
Abstract
Background
Congruent patterns in the distribution of biodiversity between regions or habitats suggest that key factors such as climatic and topographic variation may predictably shape evolutionary processes. In a number of tropical and arid biomes, genetic analyses are revealing deeper and more localised lineage diversity in rocky ranges than surrounding habitats. Two potential drivers of localised endemism in rocky areas are refugial persistence through climatic change, or ecological diversification and specialisation. Here we examine how patterns of lineage and phenotypic diversity differ across two broad habitat types (rocky ranges and open woodlands) in a small radiation of gecko lizards in the genus Gehyra (the australis group) from the Australian Monsoonal Tropics biome.
Results
Using a suite of approaches for delineating evolutionarily independent lineages, we find between 26 and 41 putative evolutionary units in the australis group (versus eight species currently recognised). Rocky ranges are home to a greater number of lineages that are also relatively more restricted in distribution, while lineages in open woodland habitats are fewer, more widely distributed, and, in one case, show evidence of range expansion. We infer at least two shifts out of rocky ranges and into surrounding woodlands. Phenotypic divergence between rocky ranges specialist and more generalist taxa is detected, but no convergent evolutionary regimes linked to ecology are inferred.
Conclusions
In climatically unstable biomes such as savannahs, rocky ranges have functioned as zones of persistence, generators of diversity and a source of colonists for surrounding areas. Phenotypic divergence can also be linked to the use of differing habitat types, however, the extent to which ecological specialisation is a primary driver or secondary outcome of localised diversification remains uncertain.
Similar content being viewed by others


Background
Contrasting patterns of biodiversity across spatial and climatic gradients spanning broad regions are a key focus of ecological and evolutionary research [1, 2]. Striking variation in the constitution, relative distribution, and evolutionary distinctiveness of biodiversity can also exist at more local scales, for instance, along elevational gradients [3,4,5] or between islands of varying size [6, 7]. Where consistent patterns of differences in biodiversity occur across habitats, regions and/or taxa, it raises the possibility that key geological, climatic or ecological factors and processes may be predictably shaping these biodiversity patterns, and may also be identifiable.
An emerging distributional pattern in several parts of the world, especially in tropical areas, is that geologically stable and exposed rocky-range systems such as escarpments, limestones or granite outcroppings are associated with localised phylogenetically divergent and/or specialised lineages [8,9,10]. In contrast, surrounding and often more continuous habitats such as woodlands or forests tend to have much more widespread lineages [11,12,13,14]. Two, not necessarily exclusive, processes may explain contrasting patterns of diversity and distribution between rocky ranges and surrounding habitats: 1) refugial persistence in microrefugia such as deep crevices, caves and/or gorges in rocky areas that are buffered from the extremes of climate [15, 16], and 2) divergent selection and localised adaptation leading to the evolution of ecological specialists that use different habitats [17, 18]. While these two processes may function synergistically, they may also generate some contrasting predictions. The refugial hypothesis would predict that the timings of isolation of saxicoline (rock-dwelling) taxa should correspond with periods of climatic stress such as aridification or increasing seasonality. These isolated taxa/lineages may also be predicted to have disjunct distributions, with relatives occurring in other putative rocky refugia, and/or other areas with more mesic climates [15, 19]. If ecological diversification and habitat specialisation is driving localised diversity, phenotypic adaptations linked to occupying different niches may be predicted [20], and ecologically divergent sister species should occur in the neighbouring habitats [21].
Here we examine temporal and spatial patterns of lineage and phenotypic diversity across two broad habitat types in a single lizard radiation in the Australian Monsoonal Tropics (AMT). The AMT spans northern Australia from the Kimberley region in the west, to Cape York in the east. The biome is characterised by a tropical savannah climate with a short, hot and high-rainfall wet season over the austral summer, and a longer warm to hot dry season with little to no rainfall [22]. The AMT landscape has only low (mostly < 500 m) ranges and plateaus and is geologically stable. However, recent palaeoecological analyses point to a highly dynamic climate history that mirrors that of the arid zone to the south – shifting from relatively arid (end Miocene) to mesic (early to mid-Pliocene), then back towards aridity with an increasingly marked monsoon (Late Pliocene and Pleistocene) [23, 24]. During the last glacial maximum, evidence also points to a weakened monsoon, increased aridity and potential contraction of woodlands to the north, to be replaced by grasslands or steppe [25, 26].
Within the AMT savannah biome there are two broad habitat types. The majority of the region consists of relatively flat open woodlands with varying densities of trees. Nested within these woodlands are numerous low (generally at most a few hundred metres above the surrounding landscape) and, to varying degrees, isolated rocky ranges and plateaus of varying size [22]. While these ranges are generally not high enough to generate their own climate, many AMT taxa nonetheless show evidence of localised and often long-term persistence in rocky ranges, especially in areas of sandstone or limestone karst [9, 27,28,29]. Comparatively, taxa in surrounding savannahs and open woodlands have wider distributions, often shallower genetic divergences and sometimes show evidence of expansion [29, 30]. Broadly, there is also an emerging pattern that lineages in higher rainfall zones of the northern AMT have smaller distributions than relatives in the more arid deserts to the south [27, 31]. Patterns of morphological variation in lineages that occur across rocky ranges and surrounding woodlands in the AMT are less well known. However, in one radiation convergent evolution of specialised rock- and tree-dwelling ecomorphs has been inferred [32], providing evidence that adaptation and ecological diversification may also have played a role in shaping differing patterns of morphological diversity across habitats.
The scansorial geckos in the australis group (genus Gehyra), currently comprise eight recognised species. Five species are only found in close association with rocky ranges and escarpments (Fig. 1; Additional file 1) and use rock crevices as daytime retreats, even though some forage on trees as well as rocks at night; we refer to these taxa as ‘saxicoline’ [30]. Three species in this complex are most often found on trees in open country with no rocky microhabitats (Fig. 1; Additional file 2), but can forage on rock surfaces in the absence of saxicoline taxa. These are referred to here as habitat ‘generalists’ following recent convention [14, 34]. Other smaller-bodied species of Gehyra co-occur in the AMT as part of the phylogenetically distant variegata group, but these are almost entirely restricted to rocky habitats and show deep phylogeographic structure [35]. Similarly, the distinct Australian Arid Zone (AAZ) radiation of the variegata group has deep phylogeographic diversity in the Pilbara craton (central Western Australia [WA]) and Central Uplands (central Australia) refugia, contrasting with widespread generalist taxa in the rest of the AAZ [14, 36]. An initial multilocus phylogeographic analysis of australis group taxa in the eastern half of the AMT (Northern Territory [NT]–Queensland [QLD]) revealed substantial phylogeographic structure in both saxicoline and generalist taxa in the rocky ranges of the Carpentarian sandstones and Selwyn Range, providing evidence that this region has functioned as an evolutionary refugium at the AMT/AAZ interface [37].

