
Abstract
Background
Triplophysa stoliczkae is the most widespread species in the genus Triplophysa and may have originated from morphological convergence. To understand the evolutionary history of T. stoliczkae, we employed a multilocus approach to investigate the phylogenetics and the morphological evolution of T. stoliczkae on the Qinghai-Tibetan Plateau.
Results
All phylogenetic analyses (two mitochondrial and five nuclear loci), a genealogical sorting index and species tree inferences suggested that T. stoliczkae consists of distinct lineages that were not closest relatives. The time estimation indicated that the divergence events between “T. stoliczkae” and other Triplophysa species occurred from approximately 0.10 to 4.51 Ma. The ancestral state analyses supported the independent evolution of T. stoliczkae morphology in distinct lineages. The morphometric analysis and convergence estimates demonstrated significant phenotypic convergence among “T. stoliczkae” lineages.
Conclusions
Triplophysa stoliczkae includes 4 different lineages with similar morphologies. The increasingly harsh environments that have occurred since the Pliocene have driven the occurrences of scrape-feeding fish in the genus Triplophysa. Morphological adaptations associated with scrape-feeding behavior resulted in convergences and the artificial lumping of four different species in the nominal taxon T. stoliczkae. A taxonomic revision for T. stoliczkae is needed.
Similar content being viewed by others


Background
Convergent evolution, the independent origination of analogous biological traits, is pervasive in the evolution of life and has long been regarded as evidence of adaptation [1,2,3,4,5]. Generally, convergent evolution is common in species-rich communities, especially when the number of species exceeds the available niches [6,7,8]. Furthermore, convergent evolution creates similar phenotypes that resulted in errors in the taxonomy [5, 9, 10]. Understanding convergent evolution may help us better understand the evolutionary history and resolve the taxonomy dilemma.
The genus Triplophysa (Cobitoidea: Nemacheilidae), a species-rich and taxonomically unstable group, is an important component of the ichthyofauna on the Qinghai-Tibetan Plateau (QTP) [11, 12]. Triplophysa stoliczkae (Steindachner, 1866) is the most widely distributed species in this genus. It lives mainly in the streams of the QTP and its peripheral regions. As a scrape-feeding fish, T. stoliczkae has the typical characteristics of a broadened sharp lower jaw, screw-like intestine and degenerated posterior chamber of its air bladder (PCAB) (Additional file 1: Figures S1 and S2). Due to morphological variations among populations, T. stoliczkae has long been a controversial subject in taxonomy [11,12,13]. Nevertheless, T. stoliczkae is currently considered to be a single, widespread species with a geographic range that covers the entire QTP. The evolutionary history of QTP ichthyofauna has been shaped by the history of river drainages [14]. Generally, the QTP fish with a late phylogenetic position have had a limited distribution area because of geographical barriers. However, the mitochondrial data have suggested that T. stoliczkae is not an ancient lineage [14,15,16]. Moreover, the current habitats of T. stoliczkae have been disconnected from each other by ancient and persistent orogeny [17]. The conflict between the late occurrence and wide distribution of T. stoliczkae could be explained by two possible scenarios. First, convergent evolution creates similar morphologies that resulted in errors in the definition of T. stoliczkae. Second, it is possible, although unlikely, that T. stoliczkae evolved once and then spread to different river systems.
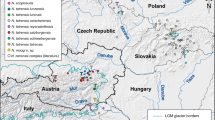
To determine the evolutionary history of T. stoliczkae, we employ a multilocus approach to analyze individuals from almost all of the distribution ranges. We also use many other Triplophysa species from various water systems (Fig. 1) to ensure robust results. By inferring the phylogenetic relationships, quantifying genealogical sorting, and dating the divergence, we attempt to answer two questions: (1) whether T. stoliczkae is monophyletic and (2) when and how many times divergence events occurred in T. stoliczkae. Moreover, under a nonmonophyly scenario, an ancestral state analysis indicates that the occurrence of T. stoliczkae morphology is based on (a) multiple independent innovations or (b) the remains of ancient traits. A morphometric analysis and convergence estimate further quantify the phenotypic convergence of “T. stoliczkae”. Finally, together with the geologic climate background, we try to understand the evolution of T. stoliczkae.

Sampling sites and the water systems. Rose-red circles denote the localities of Triplophysa stoliczkae, and gray circles represent the localities of the other Triplophysa species. Site numbers are detailed in Table 1. Light blue patches and lines depict the water systems. Rose-red dashed lines (L1 to L4) correspond to T. stoliczkae-1 to T. stoliczkae-4 in Figs. 2-5
Methods
Samples and laboratory procedures
With the permission of the fisheries departments of the Chinese government, we carried out the sampling to collect protected Triplophysa. All animal experiments were approved by the Ethics Committee of the Northwest Institute of Plateau Biology, Chinese Academy of Sciences. A total of 49 specimens of Triplophysa containing 26 specimens of T. stoliczkae and 23 other Triplophysa species from 36 sites were analyzed (Fig. 1; Table 1). Based on a previous study [16], we selected Triplophysa siluroides and Triplophysa robusta as primary the outgroup taxa and used Triplophysa rosa as the secondary outgroup species. Specimens of T. stoliczkae were collected from 8 water systems on and near the QTP: Yangtze River, Yellow River, Shiyang River, Heihe River, Ili River, Ganges River, Indus River, and Tarim River. All collected specimens were euthanized after identification. They were placed in a dry ice box for rapid hypothermic anesthesia within about 20 s. Then, they were preserved in 95% ethanol for laboratory works. Voucher specimens were archived in the collection of the Northwest Institute of Plateau Biology (NWIPB), Chinese Academy of Sciences.
