mitogenome was visualised using the CGView Web Server (http://cgview.ca/) [94], with annotations imported from Geneious.
Assembly and annotation of pectinid mitogenomes from GenBank
All currently available pectinid mitogenomes (as of January 2021) were downloaded from GenBank (Table 4) along with Ostrea lurida (Ostreidae) as an outgroup. We chose an oyster species as outgroup as there are no published mitogenomes of taxa more closely related to Pectinidae. Using only one outgroup made interpreting comparisons of mitogenome rearrangements substantially less difficult. The largest mitogenome from each species was reannotated using the MITOS2 web server with the same settings as for the Mim. varia mitogenome. New annotations of pectinid mitogenomes were reviewed manually, in the same way as it was described for Mim. varia. If there were multiple annotations for a gene, they were filtered manually based on length and sequence similarity to other species. Additionally, tRNA genes were compared with previously published annotations. Next, this reference sequence of each species was used to annotate other sequences of the same species using the “Live Annotate & Predict” feature in Geneious.
Mito-phylogenomics
Phylogenetic analysis was conducted on the 13 protein coding genes (PCGs) and two Ribosomal RNA (rRNA) genes of all available pectinid species in GenBank, Mim. varia and O. lurida as an outgroup. This approach provides the most comprehensive dataset without the ambiguities arising from tRNA annotations, non-coding regions and genome rearrangements. Every gene was extracted and aligned separately in Geneious v2021.0.3. In the case of the atp8 gene, where more copies were identified in some species, a maximum likelihood (ML) tree was built with the RAxML [95] plugin of Geneious using default parameters, and the least divergent copy was selected for further analysis. The atp8 gene of two accessions of Argopecten irradians (EU023915; NC_009687) were not included in the phylogenetic analyses given their unusual structure, and divergent sequences. The 13 PCG sequences were aligned separately based on translated amino-acids with MAFFT [96] with the FFT-NS-2 algorithm as implemented in TranslatorX [97]. As the initial alignments contained indels and poorly aligned regions, these regions were masked with Gblocks [98] as implemented in TranslatorX, with default settings (i.e., allowing smaller final blocks, gap positions within the final blocks and less strict flanking positions). The two rRNA genes were aligned with MAFFT in Geneious using the FFT-NS-2 algorithm. Regions of uncertainty in the alignment were then masked with Gblocks using the same settings as for the PCGs. Individual gene alignments were then concatenated in Geneious resulting in eight datasets used for phylogenetic analyses: (A) all PCG and rRNA genes treated with Gblocks; (B) all PCG amino acid sequences and rRNA genes treated with Gblocks; (C) all PCGs; (D) all PCGs treated with Gblocks; (E) all PCG amino acid sequences; (F) all PCG amino acid sequences treated with Gblocks; (G) rRNA genes; (H) rRNA genes treated with Gblocks. These datasets were prepared in order to investigate the effect of rRNA and protein-coding genes as well as the presence/absence of ambiguously aligned regions on the topology and branch support of the phylogeny.
Maximum likelihood phylogenetic reconstruction was achieved using IQtree v2.0.6 [99] (except for the B dataset, where IQtree v1.6.12 was used), with automatic model selection and using the best partition scheme in ModelFinder (option MFP + MERGE) [100], 10,000 Ultrafast Bootstrap replicates and 10,000 replicates of the SH-like approximate likelihood ratio test (SH-aLRT) [101] on all eight datasets. Partitioning and substitution model information for every run can be found in Additional file 6.
Bayesian analyses were done using MrBayes v3.2.7 [102]. For each dataset, two runs were performed over 1.5 million generations, with four chains each (three heated and one cold). Trees were sampled every 500 generations, and the first 25% were discarded as burn-in. Finally, a majority consensus tree was constructed to estimate posterior probabilities of branches. Convergence of each run was evaluated through effective sample size (ESS) values and trace plots in the software Tracer v.1.6 [103]. Partitioning and prior model parameter settings can be found in Additional file 7.