Representative geckos in the australis group and their habitats. a) Gehyra australis from open woodlands, b) Gehyra robusta from sandstone ranges, and c) Gehyra koira from limestone ranges. Photographs by Paul Oliver, Paul Horner and Stephen Zozaya
Here we build on this previous work by a) expanding sampling to include all taxa in the australis group from across the AMT, b) strengthening the phylogenetic framework using an exon capture approach, and c) incorporating phenotypic data into analyses of ecological diversification. Using this dataset, we first test the hypothesis that within a single radiation, rocky ranges would support more localised genetic and lineage diversity than surrounding woodlands. We then test for refugial dynamics as a potential driver of endemism within rocky ranges by estimating the pattern and timing of isolation in rocky habitats, and how this corresponds with late Quaternary climatic change. Finally, to investigate the extent to which adaptive ecological diversification has occurred we tested for phenotypic divergence between habitat types (saxicoline vs. generalist) and convergence within habitat types, trends that have both been detected in a co-distributed lizard lineage [32].
Methods
mtDNA assessments of diversity
For an initial assessment of evolutionary diversity and relationships we amplified the full mitochondrial (mtDNA) ND2 gene using primers and protocols outlined in detail elsewhere [38]. Our sampling for the australis group included 774 unique individuals (Additional file 3: Appendix 1), comprising the majority of pre-existing samples from museum collections across Australia, and an extensive collection of new material from recent fieldwork by ourselves and collaborators across the AMT. We primarily focused on the more densely sampled Gulf of Carpentaria [NT–QLD], the Top End [NT], Victoria River [NT] and the Kimberley [WA] regions, but also included samples from related taxa in more poorly sampled areas of southern New Guinea, the Torres Strait Islands, and eastern Queensland (Fig. 2). Sequence information for newly sampled specimens is lodged on GenBank (accessions MK612781 – MK613064).

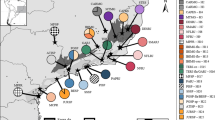
Top: Summary maximum likelihood tree (with bootstrap supports) of mitochondrial (mtDNA) lineage diversity from 774 samples in the Gehyra australis group, with lineages colour-coded by ecology (red = saxicoline [rocks only], blue = generalist). Bottom: Distribution and sampling of major mtDNA lineages in the australis group across the Australian Monsoonal Tropics (AMT): a) G. australis complex [AMT], b) CYsp/G. catenata/G. dubia clades [QLD], c) G. koira complex [Kimberley/Victoria River regions], and d) G. borroloola-G. robusta/G. pamela/Groote clades [Top End/Gulf region]. Scale bar of phylogenetic tree corresponds to substitutions per site. WA = Western Australia, NT = Northern Territory, QLD = Queensland, NSW = New South Wales, SA = South Australia
Within the mtDNA dataset, we first identified candidate lineages using the shallowest net divergence between two recognised taxa (Gehyra ipsa and G. koira) as a cut off. To provide a more objective test, we also quantified phylogeographic structure using bGMYC v1.0.2 [39]. We ran bGMYC for 100 MCC chains on a maximum likelihood tree estimated from all unique haplotypes using RAxML v8.2.8 [40] and made ultrametric using the R package phangorn v2.4.7 [41]. bGMYC provides a posterior probability that two sequences belong to the same interbreeding population, which, along with a probability threshold, can be used to determine the number of clusters present. We only recognised clusters above a relatively high posterior probability threshold of 0.9. Using lineages identified by these methods as a guide, we then selected multiple geographically dispersed individuals (mean n = 3) from each candidate lineage for genomic analysis via exon capture. Exon capture sampling was particularly focused on divergent mtDNA lineages from areas of parapatry where hybridisation and other forms of introgression would be expected if lineage divergence (speciation) were incomplete.
Exon capture
Methods for generating and processing exon capture data, and for downstream estimation of phylogenetic trees largely follow those outlined elsewhere [35]. Exon capture probes targeted four commonly used phylogenetic genes BDNF, C-mos, PDC and RAG1, and 1716 other protein-coding exon regions identified from Gehyra nana [42] and G. oceanica [43] transcriptomes. Probes were designed against target exons and synthesised as a SeqCap EZ NimbleGen in-solution capture system. Previous studies have demonstrated that such probes are highly effective for target enrichment across clades considerably older than Gehyra [44]. The pooled sample library was hybridised to these probes and amplified by ligation-mediated PCR using the SeqCap EZ Developer Library (NimbleGen) protocol, modified to include the alternative blocking oligonucleotides [45]. A quantitative PCR was run on aliquots of both pre- and post-hybridisation libraries, to ensure the hybridisation reaction had amplified the two target primers but not the non-target control. The successful post-hybridisation library was sequenced using an Illumina HiSeq2000 system at the Biomolecular Resource Facility, John Curtin School of Medical Research, ANU.
Exon selection, alignment and concatenated tree estimation
Raw sequencing reads were cleaned and trimmed using the pre-cleanup and scrubReads scripts of the SCPP pipeline [45], using E. coli (K12 MG1655 [46]) and human (GRCh37; Ensembl release 67) genome sequences as contaminant references. Regions flanking the target exons were excluded, cleaned reads were assembled into contigs using a pipeline described in [44], and filtering to base quality >20x and sequence coverage >20x, individually assembled, phased, aligned, and concatenated as described elsewhere [35]. Ambiguously aligned sequence sections and codon columns mainly composed of gap symbols were removed [35].
We obtained exon capture data for 1–11 specimens for most major lineages identified by mtDNA (the exceptions were two mtDNA lineages of G. pamela for which we did not have samples of sufficient quality for exon capture). The final exon capture dataset included 69 samples from the australis group, plus eight outgroup samples (for a total of 77 samples in the overall alignment; see Additional file 3). To gain initial insight into phylogenetic relationships, we used IQ-TREE v1.4.4 [47] to perform a maximum likelihood tree search on the 1634 gene supermatrix; 86 loci were excluded because they had insufficient variation to evaluate substitution models. We then searched for the best-fit substitution model using the option “–m TESTONLY” and applied the HKY substitution model (with 100 bootstrap replicates and random starting trees).
Exon capture lineage delineation
To identify evolutionarily divergent lineages in the exon capture dataset, we used tr2 [48] for discovery and BP&P v3.4 [49] for validation. tr2 assesses observed vs. expected rates of gene tree congruence under coalescent theory for rooted triplets of candidate taxa and a specified guide tree. For this, we used IQ-TREE gene trees generated for the coalescent-based method ASTRAL-II (556 exons with > 90% complete data; see species tree estimation below) and rooted them with the distantly related Gehyra mutilata [50]. Seven gene trees did not contain G. mutilata and were therefore discarded. The 549 rooted gene trees together with an IQ-TREE concatenated phylogeny (see above) as a guide tree were then used by tr2 for lineage discovery.