Genomic DNA was extracted from the fin or muscle using the standard 3-step phenol-chloroform method [19]. Sequences from two mitochondrial loci (mtDNA: cytochrome b, Cyt b; 16S ribosomal RNA, 16S) and exon regions of five widely used single-copy nuclear loci (nuDNA: early growth response protein 2B (EGR2B), interphotoreceptor retinoid-binding protein (IRBP), myosin heavy polypeptide 6 (myh6), recombination activating gene 1 (RAG1), and rhodopsin (RH1)) were amplified and sequenced. The primer pairs and PCR conditions are shown in Additional file 2: Table S1. The amplified fragments were sequenced from both ends using an ABI PRISM 3700 sequencing system with the PerkinElmer BigDye DNA Sequencing Kit according to the manufacturer’s protocol, with the same primers used for PCR (Beijing Tianyi Huiyuan Bioscience and Technology Incorporation, Beijing, China).
Sequences were assembled in SeqMan (DNASTAR, Madison, WI, USA), and then aligned using CLUSTAL W [20] in MEGA v6.0 [21] with default parameters. The accuracy was checked manually. Heterozygous sites were resolved using appropriate International Union of Pure and Applied Chemistry (IUPAC) ambiguity codes. All sequences were deposited in GenBank, and accession numbers are listed in Additional file 2: Table S2.
Multilocus gene tree reconstruction
Because of the linked constraint, Cyt b and 16S were concatenated as one mitochondrial locus. We analyzed the phylogenetic relationships of these Triplophysa species using maximum likelihood (ML) and Bayesian inference (BI) methods. Gene trees for the mitochondrial locus and the concatenation of the nuclear loci were inferred separately. The optimal evolutionary models for each locus (Additional file 2: Table S3) were identified by PartitionFinder 1.1.1 [22] based on the Bayesian information criterion. The ML analyses were implemented in RAxML v8.2.9 [23] using the GTRGAMMA model under the optimal partitioning scheme (Additional file 2: Table S3). Statistical supports for major nodes were estimated from 1000 bootstrap replicates. The BI analyses were performed in MrBayes v3.2.2 [24]. The posterior distributions from BI were obtained by Markov Chain Monte Carlo (MCMC) analyses with one cold chain and three heated chains using the optimal partitioning strategy as above. The samples of the trees and the parameters were drawn every 5000 steps from a total of 50,000,000 MCMC generations, and the first 25% samples were discarded as burn-in. The convergence was judged with the average standard deviation of split frequencies (< 0.01) and the effective sample size (ESS) values (> 200). Both the ML and BI analyses were implemented in the CIPRES Science Gateway (http://www.phylo.org/index.php). To assess the prominent discrepancy between mitochondrial and nuclear topologies, we performed the approximately unbiased (AU) test [25] using the CONSEL package [26].
Genealogical sorting index
According to the topology of the mtDNA, we classified T. stoliczkae into 4 groups (T. stoliczkae-1 to 4). The genealogical sorting index (gsi) [27] evaluated the degree of genealogical exclusivity for labeled groups and quantified the genealogical concordance between mtDNA and nuDNA. The normalized gsi values ranged from 0 (not monophyletic or clade structure absolutely rejected the group labels) to 1 (monophyletic or clade structure perfectly agrees on the group labels) [28]. The gsi analyses were run on each locus trees and combined loci trees. We retained the last 500 rooted trees produced from the BI for the gsi analyses. The statistical significance of the gsi analyses was tested for all trees using 1000 permutations. All analyses were completed using R package genealogicalSorting ver. 0.92 [29].
Species tree reconstruction and divergence time
Conspicuous mitonuclear discordances were detected (AU test, P < 0.001), but the nuclear results were more in line with the morphological data. In view of the potential gene flows and the disproportionately strong influence from mtDNA [30, 31], we used only the nuclear datasets to infer the species trees and date of divergence. The species designation was based on the analyses above. We treated each group as a potential species, e.g., all individuals of T. stoliczkae-1 were treated as species T. stoliczkae-1.
The *BEAST [32] species tree analysis and time estimation were simultaneously conducted in BEAST v1.8.0 [33]. The substitution models for each locus were set according to the results from PartitionFinder 1.1.1 [22] (Additional file 2: Table S3). We chose the Yule process as the species trees prior, and a piecewise linear population size with a constant root as the population size prior. A lognormal relaxed molecular clock with an uncorrelated gamma distribution was used. As no fossil record of Triplophysa fishes was available, we used one secondary calibration estimated from a previous study [34] to calibrate divergence time, constraining the time of the most recent common ancestor (TMRCA) of Triplophysa strauchii in the Ili River and Junggar River to 2.40–3.44 Ma. An accurately dated geological event, the ‘Gonghe movement,’ occurred ca. 0.15 Ma, which caused the separation of the Yellow River and Lake Qinghai [35]. Therefore, we constrained the TMRCA of Triplophysa scleroptera in Lake Qinghai and the Yellow River to 0.15 Ma. The effectiveness of these two time-calibration priors was validated in previous studies [15, 36]. We ran four independent runs for 500 million generations with a sampling frequency of every 5000 generations. The convergence of the MCMC posterior parameters was established by an effective sample size (ESS) (> 200) in Tracer 1.5 [37]. Posterior trees from the four runs were combined after removing the first 10% as burn-in in LogCombiner v1.8.0 [33]. The maximum credibility tree was created in TreeAnnotator v1.8.0 [33].