Genome rearrangements
Three approaches were used to infer mitogenome rearrangements in the Pectinidae: (1) gene collinearity analysis, which detects mosaic patterns of homology among a set of genomes, based on whole-genome sequence alignment; (2) common interval analysis, which determines pairwise rearrangement events between genomes, based on gene order data, considering several rearrangement types, but requiring identical gene content; and (3) maximum likelihood analysis of gene orders, which reconstructs phylogenies based solely on gene order data, without requiring an identical set of genes between genomes.
Gene collinearity
Gene collinearity among pectinid mitogenomes (one individual/species, 13 individuals in total) was explored using the progressiveMauve algorithm and default parameters [automatically calculating seed weight and the minimum LCB score, computing Locally Collinear Blocks (LCBs) and using full alignments] in the MAUVE Geneious plugin [104].
Common interval analysis
Two joint softwares—both relying on the same algorithm—CREx [105] and TreeREx [106] were implemented for common interval analyses. Both software consider the following types of rearrangement events: transpositions, inversions, inverse transpositions and tandem-duplication/random loss (TDRL) involving the duplication of a gene block followed by the random loss of one copy of a gene in either of the two blocks. Both tools are only able to handle identical gene sets among species. To conform to this condition, only major genes (PCGs and rRNA genes) and the MNR were included in this analysis, as tRNA gene content is variable among pectinid mitogenomes.
The MrBayes tree based on the most supported dataset (B), with only eight pectinids and one outgroup species representing unique gene orders, along with the gene order dataset described above, was used in the first step, using the TreeREx [106] software. TreeREx uses the phylogenetic information in the tree to ascertain rearrangement events among nodes of the tree, and to infer putative gene orders of common ancestors at internal nodes. TreeREx was used with default settings: strong consistency method applied (-s); weak consistency method applied (-w); parsimonious weak consistency method applied (-W); and the maximum number of inversions (-m) set to 2. Finally, the input dataset in the previous analysis, complemented with the putative gene orders inferred at internal nodes was put into CREx again, to visualize each rearrangement event between each pair of neighboring nodes.
Maximum likelihood analysis of gene orders
The MLGO (Maximum Likelihood for Gene-Order Analysis) [107] web server can be used to directly infer a phylogeny based on gene order data. The algorithm handles insertions, deletions and duplications along with transpositions. Unlike CREx/TreeREx, it is not limited to a common set of genes across taxa. Complete gene order data from 13 pectinids (one individual per species) and one outgroup (O. lurida) were used to infer a phylogeny of the Pectinidae. Branch support was computed with a bootstrap analysis of 1000 replicates.
Availability of data and materials
Raw Mimachlamys varia RNAseq reads are deposited in the NBCI Sequence Read Archive (Acc. SRP127478), and transcripts can be accessed at the Transcriptome Shotgun Assembly database (GGGO01000000). The assembled mitochondrial genome is uploaded to GenBank (MZ520326), and the GenBank accession numbers of all mitogenomes used in this study can be found in Table 4. The sequence alignments used in this article are publicly available on GitHub (https://github.com/ericpante/Malkocs_etal_2022).
References
Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–80.
Burger G, Forget L, Zhu Y, Gray MW, Lang BF. Unique mitochondrial genome architecture in unicellular relatives of animals. Proc Natl Acad Sci. 2003;100:892–7.
Dreyer H, Steiner G. The complete sequence and gene organization of the mitochondrial genome of the gadilid scaphopod Siphonondentalium lobatum (Mollusca). Mol Phylogenet Evol. 2004;31:605–17.
Feldmeyer B, Hoffmeier K, Pfenninger M. The complete mitochondrial genome of Radix balthica (Pulmonata, Basommatophora), obtained by low coverage shot gun next generation sequencing. Mol Phylogenet Evol. 2010;57:1329–33.
Hatzoglou E, Rodakis GC, Lecanidou R. Complete sequence and gene organization of the mitochondrial genome of the land snail Albinaria coerulea. Genetics. 1995;140:1353–66.
Liu YG, Kurokawa T, Sekino M, Tanabe T, Watanabe K. Complete mitochondrial DNA sequence of the ark shell Scapharca broughtonii: an ultra-large metazoan mitochondrial genome. Comp Biochem Physiol Part D Genomics Proteomics. 2013;8:72–81.