For the more computationally intensive BP&P validation method [49] we selected 50 loci that contained all individuals and were long (≥693 bp, as a proxy for information content) from the refined alignments and included one haplotype from each of two specimens per lineage (best coverage, n = 48). We focused on testing support for major groupings that were identified across all datasets. Priors mostly followed [51] (small ancestral population and shallow divergence times), however, because newer versions of BP&P use inverse gamma as priors (instead of gamma priors), we set theta: G(3, 0.002) and tau: G(3, 0.002). As recommended for any coalescent analysis, we ran two independent analyses, but with different starting seeds. To save computational time, we ran independent analyses for each clearly monophyletic species complex (G. australis, G. borroloola/robusta, G. koira and G. pamela complexes). Fine-tuning parameters were used as the default and analyses were run for a total of 5,000,000 generations, with burn-in set to 10,000 and sampling every five iterations.
Species tree estimation
Coalescent methods of phylogeny estimation have advantages over concatenation approaches, especially in young radiations [52, 53]. To take advantage of the large number of loci for which we had data, we first used the multi-individual version of ASTRAL-II v4.8.0 [54] to perform tree estimation and multilocus bootstrapping (option ‘–r’) under the multi-species coalescent model using sets of gene and bootstrap trees as input. In order to attain a more complete and informative dataset, loci with less than 90% of all samples and shorter than 300 base pairs were removed. After removal of alignments that did not meet these criteria, 569 loci remained. These were used to create gene trees and bootstrap trees (100 replicates) in IQ-TREE, selecting the HKY substitution model suggested by the IQ-TREE standard model selection (−m TESTONLY option). Due to the high number of invariant sites in some alignments, 13 loci could not be processed successfully, leaving an ASTRAL input of 556 IQ-TREE gene and bootstrap trees. We also quantified the extent of gene tree discordance for the 556 loci dataset by using the ‘-q’ option of ASTRAL-III v5.6.2 [55] to estimate the normalized quartet tree score for the existing tree, as well as the quartet scores for individual nodes.
We also inferred a species tree with StarBEAST2 v0.13.1 [56] implemented in BEAST2 [57]. Given computational constraints, we focused on the same reduced set of 50 loci as the BP&P analyses (see above). The StarBEAST2 analysis was set up in BEASTmasteR v0.1 [58] and run for 100,000,000 generations under the HKY model, calibrated Yule model, and strict lognormal clock. To calibrate the StarBEAST2 analyses we used a secondary age prior (mean 8.5 Mya, SD 2.0) on the node spanning the G. australis to G. dubia complexes taken from a fossil-calibrated analysis of a five gene dataset spanning geckos (alignment from [59], analysis from [9]). Dating analyses of other nuclear gene datasets that used different fossil calibrations, e.g. [60], have estimated similar ages for this same node in Gehyra. Nonetheless, given a suite of potential issues associated with estimating lineage divergence ages from phylogenetic data (e.g., rate variation, tip-age inflation and accuracy of fossil placement and aging), all age estimates should still be treated with caution.
Range size, genetic diversity and demographics across ecology
Species’ habitats were categorised as saxicoline (shortened to “rock”) or generalist (“gen”) following previous convention [14, 34]. This coding captures broad distributional patterns, rather than absolute microhabitat usage rules. Saxicoline taxa use rock crevices as daytime retreats but can forage on trees as well as rocks and are never found away from rocky ranges. Generalists are most typically observed on trees (or buildings) and all range widely into open woodland areas well removed from rocky ranges, but can also be observed foraging on rocks, especially when specialised saxicoline taxa are absent.
Range sizes for each candidate taxon were estimated using convex hulls estimated in ArcGIS v10 [61] around genetically (mtDNA) typed samples, and where relevant (i.e., for limestone-restricted taxa) further constrained by the extent of suitable habitat. For lineages known from very small areas (G. ipsa, plus nine newly identified lineages) we used a 5 km radius buffer around known occurrence sites.
Using the mtDNA dataset we estimated average Tamura-Nei corrected pairwise sequence divergence between and within lineages using MEGA v7.0.26 [62]. DnaSP v5.10.01 [63] was used to calculate various test statistics for population expansion – Tajima’s D [64], Fu’s Fs [65], and Ramos-Onsins and Roza’s R2 value [66], with P-values estimated from coalescent simulations of a 95% confidence interval. For nuclear data, sample sizes per lineage and locus were too small to permit robust tests for population expansion.
Following [67], we calculated the diversification rate (DR) statistic for each taxon (tip) using the StarBEAST2 tree. DR is a summary statistic of the speciation rate derived from the inverse of the branch lengths (i.e., number of splitting events) leading to the particular tip on the tree. We used Phylogenetic Generalized Least Squares (PGLS) in the R package CAPER v0.5.2 [68] and student’s T-tests to test for differences in the log-transformed DR statistic between habitat types, both within the focal radiation from the AMT (i.e., excluding poorly-sampled eastern taxa), and across the entire australis group.
Ancestral states analysis for habitat use
The evolutionary trajectory of habitat use was estimated using BioGeoBEARS v0.2.1 [69]. Six standard biogeographical models were run on the habitat dataset (i.e., saxicoline vs. generalist) in BioGeoBEARS: DEC [70], DIVALIKE [71], and BAYAREALIKE [72], and each with a “+j” variant. In addition, a Markov-k model [73] was also run; the model was constructed in BioGeoBEARS by editing the default DEC model by fixing the parameters d, e, and j to 0, setting the parameter a (for anagenetic range-switching) to be free, and eliminating from the state space the null range and any ranges comprising more than one area. All models were run on the species tree for the australis group estimated from StarBEAST2 as this provides more accurate estimation of branch lengths, especially towards the tips [52]. The outgroup to the australis group is most likely the ecologically diverse and widespread variegata group (Moritz et al. unpublished), and even this remains unconfirmed, so we restricted these analyses to the australis group alone. Models were fit using maximum likelihood, and the fit was compared with AICc (Akaike’s information criterion corrected for sample size) model weights [74]. Ancestral state estimates were made under each model.