In addition, we also used STAR [38] and MP-EST [39] methods to infer species trees. These two methods allow missing taxa in some gene trees. We performed BI analyses for each nuclear locus using the best substitution model (Additional file 2: Table S3), setting 50,000,000 MCMC generations runs and sampling every 5000 generations. After removing the first 25% trees as burn-in, we randomly sampled 1000 trees for each locus. These trees were used in the Species Tree Analysis (STRAW) online server (http://bioinformatics.publichealth.uga.edu/SpeciesTreeAnalysis/index.php) to conduct the STAR and MP-EST analyses. We ran each twice to ensure congruence.
Reconstructing ancestral state
Triplophysa stoliczkae is a scrape-feeding fish and has apomorphic character states: broadened sharp lower jaw, screw-like intestine, and degenerated PCAB [11, 12] (Additional file 1: Figure S2). In the genus Triplophysa, nearly all the scrape-feeding species share this combination of characteristics, which implies that scrape-feeding fish is a specific group in Triplophysa. To infer the occurrence of scrape-feeding fish, especially the occurrence of “T. stoliczkae”, we assigned Triplophysa into three simplified types: T. stoliczkae type, other scrape-feeding type, and normal-feeding (predatory) type. The assignment was based on the literatures [11, 12] and our field surveys.
The evolutionary trajectories of these types were reconstructed by the ML approach in Mesquite 2.75 [40]. Considering topological uncertainty, we used 10,000 *BEAST species trees (sampled the combined trees file and conducted it twice to confirm congruence) for this inference. The Markov K-state 1 model was selected, and the character transitions were considered to be disordered and reversible. The transition number between character states was calculated using the “summarize state changes over trees”.
Morphospace construction and convergence estimates
To examine phenotypic changes in the genus Triplophysa, we conducted morphological measurements and combined them with our species tree estimate. Due to typical cave-dwelling characteristics (e.g., eyes disappeared) and the lack of specimens, we removed T. rosa from the analyses. We measured 14 widely used morphometric characteristics, which can fully reflect the morphology of a species, from 176 individuals across 26 species (details in Additional file 3: Table S4). As per Blom et al. [41], we calculated residual values of each trait variation from phylogenetically corrected regressions using log-transformed traits against log-transformed standard length in R package Phytools (function phyl.resid; [42]). To eliminate multicollinearity and reduce the multidimensionality of the data, we conducted a phylogenetically corrected principal component analysis (pPCA) using the residuals of these traits. The scores on the main PC axes were retained and adopted as trait input for subsequent analyses. We then projected the significant PCs on a phylomorphospace (function phylomorphospace; [42]) to evaluate interspecific phenotypic changes.
To further quantify the convergence of “T. stoliczkae”, we calculated the amount of independently evolved similarity within PC scores by both distance-based (C1–4) and frequency-based (C5) measures of convergence in the R package convevol [43]. We performed 500 simulations using Brownian motion along the phylogeny to assess the significance of the observed C values.
Results
Sequence information
We obtained 1140 bp of Cyt b and 1025 bp of 16S rRNA. The five nuclear loci for EGR2B, IRBP, myh6, RAG1, and RH1 were 754 bp, 657 bp, 713 bp, 1505 bp, and 712 bp in final aligned length, respectively. After concatenation, the mtDNA was 2165 bp in length and consisted of 511 parsimony-informative sites. The nuDNA was 4341 bp in length and consisted of 317 parsimony-informative sites.
Phylogenetic analyses
The ML and BI analyses yielded generally congruent phylogenies. In the mtDNA results (Fig. 2), T. stoliczkae clustered into four distinct clades, all of which were monophyletic with high support values (PP = 1; BS = 100). Hence, we labeled T. stoliczkae into four groups (T. stoliczkae-1 to T. stoliczkae-4). Specifically, T. stoliczkae-1 consisted of individuals from the Yangtze River. T. stoliczkae-2 comprised individuals from the Yellow River, the Shiyang River and the Heihe River. T. stoliczkae-3 contained individuals from the Ili River. T. stoliczkae-4 included individuals from the Tarim River, the Indus River, and the Ganges River.