Snyder M, Fraser AR, Laroche J, Gartner-Kepkay KE, Zouros E, Kafatos FC. Atypical mitochondrial DNA from the deep-sea scallop Placopecten magellanicus. Proc Natl Acad Sci USA. 1987;84:7595–9.
Xin Y, Ren J, Liu X. Mitogenome of the small abalone Haliotis diversicolor Reeve and phylogenetic analysis within Gastropoda. Mar Genomics. 2011;4:253–62.
Wu X, Li X, Li L, Xu X, Xia J, Yu Z. New features of Asian Crassostrea oyster mitochondrial genomes: a novel alloacceptor tRNA gene recruitment and two novel ORFs. Gene. 2012;507:112–8.
Skibinski DOF, Gallagher C, Beynon CM. Mitochondrial DNA inheritance [11]. Nature. 1994;368:817–8.
Zouros E, Ball AO, Saavedra C, Freeman KR. Mitochondrial DNA inheritance [12]. Nature. 1994;368:818.
Zouros E. Doubly uniparental inheritance of mitochondrial DNA: might it be simpler than we thought? J Zool Syst Evol Res. 2020;58:624–31.
Ren J, Shen X, Jiang F, Liu B. The mitochondrial genomes of two scallops, Argopecten irradians and Chlamys farreri (Mollusca: Bivalvia): the most highly rearranged gene order in the family pectinidae. J Mol Evol. 2010;70:57–68.
Wu X, Xu X, Yu Z, Kong X. Comparative mitogenomic analyses of three scallops (Bivalvia: Pectinidae) reveal high level variation of genomic organization and a diversity of transfer RNA gene sets. BMC Res Notes. 2009;2:1–7.
Plazzi F, Puccio G, Passamonti M. Comparative large-scale mitogenomics evidences clade-specific evolutionary trends in mitochondrial DNAs of Bivalvia. Genome Biol Evol. 2016;8:2544–64.
Liu H, Yang Y, Sun S, Kong L, Li Q. Mitogenomic phylogeny of the Naticidae (Gastropoda: Littorinimorpha) reveals monophyly of the Polinicinae. Zool Scr. 2020;49:295–306.
Vallès Y, Boore JL. Lophotrochozoan mitochondrial genomes. Integr Comp Biol. 2006;46:544–57.
Liao D, Zhou Y, Tong J, Cao S, Yu X, Fu B, et al. Characterization and phylogenetic analysis of the complete mitochondrial genome from Rock Scallop (Crassadoma gigantea) using next-generation sequencing. Mitochondrial DNA Part B Resour. 2018;3:827–8.
Sato M, Nagashima K. Molecular characterization of a mitochondrial DNA segment from the Japanese scallop (Patinopecten yessoensis): demonstration of a region showing sequence polymorphism in the population. Mar Biotechnol. 2001;3:370–9.
Smith DR, Snyder M. Complete mitochondrial DNA sequence of the scallop Placopecten magellanicus: evidence of transposition leading to an uncharacteristically large mitochondrial genome. J Mol Evol. 2007;65:380–91.
Wu X, Li X, Yu Z. The mitochondrial genome of the scallop Mimachlamys senatoria (Bivalvia, Pectinidae). Mitochondrial DNA. 2015;26:242–4.
Marín A, Fujimoto T, Arai K. The mitochondrial genomes of Pecten albicans and Pecten maximus (Bivalvia: Pectinidae) reveal a novel gene arrangement with low genetic differentiation. Biochem Syst Ecol. 2015;61:208–17.
Xu K, Kanno M, Yu H, Li Q, Kijima A. Complete mitochondrial DNA sequence and phylogenetic analysis of Zhikong scallop Chlamys farreri (Bivalvia: Pectinidae). Mol Biol Rep. 2011;38:3067–74.
Boore JL, Medina M, Rosenberg LA. Complete sequences of the highly rearranged molluscan mitochondrial genomes of the scaphopod Graptacme eborea and the bivalve Mytilus edulis. Mol Biol Evol. 2004;21:1492–503.
Wu X, Xiao S, Li X, Li L, Shi W, Yu Z. Evolution of the tRNA gene family in mitochondrial genomes of five Meretrix clams (Bivalvia, Veneridae). Gene. 2014;533:439–46.