Morphological data and analyses
Mensural and meristic data were taken from a total of 231 specimens, 87% of which were typed for mtDNA. Some specimens from taxonomically unambiguous localities were included to increase sample sizes for four taxa: G. catenata, G. dubia, G. ipsa and G. pamela (Additional file 3). We attempted to include all lineages that were recovered with high support in the exon-based phylogenetic analyses, however, there were insufficient specimens for four lineages (Groote, pamela 4 and 5, robusta 4), and in two instances pairs of ecologically similar sister lineages were pooled to increase sample sizes (borroloola 1 + 2 and dubia 1 + 2). Twelve ecologically relevant morphological traits were measured, following [75]: snout-vent length (SVL), trunk length (TrunkL; body length between legs), trunk width (TrunkW; width between ventral skin folds of forelegs), foreleg length (ForelegL; elbow to heel of palm), hindleg length (HindlegL; knee to heel of palm), head length (HeadL; tip of snout to anterior edge of ear), head depth (HeadD; at deepest point behind eyes), head width (HeadW; at widest point behind eyes), snout length (SnoutL; anterior edge of eye to tip of snout), snout depth (SnoutD; deepest part of head in front of eyes), toe length (ToeL; base of toe to tip of toe pad), and number of lamellae on fourth right toe pad (excluding distal wedge). Tail dimensions were not recorded as tails were often damaged or missing in museum specimens. Measurements were made to the nearest 0.01 mm using digital callipers (right-hand side of the body, dorsal view).
All statistical analyses were performed using the log-transformed intra-lineage means for each trait using the R packages Picante v1.6–2, Phytools v0.5–38, GEIGER v2.0.3 and NLME v3.1–122 [76,77,78,79]. Traits other than SVL were corrected for size by regression against SVL (with the residual values being used in analyses). Previous studies have shown that sexual dimorphism is not pronounced in Gehyra [14] so we pooled data from males and females.
To initially explore patterns in the phenotypic data, non-phylogenetic principal component analyses (PCAs) were performed on the size-corrected morphological traits (SVL was excluded). PCAs were visualised using phylo-morphospace plots (Phytools), where the phylogeny is overlaid on the plot of morphological variation. Phylogenetic PCA methods were not used as their effectiveness when models of trait evolution differ has been criticised [80]. One-way analyses of variance (ANOVAs) were used on the major PCAs to compare morphology among lineages, with habitat (saxicoline vs. generalists) as the explanatory factor.
The mean values for each morphological trait were further examined using PGLS with ecology (habitat) as a predictor. These analyses included SVL as this is an important trait in Gehyra [35], and size-corrected scores for all other variables. For each trait we tested the fit of either Brownian Motion (BM) or Ornstein-Uhlenbeck (OU) models of trait evolution (comparing AICc scores in GEIGER), and ran PGLS with the model that had the best fit.
Finally, using two comparative approaches implemented in R – SURFACE [81] and l1OU [82] – we evaluated whether lineages that have independently shifted into similar habitats have also converged on similar phenotypic optima. We ran both analyses using species’ means for log-transformed SVL and the size-corrected residuals for each log-transformed trait. SURFACE uses a stepwise corrected AICc approach to fit Hansen models and evaluates the most optimal set of evolutionary regimes and regime shifts. The package l1OU employs LASSO (least absolute shrinkage and selector operator) to determine the optimal number of selective regimes in a phylogeny. l1OU applies different Ornstein–Uhlenbeck models to the phylogeny to determine how many different selection regimes are needed to explain the data, and then tries to collapse regimes together. Convergence is indicated by either identical (collapsed) or very similar sets of OU parameters in distantly related taxa.
Results
Lineage delimitation from mtDNA and exon datasets
Within the well-sampled Gulf, Top End and Kimberley regions, we provisionally identified 22 candidate lineages in the mtDNA dataset that showed equal or greater divergence than recognised taxa (Fig. 2). Net sequence divergence among intraspecific lineages varied from 6.6 to 14.0% (mean = 9.3%), in comparison to 6.0% between G. ipsa and its sister lineage – koira 4 within G. koira (Additional file 4: Table S1). All but one of these divergent lineages fall within species complexes that have previously been described and circumscribed: G. australis complex – four lineages, all generalist; G. borroloola – two lineages, both saxicoline; G. koira complex – five lineages, four saxicoline, one generalist; G. pamela complex – five lineages, all saxicoline; and G. robusta complex – five lineages, all saxicoline. One additional divergent saxicoline lineage from Groote Eylandt (hereafter “Groote”) off the east coast of the Top End (NT) was not obviously associated with any currently recognised species or species complex (Fig. 2).
In the eastern distribution of the australis group our sampling was relatively sparse and we conservatively identified only four generalist lineages: one unrecognised taxon “CYsp” (Cape York and the Trans-Fly region of southern New Guinea), G. catenata and G. dubia (two lineages).
Lineage delineation using bGMYC on mtDNA identified 41 distinct populations across the entire australis group. Populations identified in the G. australis (n = 4 lineages), G. borroloola (n = 2) and G. pamela (n = 5) complexes were identical to those from the ad hoc method outlined above (Additional file 4: Table S2). One additional lineage was delineated in the G. robusta complex (n = 6). Much finer structuring was identified within the G. koira complex, specifically koira 1 (n = 7), and the more poorly sampled taxa from eastern Australia – G. dubia (n = 5) and CYsp (n = 7) complexes.
In all trees based on the extensive exon sequences sampled from across the range of the australis group (Additional file 5: Figure S1), all recognised species are monophyletic, apart from G. koira which is paraphyletic with respect to G. ipsa (concatenated and species trees; Fig. 3). Furthermore, samples from each of the major mtDNA lineages described above were also monophyletic in the concatenated exon capture phylogeny (Fig. 2a). Species delimitation tests using tr2 on 549 gene trees identified 36 distinct lineages in the australis group (Additional file 4: Table S2). In two complexes lineages were entirely congruent with the major mtDNA lineages: G. borroloola complex, n = 2 lineages; G. robusta complex, n = 5 lineages. Additional lineages were identified by tr2 in two other species complexes: G. australis complex (n = 5 vs. 4), and G. pamela complex (n = 5 vs. 3; noting that two mtDNA lineages were missing from the exon capture dataset for this species). However, in the G. koira complex the number of tr2 exon capture lineages greatly exceeded the number of major mtDNA lineages: n = 14 vs. 5, including n = 7 in koira 1, and n = 4 in koira 4. BP&P species delimitation using the exon data supported the distinctiveness of all 22 major candidate lineages identified in the mtDNA dataset with high support (= 1.000). There is also congruence between the mtDNA and the nuclear genome in that no lineages supported by the tr2 analysis of the 549 exons rendered major mtDNA groupings paraphyletic.

Estimates of phylogenetic relationships within the australis group based on exon capture data: a) maximum likelihood phylogeny with bootstrap support values estimated using IQ-TREE and 1634 loci for all individuals (scale bar = substitutions per site), and b) chronogram with Bayesian support values for major lineages estimated using StarBEAST2 and 50 loci, with nodes also strongly supported by ASTRAL (bootstrap support > 95) demarcated with an asterisk (axis = millions of years before present)
In the better sampled Gulf, Top End and Kimberley regions all methods of lineage delimitation identified more evolutionarily distinctive lineages in rocky ranges than surrounding woodlands: 17 saxicoline lineages vs. 5 generalist lineages detected by the ad hoc mtDNA lineages method; 23 vs. 8 using bGMYC of mtDNA; and 22 vs. 9 from exons and tr2. Lineages utilising the same habitat type almost never occurred in sympatry, with the possible exception of the distantly related australis 1 and koira 4 in the Kimberley (Fig. 2). Conversely, saxicoline taxa overlapped with generalist taxa extensively.