Phylogenetic reconstructions based on mtDNA (left) and nuDNA (right). Both trees are generated from a Bayesian inference. Values on the key nodes indicate the posterior probabilities (PPs) obtained in the Bayesian inference and bootstrap support (BS) from the maximum likelihood analyses (PP/BS). The background colors are denoted as those in Fig. 1. Triplophysa stoliczkae individuals are labeled into 4 groups corresponding to the mtDNA clades, with circles for mtDNA and squares for nuDNA. The genealogical concordance between mtDNA and nuDNA trees are evaluated by the genealogical sorting index (gsi) values listed in the box
The results based on nuDNA recovered similar topologies for T. stoliczkae as those based on mtDNA (Fig. 2). T. stoliczkae-2, T. stoliczkae-3, T. stoliczkae-4 were distinct monophyletic clades with high support values (PP = 1; BS ranged from 91 to 100), while T. stoliczkae-1 formed a clade with two other Triplophysa species (PP = 1; BS = 100), T. chondrostoma and T. rotundiventris. The conservative AU test detected a significant conflict between mtDNA and nuDNA topologies (P < 0.001). The conflict was mainly due to the relationships among other Triplophysa species.
Genealogical sorting indices
We set an accepted priori value of gsi > 0.80. Four labeled Triplophysa stoliczkae groups reached monophyly in the mtDNA test (Table 2). The nuDNA results indicated significant monophyly in the groups except for T. stoliczkae-1 (gsi = 0.64). Individual nuclear loci suggested moderate genealogical divergence in T. stoliczkae-1 (gsi ranged from 0.66 to 0.73). T. stoliczkae-2 reached or was close to reaching monophyly (gsi ranged from 0.78 to 1.00). T. stoliczkae-3 reached monophyly at the EGR2B locus (gsi = 0.99), and moderate genealogical divergence at other nuclear loci (gsi ranged from 0.45 to 0.73). T. stoliczkae-4 recovered robust genealogical exclusivity at all nuclear loci (gsi ranged from 0.90 to 1.00). All analyses rejected the monophyly of T. stoliczkae (gsi ranged from 0.35 to 0.56). The discordance between mtDNA and nuDNA in T. stoliczkae-1 was quantified by relatively low gsi values (from 0.55 to 0.66, Fig. 2).
Species trees and divergence times
The interspecific relationships among the Triplophysa are not easily resolved. Although low support values occurred in a certain number of nodes, we noted that the three coalescent methods recovered generally congruent topologies (Fig. 3). The four T. stoliczkae groups were nested into distinct lineages, but all had moderate support values with their sibling clades. Nevertheless, T. stoliczkae-1 and T. stoliczkae-3 were strongly distinct from the other two T. stoliczkae groups because of strong support at the base of the clades (red dot in Fig. 3). The TMRCA for clade M was approximately 8.25 Ma (95% highest posterior density (HPD): 4.21–14.36 Ma) (Fig. 4a and Additional file 1: Figure S3). The divergence events between “T. stoliczkae” and the other species occurred from approximately 0.10 to 4.51 Ma. The earliest split emerged between T. stoliczkae-4 and its sibling group (c. 4.51 Ma; 95% HPD: 2.12–8.04 Ma), and the latest split occurred between T. stoliczkae-1 and its sibling species (c. 0.10 Ma; 95% HPD: 0.00–0.41 Ma).

Species tree reconstructions of Triplophysa using three different methods based on five nuclear loci. Trees are maximum clade credibility cladograms (w/o branch lengths). Numbers on the nodes denote the posterior support for *BEAST and bootstrap support for STAR and MP-EST.

The evolutionary trajectory of the three simplified types in Triplophysa. a Time-calibrated Bayesian species tree from the *BEAST analysis. Circles on the tips indicate three simplified types, and pie charts at ancestral nodes represent their relative ML supports. The triangle with arrow indicates the direction with unequivocal transition numbers (average). N1-N4 denote four nodes. b Three characteristics of Triplophysa. Lower jaw: broadened, sharp and uncovered by lips (rose red); spoon-like, sharp and uncovered by lips (black red); spoon-like, blunt and covered by lips (gray). PCAB (posterior chamber of air bladder): developed (gray) and degenerated (rose red). Intestine: screw shape (rose red) and zig-zag shape (gray). Details are depicted in Additional file 1: Figure S5. c Climatic events (modified from Favre et al. [17] and Zachos et al. [18]): the red dotted line denotes the major global temperature trends; tildes (∼) represent the Quaternary climate oscillations
Evolution of ancestral state
We considered a state to be reconstructed when the relative likelihood (RL) was greater than 0.87. Although normal-feeding was slightly equivocal (RL = 0.84) for the ancestral (N1) state of Triplophysa fish, it was reconstructed at basal nodes N2 and N3 (RL = 0.88 and 0.99, respectively; Fig. 4a). The states of the remaining nodes were well reconstructed except for N4. In the N4 node, the RL for normal-feeding was 0.51, and the RLs of the other two scrape-feeding types were 0.24 and 0.25. The reconstruction analyses indicated that scrape-feeding most likely stemmed from the normal-feeding at least four times and that T. stoliczkae-1 further experienced morphological convergence with other “T. stoliczkae”-like species (T. stoliczkae-3 and T. stoliczkae-4 and possibly T. stoliczkae-2). The evolutionary tendencies were supported by the ML number of unequivocal transitions (arrows triangle in Fig. 4a).
Morphometric and convevol analyses
Triplophysa displayed considerable morphological diversity (Fig. 5). The pPCA yielded two significant axes responsible for nearly 80% of the variance within the morphological dataset. PC1 and PC2 explained approximately 48 and 31.8% of the variance, respectively. Four lineages of “T. stoliczkae” clustered together in morphospace and represented a morphological type different from the others (Fig. 5 and Additional file 1: Figure S4). Moreover, these four lineages showed closer phenotypic distances than that of their ancestors.