Yamazaki N, Ueshima R, Terrett JA, Yokobori S, Kaifu M, Segawa R, et al. Evolution of pulmonate gastropod mitochondrial genomes: comparisons of gene organizations of Euhadra, Cepaea and Albinaria and implications of unusual tRNA secondary structures. Genetics. 1997;145:749–58.
Yao L, Yu H, Liu Y, Zhang Y, Li Y. The complete mitochondrial genome and phylogenetic analysis of Amusium pleuronectes. Mitochondrial DNA Part B. 2020;5:2318–9.
Bettinazzi S, Plazzi F, Passamonti M. The complete female- and male-transmitted mitochondrial genome of Meretrix lamarckii. PLoS ONE. 2016;11:e0153631.
Gaitán-Espitia JD, Quintero-Galvis JF, Mesas A, D’Elía G. Mitogenomics of southern hemisphere blue mussels (Bivalvia: Pteriomorphia): insights into the evolutionary characteristics of the Mytilus edulis complex. Sci Rep. 2016;6:1–10.
Plazzi F, Ribani A, Passamonti M. The complete mitochondrial genome of Solemya velum (Mollusca: Bivalvia) and its relationships with Conchifera. BMC Genomics. 2013;14:1–15.
Alejandrino A, Puslednik L, Serb JM. Convergent and parallel evolution in life habit of the scallops (Bivalvia: Pectinidae). BMC Evol Biol. 2011;11:1–9.
Canapa A, Barucca M, Marinelli A, Olmo E. Molecular data from the 16s rRNA gene for the phylogeny of Pectinidae (Mollusca: Bivalvia). J Mol Evol. 2000;50:93–7.
Saavedra C, Peña JB. Phylogenetics of American scallops (Bivalvia: Pectinidae) based on partial 16S and 12S ribosomal RNA gene sequences. Mar Biol. 2006;150:111–9.
Feng Y, Li Q, Kong L, Zheng X. DNA barcoding and phylogenetic analysis of Pectinidae (Mollusca: Bivalvia) based on mitochondrial COI and 16S rRNA genes. Mol Biol Rep. 2011;38:291–9.
López-Piñón MJ, Freire R, Insua A, Méndez J. Sequence characterization and phylogenetic analysis of the 5S ribosomal DNA in some scallops (Bivalvia: Pectinidae). Hereditas. 2008;145:9–19.
Mahidol C, Na-Nakorn U, Sukmanomon S, Yoosuk W, Taniguchi N, Nguyen TTT. Phylogenetic relationships among nine scallop species (Bivalvia: Pectinidae) inferred from nucleotide sequences of one mitochondrial and three nuclear gene regions. J Shellfish Res. 2007;26:25–32.
Malkowsky Y, Klussmann-Kolb A. Phylogeny and spatio-temporal distribution of European Pectinidae (Mollusca: Bivalvia). Syst Biodivers. 2012;10:233–42.
Matsumoto M. Phylogenetic analysis of the subclass Pteriomorphia (Bivalvia) from mtDNA COI sequences. Mol Phylogenet Evol. 2003;27:429–40.
Pujolar JM, Marčeta T, Saavedra C, Bressan M, Zane L. Inferring the demographic history of the Adriatic Flexopecten complex. Mol Phylogenet Evol. 2010;57:942–7.
Puslednik L, Serb JM. Molecular phylogenetics of the Pectinidae (Mollusca: Bivalvia) and effect of increased taxon sampling and outgroup selection on tree topology. Mol Phylogenet Evol. 2008;48:1178–88.
Saavedra C, Peña JB. Phylogenetic relationships of commercial European and Australasian king scallops (Pecten spp.) based on partial 16S ribosomal RNA gene sequences. Aquaculture. 2004;235:153–66.
Brugler MR, France SC. The complete mitochondrial genome of the black coral Chrysopathes formosa (Cnidaria:Anthozoa:Antipatharia) supports classification of antipatharians within the subclass Hexacorallia. Mol Phylogenet Evol. 2007;42:776–88.
Lavrov DV, Wang X, Kelly M. Reconstructing ordinal relationships in the Demospongiae using mitochondrial genomic data. Mol Phylogenet Evol. 2008;49:111–24.