Exon capture phylogeny
Topology and nodes with high support were consistent across all analyses of the nuclear datasets (IQ-TREE, ASTRAL-II, StarBEAST2; Fig. 3; Additional file 5: Figure S2). Two lineages from Queensland—CYsp and G. catenata/G. dubia—were always recovered as, respectively, the first and second diverging lineage from the remainder of the australis group. Remaining taxa from across the Gulf, Top End and Kimberley regions formed a strongly supported clade, within which G. pamela from the Arnhem Escarpment (NT) was universally recovered as the sister to a clade comprising the remaining four groups (G. australis complex, G. borroloola + G. robusta complexes, G. koira complex, and Groote). Relationships amongst these four groups tended to be more weakly supported and/or inconsistent across methods (Fig. 3; Additional file 5: Figure S2). Patterns of relationships within species complexes were generally consistent and highly supported (Fig. 3). The one exception was three lineages from the southern Kimberley in the G. koira complex that formed a polytomy (koira 3, koira 4 and G. ipsa), which further contrasted against strong mtDNA support for a close relationship between koira 4 and G. ipsa.
The normalized quartet tree score (a potential measure of incomplete lineage sorting [ILS]) for the whole tree calculated in ASTRAL-III was 0.64. For this measure scores closer to 1.00 suggest low levels of ILS. Quartet scores (i.e. percentage of quartets that agree with branch) for nodes varied but were generally high for deeper nodes in the overall australis group (> 60%), and lower for nodes at the base of the clade including the G. australis complex, G. borroloola + G. robusta complexes, G. koira complex and Groote (Additional file 5: Figure S3).
Based on the secondary age priors we used, three major lineages are estimated to have progressively diverged from the remainder of the australis group during the late Miocene: CYsp (mean 9.2 Mya, 95% credible interval [CI] 6.0–12.7 Mya); G. catenata + G. dubia complex (mean 8.5 Mya, 95% CI 5.5–11.4 Mya); and the G. pamela complex (mean 6.3 Mya, 95% CI 4.0–8.7 Mya). More recent Plio-Pleistocene divergences are inferred for initial, and seemingly relatively rapid, radiation of the clade containing all other lineages (mean 4.1 Mya, 95% CI 2.6–5.5 Mya).
Range sizes, habitat transitions, and demographic history
For the major lineages identified from congruence across mtDNA and exons, saxicoline taxa consistently had ranges an order of magnitude smaller than generalists (Additional file 4: Table S3; mean for all major lineages 12,538 vs. 245,646 km2; mean after excluding singleton site lineages with arbitrary ranges 30,541 vs. 245,646 km2).
In analyses of habitat shifts, the Markov-k (MK) model had the best fit (Additional file 4: Table S4). In all analyses multiple shifts between states were inferred (Fig. 4). Habitat preference at the base of the australis group was ambiguous, however the saxicoline ecology was supported as basal to the clade containing most species from the Top End to Kimberley regions (Groote plus the G. australis, G. borroloola, G. koira, G. pamela and G. robusta complexes). Within this radiation at least two more recent and independent shifts from saxicoline to a more generalist habitat use were also inferred: first, from saxicoline lineages to the generalist (and mostly arboreal) G. australis complex during the Pliocene; and another more recent (Pleistocene) and more clearly resolved shift from saxicoline to generalist (again mainly arboreal) in a lineage largely endemic to the Kimberley (koira 4; Fig. 4).

Estimated ancestral states, and number and trajectory of habitat shifts within the Gehyra australis group under the Markov-k (MK) model as implemented in BioGeoBEARS (red = saxicoline [rocks only], blue = generalist). Note inferred shifts from saxicoline to generalist ecology at base of G. australis complex and within the G. koira complex
Using sequence variation for the better-sampled mtDNA, a genetic signature of population expansion was only found in the G. australis complex (generalist), and more specifically, the most widespread lineage within it (australis 1) (P-value for Fu’s Fs < 0.001; Additional file 4: Table S5).
Across all combinations of taxa (all lineages, eastern lineages excluded) and all statistical tests (PGLS and T-tests) there was no significant difference between saxicoline and generalist habitats in the rate of lineage diversification, as estimated using the DR statistic (Additional file 4: Table S6).
Morphological variation and evolution
Size-corrected PCA based on residuals (rPCA) suggested differentiation by habitat along PC axis 1 (rPC1; heavily loaded to toe length and head length; Additional file 4: Table S7). ANOVA also detected significant differences between saxicoline and generalist taxa in rPC1 (-HeadL and +ToeL; phyloANOVA P-value = 0.015), but not other axes: rPC2 (+HindlegL and -TrunkW) and rPC3 (-SnoutD, -HeadD and -ForelegL; Additional file 4: Tables S7–8).
Univariate analyses of traits using PGLS under either Brownian (BM) or Ornstein-Uhlenbeck (OU) models (whichever was identified as best fit by AICc) found significant differences between the two habitat groups (Additional file 4: Table S9; Additional file 5: Figure S4), with saxicoline taxa tending to be larger (SVL, OU model, P = 0.028); with (after size-correction) relatively longer forelimbs (OU, P = 0.039); relatively longer heads (BM, P = 0.040) and snouts (OU, P = 0.008); relatively shallower heads (OU, P = 0.031) and snouts (BM, P = 0.010); and relatively shorter toes (BM, P = 0.008).
In phylo-morphospace plots of the most variable axes across both univariate and multivariate datasets (logSVL against rPC1; Fig. 5), the G. koira complex was the most dispersed, driven by striking divergence in body size (G. ipsa large, koira 4 small). Two distantly related saxicoline clades (G. borroloola + G. robusta complexes, and G. pamela complex) overlapped, while saxicoline taxa in the G. koira complex were more divergent (much larger and tending to have shorter heads and longer toes). Amongst the generalist taxa the most striking similarity was between koira 4 and relatively distantly related members of the G. australis complex (Fig. 5).

Phylo-morphospace plot of trait means for lineages in the Gehyra australis group based on Principal Component Axis 1 (residuals of size-corrected data; rPC1) against log-SVL. Lineages are colour-coded by ecology (red = saxicoline, blue = generalist) and connected by the StarBEAST2 phylogeny
SURFACE analyses for convergence based on the entire dataset inferred only two regime shifts, both in saxicoline lineages: a) the relatively long-headed and short-toed G. borroloola and G. robusta complexes, and b) the very large species G. ipsa. No convergent regime shifts were identified (Additional file 5: Figure S5). l1OU analyses identified seven regime shifts, comprising the two above, plus additional candidate lineage-specific shifts in the G. australis (n = 1), G. koira (n = 2), G. pamela (n = 1) and G. robusta (n = 1) complexes (Additional file 5: Figure S6). No convergent regimes were identified in either method.