Phylomorphospace plot of Triplophysa, superimposing the branching patterns of the phylogeny (black lines) onto the first two PC axes. Each dot represents a species phenotypic value (rose red and gray) or a hypothesized ancestral phenotype (black). Rose-red dots denote T. stoliczkae-1 to T. stoliczkae-4 and gray dots indicate other Triplophysa species. Scrape-feeding species are highlighted with a dotted circle
Both distance-based and frequency-based measures found significantly convergent among the “T. stoliczkae” lineages (P ≤ 0.012; Table 3). Analyses revealed a C1 of 0.77, indicating that evolution has closed 77% of the distance among “T. stoliczkae” lineages. Moreover, convergence accounted for 4% of the total evolution in the smallest clade containing four “T. stoliczkae” lineages (C4 = 0.04). These four “T. stoliczkae” lineages were also found to be significantly convergent using the frequency-based measure (C5 = 4; Additional file 1: Figure S4).
Discussion
Convergent evolution in Triplophysa stoliczkae
Conspicuous mitonuclear discordances (Fig. 2; AU test P < 0.001) suggest that the genus Triplophysa presumably has experienced extensive introgression and/or incomplete lineage sorting (ILS), which may also account for the weak statistical support for most branches in the coalescent analyses (Fig. 3). Introgressive events always interfere with a coalescent analysis and cause low statistical support [31, 36], which has been reported in some Triplophysa species [36]. Moreover, although the coalescent analysis can accommodate the ILS, insufficient information in nuclear loci will also cause moderate support values. Both introgression and ILS generally cause counterintuitive phylogenetic relationships in intra- and interspecies [31, 44]. These two factors can explain the moderate coalescent support for the four T. stoliczkae lineages but not the source of the nonmonophyly of T. stoliczkae for two main reasons: (1) introgression has striking impacts on the mtDNA tree, while ILS is prone to give misleading nuDNA results [30, 45]. In our case, the discordances were mainly due to the relationships among other Triplophysa species and not those within the T. stoliczkae (T. stoliczkae-1 see below). Both the mtDNA and the nuDNA trees indicate that “T. stoliczkae” includes four distinct species, and mtDNA results described a clearer and more reasonable topology (Fig. 2). (2) All three species tree methods can accommodate the ILS [32, 38, 39]. However, none of these methods recovered the monophyly of T. stoliczkae (Fig. 3). Hence, we reject the scenario that T. stoliczkae originated only once from a single ancestor. T. stoliczkae-1 forms a monophyletic group with T. chondrostoma and T. rotundiventris in the nuDNA tree (Fig. 2), which may result from the ILS (not introgression). Due to the lower sorting rates of nuDNA, the ILS is more prone to influence nuDNA than mtDNA results, especially among closely related species [30, 45]. Accommodated the ILS, coalescent analyses recover congruent interspecific relationships with the mtDNA tree for these three species (Figs. 2 and 3). In conclusion, T. stoliczkae consists of four distinct lineages that are not closest relatives.
The ancestral state reconstruction analysis identified the independent innovations of the “T. stoliczkae” type in distinct lineages (Fig. 4a). Morphometric and convevol analyses yielded significant phenotypic convergence between the “T. stoliczkae” lineages (Fig. 5; Table 3). These lines of evidence strongly support that T. stoliczkae was the product of convergent evolution. The occurrences of “T. stoliczkae” (c. 4.51–0.10 Ma) were just in the period of intense tectonic movements and dramatic climatic change on the QTP and its peripheral regions [46, 47]. In addition, three typical characteristics of Triplophysa occurred “randomly” in the phylogeny (Fig. 4b). The typical characteristic combination associated with scrape feeding behavior indicated ecological adaptation. Therefore, we speculate that these historical upheavals created similar ecological conditions, which led to the repeated and directed evolution in “T. stoliczkae”.
The TMRCA of clade M occurred at approximately 8.25 Ma (Fig. 4a). Geological studies have suggested that the large-scale intense uplift of QTP began at approximately 8 Ma [46, 47], from which the macro-environment became more severe on the QTP (Fig. 4c) [17, 18]. Palynological studies have indicated that a marked ecological shift towards a drier climate occurred c. 4.5 Ma on the QTP [48] when the earliest split between “T. stoliczkae” and its sibling group was occurring (Fig. 4a). Therefore, it was reasonable to believe that the intense tectonic movements caused radiation of the species. In addition, due to the uplift and the increasingly harsh climate, the species on the QTP experienced high levels of extinction. Rivers and lakes became increasingly oligotrophic. To cope with the growing food shortage, some populations of Triplophysa fishes had to feed on algae with low-nutrient contents on the surfaces of stones and mud [12]. Eventually, the scrape-feeding behavior was fixed in certain populations that had been engaging in normal-feeding (predatory) behavior. A sharp lower jaw and long intestine were required for scrape-feeding fish to consume the algae [12]. The long intestine formed a screw shape. Moreover, scrape-feeding fish lay on stone, scraping with the force produced by the body’s swing, without vertical movement. A degenerated PCAB is more suitable for scrape-feeding fish to maintain a balanced body during feeding. Therefore, these typical features are found in all scrape-feeding Triplophysa fishes. Furthermore, without vertical movement, in comparison to an inflated and lateral opening of an air bladder, a moderate and lateral closed bony capsule of an air bladder is more suitable for scrape-feeding fish because of the reduced demand for sensing water pressure changes [12]. Both a thick caudal peduncle and emarginate caudal fin are also suitable for scrape-feeding behavior [11, 12]. The optimization of morphological structure strengthens convergent evolution (such as T. stoliczkae-1). The innovation of scrape feeding, coupled with associated morphological adaptations, led to convergent evolution in T. stoliczkae.