Kayal E, Roure B, Philippe H, Collins AG, Lavrov DV. Cnidarian phylogenetic relationships as revealed by mitogenomics. BMC Evol Biol. 2013;13:1–18.
Brand AR. Chapter Scallop ecology: distributions and behaviour. Dev Aquac Fish Sci. 2006;35 C:651–744.
Breitwieser M, Viricel A, Graber M, Murillo L, Becquet V, Churlaud C, et al. Short-term and long-term biological effects of chronic chemical contamination on natural populations of a marine bivalve. PLoS ONE. 2016;11:150184.
Breitwieser M, Viricel A, Churlaud C, Guillot B, Martin E, Stenger PL, et al. First data on three bivalve species exposed to an intra-harbour polymetallic contamination (La Rochelle, France). Comp Biochem Physiol Part C Toxicol Pharmacol. 2017;199:28–37.
Milinkovitch T, Bustamante P, Huet V, Reigner A, Churlaud C, Thomas-Guyon H. In situ evaluation of oxidative stress and immunological parameters as ecotoxicological biomarkers in a novel sentinel species (Mimachlamys varia). Aquat Toxicol. 2015;161:170–5.
Breitwieser M, Becquet V, Thomas-Guyon H, Pillet V, Sauriau PG, Graber M, et al. Population structure and genetic diversity in the variegated scallop, Mimachlamys varia (Linnaeus, 1758), a novel bioindicator of chemical pollution on the French coastline. J Molluscan Stud. 2018;84:417–25.
Viricel A, Becquet V, Dubillot E, Pante E. De novo assembly and functional annotation of the transcriptome of Mimachlamys varia, a bioindicator marine bivalve. Mar Genomics. 2018;41:42–5.
Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;101:301–20.
Papakonstantinou T, Law RHP, Nagley P, Devenish RJ. Non-functional variants of yeast mitochondrial ATP synthase subunit 8 that assemble into the complex. Biochem Mol Biol Int. 1996;39:253–60.
Stephens AN, Khan MA, Roucou X, Nagley P, Devenish RJ. The molecular neighborhood of subunit 8 of yeast mitochondrial F1F0-ATP synthase probed by cysteine scanning mutagenesis and chemical modification. J Biol Chem. 2003;278:17867–75.
Bojko J. The mitochondrial genome of UK (non-native) Dikerogammarus haemobaphes (Amphipoda: Gammaridae) informs upon Dikerogammarus evolution, invasions and associated microparasites. Hydrobiologia. 2019;847:229–42.
Yokobori S, Fukuda N, Nakamura M, Aoyama T, Oshima T. Long-term conservation of six duplicated structural genes in cephalopod mitochondrial genomes. Mol Biol Evol. 2004;21:2034–46.
Kawashima Y, Nishihara H, Akasaki T, Nikaido M, Tsuchiya K, Segawa S, et al. The complete mitochondrial genomes of deep-sea squid (Bathyteuthis abyssicola), bob-tail squid (Semirossia patagonica) and four giant cuttlefish (Sepia apama, S. latimanus, S. lycidas and S. pharaonis), and their application to the phylogenetic analysis of Decapodiformes. Mol Phylogenet Evol. 2013;69:980–93.
Guerra D, Bouvet K, Breton S. Mitochondrial gene order evolution in Mollusca: inference of the ancestral state from the mtDNA of Chaetopleura apiculata (Polyplacophora, Chaetopleuridae). Mol Phylogenet Evol. 2018;120:233–9.
Ghiselli F, Gomes-Dos-Santos A, Adema CM, Lopes-Lima M, Sharbrough J, Boore JL. Molluscan mitochondrial genomes break the rules. Philos Trans R Soc B Biol Sci. 2021;376:20200159.
Grande C, Templado J, Zardoya R. Evolution of gastropod mitochondrial genome arrangements. BMC Evol Biol. 2008;8:1–15.
Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem. 1997;66:409–36.
Voigt O, Erpenbeck D, Wörheide G. A fragmented metazoan organellar genome: the two mitochondrial chromosomes of Hydra magnipapillata. BMC Genomics. 2008;9:1–10.