Discussion
Comprehensive mtDNA sampling of diversity in geckos from the australis group (Gehyra) combined with a well-resolved tree from an exon capture dataset shows that lineage diversity is higher in rocky ranges than in surrounding woodlands. Saxicoline taxa also occupy geographic ranges that are an order of magnitude smaller than those of generalists, with more than half these saxicoline taxa being short-range endemics (ranges less than 10,000 km2) [83]. This result accords with other studies in rainforest, savannah and arid habitats that have identified that localised lizard endemism and diversity is often high in lineages associated with rocky ranges [84,85,86], with taxa in surrounding habitats often tending to be more genetically homogeneous and/or widespread [8, 13, 14]. Below, we focus on two, not necessarily exclusive, processes that may underpin these contrasting patterns of diversity and distribution across these interdigitated habitat types: 1) refugial persistence in rocky ranges through climatic change, and 2) localised evolution of habitat specialists across striking habitat gradients.
Refugia – On and off the rocks
Past climatic change has played a critical role in shaping contrasting patterns of distribution and diversity across Australian biomes, regions and habitats [87, 88]. Recent palaeoclimatic data point towards the AMT being a young biome with a dynamic climatic history of arid-mesic-arid cycling that parallels that of the arid zone to the south, shifting from relatively arid at late Miocene, to mesic in the early to mid-Pliocene, then towards increasing aridity and a marked monsoon through the Late Pliocene and Pleistocene [23]. Our estimate of crown age for the radiation of most of the australis group across the Gulf, Top End and Kimberley regions is congruent with this mesic pulse in the early Pliocene. Other phylogenetic studies also point to endemic AMT savannah lineages radiating over this timeframe [27, 30, 31, 89], while older relicts, potentially persisting remnants from an earlier (mid-Miocene) wet phase, tend to have highly disjunct distributions and very low diversity [28]. Critically, the majority of lineages of Gehyra that we estimate to have diversified through major climatic changes since the Late Miocene are restricted to rocky ranges.
Periods of increasing aridity also seem to have reinforced an overall south-to-north gradient of increasingly old and/or finely structured diversity, even within the rock-associated lineages in both the australis group, and other AMT radiations [31, 90]. At one end of the temporal spectrum, to the north a deep (pre-Pliocene) lineage within the main AMT radiation (G. pamela) is restricted to the rocky Arnhem Escarpment of the Top End. This topographically complex, relatively high rainfall region is characterised by high phylogenetic endemism in other taxa [8] including isolated lineages that are either absent, or highly relictual across the remainder of the AMT [91, 92]. In the more arid southern AMT, isolated populations of saxicolous lineages from topographically complex ranges in both the southern Kimberley (G. ipsa) and the Gulf region (robusta 5), suggest more recent (late Pliocene – early Pleistocene) isolation. The apparently disjunct distribution of koira 3 across the southern and eastern Kimberley limestones implies an even more recent range contraction over relatively large areas, perhaps linked to Pleistocene expansion of desert sand dunes [93] from the south over formerly continuous limestone habitats.
Comparisons of mtDNA diversity in the two widespread taxa in the western AMT provide evidence that ecological differences may have shaped contrasting patterns of persistence. The saxicoline koira 1 has strong internal phylogeographic structure, implying multiple areas of persistence. By contrast, the geographically overlapping generalist (mostly arboreal) australis 1 lineage has effectively no internal structure and shows evidence of recent range expansion (Additional file 4: Table S5). In some other low relief tropical landscapes, rocky ranges appear to have functioned as critical refugia during periods of relative aridity over evolutionary timescales [9, 85, 94]. Localised and higher genetic diversity in rocks also suggests that contemporary variation in endemism across habitats may be underpinned by differing probabilities of extinction in response to climatic change [5]. Accordingly, estimation of species' responses to future climatic change in these habitats will also increasingly need to incorporate the potential role and accessibility of microrefugia [95].
In addition to being refuges, our ancestral state analyses further imply that rocky ranges may be sources of recolonisation into surrounding habitats. In the G. koira complex four out of five species are saxicoline, while koira 4, which is deeply nested within the complex, is largely arboreal. Strikingly, koira 4 is similar to co-occurring members of the generalist G. australis complex in body size and also shares a plain dorsal pattern, to the extent that fieldworkers consistently cannot distinguish these two taxa. This seemingly recent ecological and phenotypic shift out of rocky ranges occurs in the west Kimberley, a region that marks the eastern distributional limits of many mesic-dependent taxa and is a recognised hotspot of endemism [96]. Estimation of the trajectories of ecological evolution in Gehyra elsewhere in Australia [14], and other geckos from Australia [28] and overseas [97] also imply that contraction into rocky refugia may not necessarily be an endpoint. Instead, these habitats are inferred to function as sources of diversity for surrounding habitats when (or where) climates are less challenging. Going forward, a major challenge to testing such a model will be controlling for how non-random or differing rates of extinction and anagenesis across habitat types may bias ancestral state estimation [98].
Specialisation on the rocks
Ecological differentiation and adaptation to distinctive microhabitats is an alternative process that could generate localised endemism, even in the absence of population isolation and persistence [17]. One prediction of ecological divergence is that taxa in different habitats may show evidence of associated adaptive divergence or specialisation [21]. In overall univariate analyses we found evidence that the saxicoline taxa in the australis group were larger than generalists, and had longer and shallower heads, longer forelimbs, and shorter toes. Phylo-morphospace plots also suggested that the distantly related saxicoline G. borroloola/robusta and G. pamela complexes tend to have shorter toes and longer heads. Some of these phenotypic shifts, such as flattened body shape, typify rock-dwelling taxa in other lineages of Gehyra [14], other geckos [18], and other radiations of lizards [34, 99, 100]. However, while follow-up SURFACE and l1OU analyses identified divergent morphological regimes, they did not suggest convergence. This contrasts with a geographically overlapping radiation of Australian skinks in the genus Cryptoblepharus that shows a very strong link between body shape and ecology, coupled with clear evidence of convergent evolutionary regimes leading to distinct rock and tree ecomorphs [32]. However, recent work does suggest that in Gehyra, and at least some other lizard radiations, the link between ecology, phenotype (at least as we have scored these data) and lineage diversity may be complex [101, 102], and not as overt as in some classical adaptive radiations of lizards [103]. In this context, it is safe to argue that some phenotypic diversification linked to ecology has occurred in the australis group, but the extent to which ecological diversification may have been a primary driver of higher diversity and localised endemism in rocky ranges in this, and many other systems, remains difficult to test.
Biotic interactions?