Implications for taxonomy
Triplophysa stoliczkae is the most controversial species in the genus Triplophysa [11,12,13]. Because of the cursory morphological description, many species (T. tibetana, T. brevicauda, T. leptosoma, T. stenura, T. tenuicauda, T. stewarti) were once mistaken as T. stoliczkae [11, 12, 49, 50]. Currently, we refer to the revised classification [11, 12], which still considers T. stoliczkae as a single species spreading throughout the QTP. However, our results show that T. stoliczkae should be reclassified into four separate species. In addition, previous studies have also described subtle morphological differences among different “T. stoliczkae” populations [11, 12]. T. stoliczkae was first described from Lake Tsumuriri in the upper Indus River system [51]. Therefore, we suggest retaining the name T. stoliczkae for populations in the Indus River system, as well as the populations in the Ganges River and the Tarim River. Triplophysa dorsonotatus has been described from the Kunes River (Ili River system) but was later synonymized with T. stoliczkae [11, 12, 52]. We suggest recommissioning the name T. dorsonotatus for the “T. stoliczkae”-like species from the Ili River system. The population in the Yangtze River is a distinct species and populations in the Yellow River, the Shiyang River, and the Heihe River are from another separate species. Further descriptions are needed to redefine the two species. Our work, with a limited number of individuals per species, represents a conservative study. Additional samples might reveal additional T. stoliczkae lineages.
Conclusions
In summary, T. stoliczkae presents a striking case of morphological convergence. The harsh climate that has occurred since the Pliocene forced some populations of the Triplophysa fishes to evolve from predatory feeding to scrape feeding. This change in feeding behavior together with associated morphological adaptations caused morphological convergence and created the observed widespread “T. stoliczkae” species. A further taxonomic revision of T. stoliczkae is advisable.
Availability of data and materials
All nucleotide sequences are available in GenBank [MG725380-MG725614, MG735502-MG735539].
References
McGhee GR. Convergent evolution: limited forms most beautiful. Cambridge: MIT Press; 2011.
Alejandrino A, Puslednik L, Serb JM. Convergent and parallel evolution in life habit of the scallops (Bivalvia: Pectinidae). BMC Evol Biol. 2011;11:164.
Harmon LJ, Kolbe JJ, Cheverud JM, Losos JB. Convergence and the multidimensional niche. Evolution. 2005;59(2):409–21.
Blackledge TA, Gillespie RG. Convergent evolution of behavior in an adaptive radiation of Hawaiian web-building spiders. Proc Natl Acad Sci U S A. 2004;101(46):16228–33.
Ellingson RA, Swift CC, Findley LT, Jacobs DK. Convergent evolution of ecomorphological adaptations in geographically isolated bay gobies (Teleostei: Gobionellidae) of the temperate North Pacific. Mol Phylogenet Evol. 2014;70:464–77.
Muschick M, Indermaur A, Salzburger W. Convergent evolution within an adaptive radiation of cichlid fishes. Curr Biol. 2012;22(24):2362–8.
Scheffer M, van Nes EH. Self-organized similarity, the evolutionary emergence of groups of similar species. Proc Natl Acad Sci U S A. 2006;103(16):6230–5.
Miller TE, Powell E. When can competition for resources lead to ecological equivalence? Evol Ecol Res. 2010;12(7):843–54.
Miyazaki JI, Dobashi M, Tamura T, Beppu S, Sakai T, Mihara M, Hosoya K. Parallel evolution in eight-barbel loaches of the genus Lefua (Balitoridae, Cypriniformes) revealed by mitochondrial and nuclear DNA phylogenies. Mol Phylogenet Evol. 2011;60(3):416–27.
Ukuwela KD, de Silva A, Fry BG, Lee MS, Sanders KL. Molecular evidence that the deadliest sea snake Enhydrina schistosa (Elapidae: Hydrophiinae) consists of two convergent species. Mol Phylogenet Evol. 2013;66(1):262–9.
Wu Y, Wu C. The fishes of the Qinghai-Xizang plateau. Chengdu: Sichuan Publishing House of Science and Technology; 1992.
Zhu S. The loaches of the subfamily Nemacheilinae in China (Cypriniformes: Cobitidae). Nanjing: Jiangsu Science and Technology Publishing House; 1989.
Prokofiev A. Materials towards the Revision of the Genus Triplophysa Rendahl, 1933 (Cobitoidea: Balitoridae: Nemacheilinae): A Revision of Nominal Taxa of Herzenstein (1888) Described within the Species “Nemachilus” stoliczkae and “N.” dorsonotatus, with the Description of the New Species T. scapanognatha sp. nova. J Ichthyol. 2007;47(1):1–20.