Zhang J, Zhou C, Gai Y, Song D, Zhou K. The complete mitochondrial genome of Parafronurus youi (Insecta: Ephemeroptera) and phylogenetic position of the Ephemeroptera. Gene. 2008;424:18–24.
Gjetvaj B, Cook DI, Zouros E. Repeated sequences and large-scale size variation of mitochondrial DNA: a common feature among scallops (Bivalvia: Pectinidae). Mol Biol Evol. 1992;9:106–24.
Rigaa A, Monnerot M, Sellos D. Molecular cloning and complete nucleotide sequence of the repeated unit and flanking gene of the scallop Pecten maximus mitochondrial DNA: putative replication origin features. J Mol Evol. 1995;41:189–95.
Fuller KM. Polymorphism and heteroplasmy of mitochondrial DNA in the scallop Placopecten magellanicus (Gmelin). 1991.
Lonsdale DM, Brears T, Hodge TP, Melville SE, Rottmann WH. The plant mitochondrial genome: homologous recombination as a mechanism for generating heterogeneity. Philos Trans R Soc Lond B Biol Sci. 1988;319:149–63.
Palmer JD, Herbon LA. Plant mitochondrial DNA evolved rapidly in structure, but slowly in sequence. J Mol Evol. 1988;28:87–97.
Van de Paer C, Bouchez O, Besnard G. Prospects on the evolutionary mitogenomics of plants: a case study on the olive family (Oleaceae). Mol Ecol Resour. 2018;18:407–23.
Pozzi A, Plazzi F, Milani L, Ghiselli F, Passamonti M. SmithRNAs: could mitochondria “bend” nuclear regulation? Mol Biol Evol. 2017;34:1960–73.
Passamonti M, Calderone M, Delpero M, Plazzi F. Clues of in vivo nuclear gene regulation by mitochondrial short non-coding RNAs. Sci Rep. 2020;10:1–12.
Smedley GD, Audino JA, Grula C, Porath-Krause A, Pairett AN, Alejandrino A, et al. Molecular phylogeny of the Pectinoidea (Bivalvia)indicates Propeamussiidae to be a non-monophyletic family with one clade sister to the scallops (Pectinidae). Mol Phylogenet Evol. 2019;137:293–9.
Biscotti MA, Canapa A, Olmo E, Barucca M, Teo CH, Schwarzacher T, et al. Repetitive DNA, molecular cytogenetics and genome organization in the King scallop (Pecten maximus). Gene. 2007;406:91–8.
Ojala D, Montoya J, Attardi G. TRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290:470–4.
D’Souza AR, Minczuk M. Mitochondrial transcription and translation: overview. Essays Biochem. 2018;62:309–20.
Tyagi K, Chakraborty R, Cameron SL, Sweet AD, Chandra K, Kumar V. Rearrangement and evolution of mitochondrial genomes in Thysanoptera (Insecta). Sci Rep. 2020;10:1–16.
Yoshizawa K, Johnson KP, Sweet AD, Yao I, Ferreira RL, Cameron SL. Mitochondrial phylogenomics and genome rearrangements in the barklice (Insecta: Psocodea). Mol Phylogenet Evol. 2018;119:118–27.
Xu W, Jameson D, Tang B, Higgs PG. The relationship between the rate of molecular evolution and the rate of genome rearrangement in animal mitochondrial genomes. J Mol Evol. 2006;63:375–92.
Delargy A. Quantitative methods for producing evidence to support sustainable King Scallop Management. Bangor: Bangor University; 2020.
Stewart BD, Howarth LM. Quantifying and managing the ecosystem effects of scallop dredge fisheries. Dev Aquac Fish Sci. 2016;40:585–609.
Rustin P. Mitochondria, from cell death to proliferation. Nat Genet. 2002;30:352–3.
Quirós PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol. 2016;17:213–26.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017;45:e18.
Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
La Roche J, Snyder M, Cook DI, Fuller K, Zouros E. Molecular characterization of a repeat element causing large-scale size variation in the mitochondrial DNA of the sea scallop Placopecten magellanicus. Mol Biol Evol. 1990;7:45–64.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Dierckxsens N, Mardulyn P, Smits G. Unraveling heteroplasmy patterns with NOVOPlasty. NAR Genomics Bioinform. 2020;2:1–10.