While we focus on refugial dynamics and specialisation above, a third process, biotic interactions, may also play an important role in shaping communities and distributions. For instance, in lizards, specialisations to utilise habitats such as rocks, have been linked to a lowered ability to compete in other habitats via mechanisms such as reduced reproductive output or dietary breadth [34]. Notably, we found that australis group taxa with similar habitat preferences almost never overlap geographically, while conversely, saxicoline and generalist taxa are frequently sympatric, even in closely-related lineages such as koira 4 and koira 1. Furthermore, generalist taxa usually use rocks only when saxicolous taxa are absent ([33], Moritz, Oliver, Tedeschi pers. obs.). Recent studies have pointed to rapid shifts in body size and the likely importance of size-based sorting of local assemblages in other lineages of Gehyra [35, 75]. In the west Kimberley, the large saxicoline koira 1 lineage also abuts with the distributional range edge of another saxicoline (and large) taxon, Gehyra xenopus, while the generalist (and much smaller) koira 4 widely overlaps with G. xenopus (authors pers. obs.). These data all support an established pattern that similar-sized and ecologically undifferentiated lizard taxa rarely persist in the same communities [35, 103, 104], underlining the argument that interactions between species play an important role in shaping extant distributional limits [105]. However, testing the extent to which this ecological differentiation is a secondary outcome of speciation events or a primary driver of them remains challenging.
Conclusions
Across a mosaic of habitats in the savannah of northern Australia, rocky ranges appear to have provided climatically-buffered microrefugia through late Cenozoic aridification, mediating localised persistence, diversification and even recolonisation of surrounding woodlands. This result emphasises the likely importance of microrefugia within rocky habitats for persistence through past – and future – climatic changes. Patterns of phenotypic variation and geographic distributions suggest that ecological specialisation and species interactions may have played a complementary role in shaping contemporary diversity patterns; however, the extent to which these latter processes may be primary drivers of lineage divergence remains ambiguous. Going forward, the challenge remains how to find ways to convincingly disentangle the roles these key processes have, and will, play in shaping diversity patterns through changing climates. Integrative studies linking genetic patterns with a detailed understanding of physiology, habitat preferences and biotic interactions offer the potential to meet this challenge [106].
Abbreviations
- AAZ:
-
Australian Arid Zone
- AICc:
-
Akaike’s Information Criterion corrected
- AMT:
-
Australian Monsoonal Tropics
- ANOVA:
-
Analysis of Variance
- BM:
-
Brownian Motion
- DR:
-
Diversification Rate
- mtDNA:
-
mitochondrial DNA
- NSW:
-
New South Wales
- NT:
-
Northern Territory
- OU:
-
Ornstein-Uhlenbeck
- PCA:
-
Principal Component Analysis
- QLD:
-
Queensland
- SA:
-
South Australia
- SVL:
-
snout-vent length
- WA:
-
Western Australia
References
Janzen DH. Why mountain passes are higher in the tropic. Am Nat. 1967;101:233–49.
Willig MR, Kaufman DM, Stevens RD. Latitudinal gradients of biodiversity: pattern, process, scale, and synthesis. Annu Rev Ecol Evol Syst. 2003;34:273–309.
Rahbek C. The elevational gradient of species richness: a uniform pattern? Ecography. 1995;18:200–5.
Tallowin O, Allison A, Algar AC, Kraus F, Meiri S. Papua New Guinea terrestrial-vertebrate richness: elevation matters most for all except reptiles. J Biogeogr. 2017;44:1734–44 Available from: http://doi.wiley.com/10.1111/jbi.12949.
Quintero I, Jetz W. Global elevational diversity and diversification of birds. Nature. 2018;555:246–50 Available from: http://www.nature.com/doifinder/10.1038/nature25794.
MacArthur RM, Wilson E. The theory of island biogeography. Monographs in population biology. Vol. 1. Princeton, NJ: Princeton University Press; 1967.
Lavery TH, Olds AD, Seddon JM, Leung LKP. The mammals of northern Melanesia: speciation, ecology, and biogeography. Mamm Rev. 2016;46:60–76.
Rosauer DF, Blom MPK, Bourke G, Catalano S, Donnellan S, Gillespie G, et al. Phylogeography, hotspots and conservation priorities: an example from the top end of Australia. Biol Conserv. 2016;204:83–93 Available from: https://doi.org/10.1016/j.biocon.2016.05.002.
Oliver PM, Laver RJ, De Mello MF, Pratt RC, Hunjan S, Moritz CC. A novel hotspot of vertebrate endemism and an evolutionary refugium in tropical Australia. Divers Distrib. 2017;23:53–66 Available from: http://doi.wiley.com/10.1111/ddi.12506.
Werneck FP, Nogueira C, Colli GR, Sites JW, Costa GC. Climatic stability in the Brazilian Cerrado: implications for biogeographical connections of south American savannas, species richness and conservation in a biodiversity hotspot. J Biogeogr. 2012;39:1695–706.
Werneck FP, Gamble T, Colli GR, Rodrigues MT, Sites JW. Deep diversification and long-term persistence in the south american “dry diagonal”: integrating continent-wide phylogeography and distribution modeling of geckos. Evolution. 2012;66:3014–34.
Oliver PM, Laver RJ, Melville J, Doughty P. A new species of velvet gecko (Oedura: Diplodactylidae) from the limestone ranges of the southern Kimberley. Western Australia Zootaxa. 2014;3873:49–61.
Pepper M, Ho SYW, Fujita MK, Scott KJ. The genetic legacy of aridification: climate cycling fostered lizard diversification in Australian montane refugia and left low-lying deserts genetically depauperate. Mol Phylogenet Evol. 2011;61:750–9 Available from: https://doi.org/10.1016/j.ympev.2011.08.009.
Ashman LG, Bragg JG, Doughty P, Hutchinson M, Bank S, Matzke N, et al. Diversification across biomes in a continental lizard radiation. Evolution. 2018;72:1553–69.
Couper PJ, Hoskin CJ. Litho-refugia: the importance of rock landscapes for the long-term persistence of Australian rainforest fauna. Aust Zool. 2008;34:554–60.
Kearney M. Hot rocks and much-too-hot rocks: seasonal patterns of retreat-site selection by a nocturnal ectotherm. J Therm Biol. 2002;27:205–18.
Endler JA. Gene flow and population differentiation. Science. 1973;179:243–50.
Nielsen SV, Oliver PM. Morphological and genetic evidence for a new karst specialist lizard from New Guinea (Cyrtodactylus: Gekkonidae). R Soc Open Sci. 2017;4:170781.
Grandcolas P, Nattier R, Trewick S. Relict species: a relict concept? Trends Ecol Evol. 2014;29:655–63 Available from: http://www.sciencedirect.com/science/article/pii/S0169534714002110.
Schluter D. The Ecology of Adaptive Radiation. Oxford Ser. Ecol. Evol. 2000:300.