He D, Chen Y, Chen Y. The molecular phylogeny and biogeography of genus Triplophysa. Prog Nat Sci. 2006;16:1395–404.
Feng C, Wu Y, Tian F, Tong C, Tang Y, Zhang R, Li G, Zhao K. Elevational diversity gradients of Tibetan loaches: the relative roles of ecological and evolutionary processes. Ecol Evol. 2017;7:9970–7.
Wang Y, Shen Y, Feng C, Zhao K, Song Z, Zhang Y, Yang L, He S. Mitogenomic perspectives on the origin of Tibetan loaches and their adaptation to high altitude. Sci Rep. 2016;6:1–10.
Favre A, Päckert M, Pauls SU, Jähnig SC, Uhl D, Michalak I, Muellner-Riehl AN. The role of the uplift of the Qinghai-Tibetan plateau for the evolution of Tibetan biotas. Biol Rev. 2015;90(1):236–53.
Zachos J, Pagani M, Sloan L, Thomas E, Billups K. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science. 2001;292(5517):686–93.
Sambrook J, Fritsch EF, Maniatis T. Molecular cloning, vol. 2. New York: Cold Spring Harbor Laboratory Press; 1989.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9. https://doi.org/10.1093/molbev/mst197.
Lanfear R, Calcott B, Ho SY, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29(6):1695–701.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42.
Shimodaira H. An approximately unbiased test of phylogenetic tree selection. Syst Biol. 2002;51(3):492–508.
Shimodaira H, Hasegawa M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001;17(12):1246–7.
Cummings MP, Neel MC, Shaw KL. A genealogical approach to quantifying lineage divergence. Evolution. 2008;62(9):2411–22.
Chen G, Hare MP. Cryptic diversity and comparative phylogeography of the estuarine copepod Acartia tonsa on the US Atlantic coast. Mol Ecol. 2011;20(11):2425–41.
Bazinet AL, Neel MC, Shaw KL, Cummings MP. The genealogical sorting index: software and web site for quantifying the exclusivity of lineages. 2008. http://www.genealogicalsorting.org.
McLean BS, Jackson DJ, Cook JA. Rapid divergence and gene flow at high latitudes shape the history of Holarctic ground squirrels (Urocitellus). Mol Phylogenet Evol. 2016;102:174–88.
Leaché AD, Harris RB, Rannala B, Yang Z. The influence of gene flow on species tree estimation: a simulation study. Syst Biol. 2014;63(1):17–30.
Heled J, Drummond AJ. Bayesian inference of species trees from multilocus data. Mol Biol Evol. 2010;27(3):570–80.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–73.
Li G, Peng Z, Zhang R, Tang Y, Tong C, Feng C, Zhang C, Zhao K. Mito-nuclear phylogeography of the cyprinid fish Gymnodiptychus dybowskii in the arid Tien Shan region of Central Asia. Biol J Linn Soc. 2016;118(2):304–14.
Li J, Fang X, Pan B, Zhao Z, Song Y. Late Cenozoic intensive uplift of Qinghai-Xizang plateau and its impacts on environments in surrounding area. Quaternary Sci. 2001;21:381–91.
Feng C, Zhou W, Tang Y, Gao Y, Chen J, Tong C, Liu S, Wanghe K, Zhao K. Molecular systematics of the Triplophysa robusta (Cobitoidea) complex: extensive gene flow in a depauperate lineage. Mol Phylogenet Evol. 2019;132:275–83.
Rambaut A, Drummond A. TRACER version 1.5. 2007. http://beast.bio.ed.ac.uk/Tracer.
Liu L, Yu L, Pearl DK, Edwards SV. Estimating species phylogenies using coalescence times among sequences. Syst Biol. 2009;58(5):468–77.
Liu L, Yu L, Edwards SV. A maximum pseudo-likelihood approach for estimating species trees under the coalescent model. BMC Evol Biol. 2010;10(1):302.
Maddison WP, Maddison DR. Mesquite: A Modular System for Evolutionary Analysis. 2.75 ed; 2011. http://mesquiteproject.org
Blom MPK, Horner P, Moritz C. Convergence across a continent: adaptive diversification in a recent radiation of Australian lizards. Proc Biol Sci. 2016;283(1832):20160181.
Revell LJ. Size-correction and principal components for interspecific comparative studies. Evolution. 2009;63(12):3258–68.
Stayton CT. The definition, recognition, and interpretation of convergent evolution, and two new measures for quantifying and assessing the significance of convergence. Evolution. 2015;69(8):2140–53.
Joly S, McLenachan PA, Lockhart PJ. A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am Nat. 2009;174(2):E54–70.
Leavitt DH, Marion AB, Hollingsworth BD, Reeder TW. Multilocus phylogeny of alligator lizards (Elgaria, Anguidae): testing mtDNA introgression as the source of discordant molecular phylogenetic hypotheses. Mol Phylogenet Evol. 2017;110:104–21.
Li J, Fang X, Song C, Pan B, Ma Y, Yan M. Late Miocene–quaternary rapid stepwise uplift of the NE Tibetan plateau and its effects on climatic and environmental changes. Quat Res. 2014;81(3):400–23.