Donath A, Jühling F, Al-Arab M, Bernhart SH, Reinhardt F, Stadler PF, et al. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019;47:10543–52.
Al Arab M, Höner zu Siederdissen C, Tout K, Sahyoun AH, Stadler PF, Bernt M. Accurate annotation of protein-coding genes in mitochondrial genomes. Mol Phylogenet Evol. 2017;106:209–16.
Laslett D, Canbäck B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172–5.
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80.
Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15.
Grant JR, Stothard P. The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008;36 Web server issue:181–4.
Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90.
Katoh K, Asimenos G, Toh H. Multiple alignment of DNA sequences with MAFFT. Methods Mol Biol. 2009;537:39–64.
Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38(SUPPL. 2):W7-13.
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–52.
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Kalyaanamoorthy S, Minh BQ, Wong TKF, Von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Rambaut A, Drummond AJ. Tracer v1.6. 2013. http://tree.bio.ed.ac.uk/software/tracer/.
Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–403.
Bernt M, Merkle D, Ramsch K, Fritzsch G, Perseke M, Bernhard D, et al. CREx: inferring genomic rearrangements based on common intervals. Bioinformatics. 2007;23:2957–8.
Bernt M, Merkle D, Middendorf M. An algorithm for inferring mitogenome rearrangements in a phylogenetic tree. In: Lecture notes in computer science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics). Berlin: Springer; 2008. p. 143–57.
Hu F, Lin Y, Tang J. MLGO: Phylogeny reconstruction and ancestral inference from gene-order data. BMC Bioinform. 2014;15:354.
Acknowledgements
We thank Nathalie Imbert and Denis Fichet for coordinating axe 2 of the DYPOMAR project and Benoit Simon-Bouhet for additional funding. RNA extractions were prepared at the Molecular Core Facility at the University of La Rochelle. We thank the bioinformatics platform Toulouse Midi-Pyrenees (Bioinfo GenoToul).
Funding
The first author thanks the Erasmus+ programme of the European Union for funding his mobility and stay at the LIENSs laboratory in La Rochelle. This work was supported by the Contrat de Plan Etat-Région (CPER/FEDER) ECONAT (RPC DYPOMAR).
Author information
Authors and Affiliations
Contributions
ED and VB collected the samples, AV and VB performed laboratory analyses, TM, EP and LE performed data analyses, TM wrote the manuscript, AV and EP reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No vertebrates or regulated invertebrates were involved in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Putative secondary structures of mitochondrial tRNAs in Mimachlamys varia. Amino acids are represented by their one-letter code. (*: structure predicted by RNAfold, all other structures were predicted by MITOS2)
Additional file 2.
Putative secondary structure of the Major Non-coding Region of Mimachlamys varia. There is a 100 basepair overlap kept between parts. 1: 5484–6439; 2: 6339–7805; 3: 7705–9689.
Additional file 3.
Annotation tables for species involved in this study.
Additional file 4.
Common interval analysis results presented on a Bayesian tree, where each tip represents a unique gene order. Internal nodes are named as An, where n is a number between 0 and 7, and represent ancestral gene orders. C and P indicate consistency and parsimony values, respectively. The colors code consistent (green; highest level of certainty), k-consistent (yellow; intermediate level of certainty) and inconsistent (red; low level of certainty) nodes. Mutation types are indicated on corresponding branches, T: transposition, TDRL: tandem duplication and random loss. Arrows point to diagrams showing the two gene orders, and the mutation steps leading from the more ancestral to the younger node. Frames connect gene blocks affected by a rearrangement event, and parts of the mitogenome that is affected by a rearrangement event.
Additional file 5.
Tandem repeats detected in pectinid mitogenomes.
Additional file 6.
Partitioning and substitution model information for all eight datasets used in the Maximum Likelihood analyses.
Additional file 7.
Partitioning schemes and prior model parameter settings for all eight datasets used in the Bayesian analyses.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Malkócs, T., Viricel, A., Becquet, V. et al. Complex mitogenomic rearrangements within the Pectinidae (Mollusca: Bivalvia). BMC Ecol Evo 22, 29 (2022). https://doi.org/10.1186/s12862-022-01976-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-022-01976-0