Li J, Fang X. Uplift of the Tibetan plateau and environmental changes. Chin Sci Bull. 1999;44(23):2117–24.
Li J, Wang L, Pei Y, Peng S. Significant paleovegetation shift at ~415 Ma recorded in the red clay sequence of the Central China loess plateau and its link with the Qinghai-Tibet uplift. Earth Sci Front. 2006;13(5):3422348.
Herzenstein SM. Fische. Band iii, Abth 2. Zoologischer Theil; 1888.
Lloyd R. Report on the fish collected in Tibet by Capt. FH Stewart, IMS. Rec Indian Mus. 1908;2(4):341–4.
Ichthyologische SF, Mittheilungen VI. Zur fisch-fauna Kaschmirs und benachbarten Landerstriche. Verh Zool Bot Gesell Wien. 1866;16:761–96.
Kessler KF. Beitrare zur Ichthylogie von Central-Asien. Mèl Biol Bull Acad Sci St Petersb. 1879;25:282–310.
Acknowledgements
We extend our sincere gratitude to the editor and anonymous reviewers for their constructive comments that helped to improve the manuscript. We appreciate Drs. Tim Shaw, Liang Liu, Bryan McLean, Ning Ding, and Jinmin Chen for their kind assistance and suggestions. We also thank two free public resources, ArcGIS Online service, and Natural Earth database, for providing the raw maps in Fig. 1.
Funding
This work was supported by grants from the National Natural Science Foundation of China [31870365 and 31572258 to K.Z., 31500306 to C.Z. and 31700325 to F.T.]. Funds 31870365 and 31572258 funded the sampling work. Funds 31572258, 31500306, and 31700325 supported laboratory works and sequencing. Funds 31870365 funded the English language editing service.
Author information
Authors and Affiliations
Contributions
C.F. and K.Z. conceived the ideas and led the writing. C.F., Y.T., and C.Z. conducted sample measurement and laboratory works (e.g. DNA extraction, PCR, etc.). C.F., Y.T., and S.L. performed the analyses. F.T., Y.T., and S.L. revised the manuscript. C.F., Y.T., S.L., and K.Z. took part in the field works. K.Z., C.Z. and F.T. funded this work. All authors gave the final approval for publication.
Corresponding author
Ethics declarations
Ethics approval
All animal experiments for this project were approved by the Ethics Committee of the Northwest Institute of Plateau Biology, Chinese Academy of Sciences [NWIPB201503018]. No human subjects were used in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Photo of the study species, Triplophysa stoliczkae in Lake Pangong, which is part of the Indus River system. Figure S2. Triplophysa stoliczkae. a Lateral view. b Ventral view (mouth). c Air bladder. d Intestine. c–d Data from Wu and Wu [1]. Figure S3. A time-calibrated Bayesian species tree from the *BEAST analysis. Numbers near the nodes indicate mean values (95% highest posterior density) of divergence time (Ma). Figure S4. A frequency-based measure of convergence. A phylomorphospace of Triplophysa based on the first two PC axes (pPCA, Fig. 4a) is shown. Dots in the circle represent the four focal taxa of interest (T. stoliczkae-1 to T. stoliczkae-4). The purple ellipse indicates the phenotypic space of these focal taxa. Red arrows indicate four lineages that cross into this ellipse. Figure S5. a–b Intestine: a. screw shape (Triplophysa stoliczkae), b. zig-zag shape (T. siluroides). c–e Air bladder: c. PCAB (posterior chamber of air bladder) completely degenerated (T. stoliczkae), d. PCAB degenerated and almost invisible to the eye (T. robusta), e. PCAB developed (T. tibetana). f–h Lower jaw: f. broadened, sharp and uncovered (T. stoliczkae), g. spoon-like, sharp and uncovered (T. stenura) and h. spoon-like, blunt and covered by lips (T. siluroides). Data a–d from Wu and Wu [1]. Data e from Zhu [2]. Data g–h from Wu and Wu [1]. (DOCX 872 kb) (DOCX 871 kb)
Additional file 2:
Table S1. Primers used in this study and PCR conditions. Table S2. GenBank accession numbers for the specimens included in this study. Sample ID corresponds to Fig. 1. Table S3. The optimal partitioning scheme and substitution models for each locus. (DOCX 45 kb) (DOCX 44 kb)
Additional file 3:
Table S4. 14 morphometric characters used in the morphospace construction. Including standard length (SL), preanal length (PreanalL), preanus length (PreanusL), prepelvic length (PrepelvicL), caudal peduncle length (CPL), head length (HeadL), prepectoral length (PrepectoralL), snout length (SnoutL), postorbital length (PostorbitalL), interorbital width (InterorbitalW), eye diameter (EyeD), head depth (HeadD), body depth (BodyD), and caudal peduncle depth (CPD). (CSV 15 kb) (CSV 14 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Feng, C., Tang, Y., Liu, S. et al. Multiple convergent events created a nominal widespread species: Triplophysa stoliczkae (Steindachner, 1866) (Cobitoidea: Nemacheilidae). BMC Evol Biol 19, 177 (2019). https://doi.org/10.1186/s12862-019-1503-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-019-1503-3