Moritz C, Patton JL, Schneider CJ, Smith TB. Diversification of rainforest faunas: An integrated molecular approach. Annu Rev Ecol Syst. 2000;31:533–63 Available from: http://www.annualreviews.org/doi/10.1146/annurev.ecolsys.31.1.533.
Bowman DMJS, Brown GK, Braby MF, Brown JR, Cook LG, Crisp MD, et al. Biogeography of the Australian monsoon tropics. J Biogeogr. 2010;37:201–16.
Christensen BA, Renema W, Henderiks J, De Vleeschouwer D, Groeneveld J, Castañeda IS, et al. Indonesian Throughflow drove Australian climate from humid Pliocene to arid Pleistocene. Geophys Res Lett. 2017;44:6914–25.
Andrae JW, McInerney FA, Polissar PJ, Sniderman JMK, Howard S, Hall PA, et al. Initial expansion of C4 vegetation in Australia during the late Pliocene. Geophys Res Lett. 2018;45:1–10.
O’conner S, Frawley S. A 40,000 year wood charcoal record from Carpenter’s Gap 1: New insights into palaeovegetation change and indigenous foraging strategies in the Kimberley, Western Australia. In: Haberle SG, Steveson J, Preble M, editors. Altered Ecol Fire, Clim Hum Influ Terr landscapes (Terra Aust 32); 2010. p. 299–321.
Denniston RF, Asmerom Y, Lachniet M, Polyak VJ, Hope P, An N, et al. A last glacial maximum through middle Holocene stalagmite record of coastal Western Australia climate. Quat Sci Rev. 2013;77:101–12 Available from: http://linkinghub.elsevier.com/retrieve/pii/S0277379113002539.
Laver RJ, Doughty P, Oliver P. Origins and patterns of endemic diversity in two specialized lizard lineages from the Australian Monsoonal Tropics (Oedura spp.). J Biogeogr. 2018;45:142–53.
Oliver PM, Smith KL, Laver RJ, Doughty P, Adams M. Contrasting patterns of persistence and diversification in vicars of a widespread Australian lizard lineage (the Oedura marmorata complex). J Biogeogr. 2014;41:1–12.
Afonso Silva AC, Bragg JG, Potter S, Fernandes C, Coelho MM, Moritz C. Tropical specialist vs. climate generalist: diversification and demographic history of sister species of Carlia skinks from northwestern Australia. Mol Ecol. 2017;26:4045–58.
Potter S, Xue AT, Bragg JG, Rosauer DF, Roycroft EJ, Moritz C. Pleistocene climatic changes drive diversification across a tropical savanna. Mol Ecol. 2017;27:520–3 Available from: http://doi.wiley.com/10.1111/mec.14441.
Laver RJ, Nielsen SV, Rosauer DF, Oliver PM. Trans-biome diversity in Australian grass-specialist lizards (Diplodactylidae: Strophurus). Mol Phylogenet Evol. Elsevier. 2017;115:62–70 Available from: https://doi.org/10.1016/j.ympev.2017.07.015.
Blom MPK, Horner P, Moritz C. Convergence across a continent: adaptive diversification in a recent radiation of Australian lizards. Proc R Soc B Biol Sci. 2016;283:20160181.
Greer AE. The biology and evolution of Australian lizards. Surrey Beatty and Sons: Sydney; 1989.
Pelegrin N, Mesquita DO, Albinati P, Caldas FLS, de Queiroga Cavalcanti LB, Costa TB, et al. Extreme specialization to rocky habitats in Tropidurus lizards from Brazil: trade-offs between a fitted ecomorph and autoecology in a harsh environment. Austral Ecol. 2017;42:677–89.
Moritz C, Pratt RC, Bank S, Bourke G, Bragg JG, Doughty P, et al. Cryptic lineage diversity, body size divergence and sympatry in a species complex of Australian lizards (Gehyra). Evolution. 2018;72:54–66.
Sistrom M, Donnellan SC, Hutchinson MN. Delimiting species in recent radiations with low levels of morphological divergence: a case study in Australian Gehyra geckos. Mol Phylogenet Evol. 2013;68:135–43 Available from: https://doi.org/10.1016/j.ympev.2013.03.007.
Noble C, Laver RJ, Rosauer DF, Ferrier S, Moritz CC. Phylogeographic evidence for evolutionary refugia in the Gulf sandstone ranges of northern Australia. Aust J Zool. 2018;65:408–16.
Sistrom MJ, Hutchinson MN, Hutchinson RG, Donnellan SC. Zootaxa, molecular phylogeny of Australian Gehyra (Squamata: Gekkonidae) and taxonomic revison of Gehyra variegata in South-Eastern Australia. Zootaxa. 2009;32:14–32.
Reid NM, Carstens BC. Phylogenetic estimation error can decrease the accuracy of species delimitation: a Bayesian implementation of the general mixed yule-coalescent model. BMC Evol Biol. 2012;12:196.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Schliep KP. Phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–3.
Bragg JG, Potter S, Bi K, Catullo R, Donnellan SC, Eldridge MDB, et al. Resources for phylogenomic analyses of Australian terrestrial vertebrates. Mol Ecol Resour. 2017;17:869–76.
Tonione MA, Fisher RN, Zhu C, Moritz C. Deep divergence and structure in the tropical oceanic Pacific: a multilocus phylogeography of a widespread gekkonid lizard (Squamata: Gekkonidae: Gehyra oceanica). J Biogeogr. 2016;43:268–78.
Bragg JG, Potter S, Bi K, Moritz C. Exon capture phylogenomics: efficacy across scales of divergence. Mol Ecol Resour. 2016;16:1059–68.
Peñalba JV, Smith LL, Tonione MA, Sass C, Hykin SM, Skipwith PL, et al. Sequence capture using PCR-generated probes: a cost-effective method of targeted high-throughput sequencing for nonmodel organisms. Mol Ecol Resour. 2014;14:1000–10.
Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–62 Available from: http://science.sciencemag.org/content/277/5331/1453.
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Fujisawa T, Aswad A, Barraclough TG. A rapid and scalable method for multilocus species delimitation using Bayesian model comparison and rooted triplets. Syst Biol. 2016;65:759–71.
Yang Z. The BPP program for species tree estimation and species delimitation. Curr Zool. 2015;61:854–65.
Heinicke MP, Greenbaum E, Jackman TR, Bauer AM. Phylogeny of a trans-Wallacean radiation (Squamata, Gekkonidae, Gehyra) supports a single early colonization of Australia. Zool Scr. 2011;40:584–602.
Leaché AD, Fujita MK. Bayesian species delimitation in west African forest geckos (Hemidactylus fasciatus). Proc R Soc B Biol Sci. 2010;277:3071–7 Available from: http://rspb.royalsocietypublishing.org/cgi/doi/10.1098/rspb.2010.0662.
Ogilvie HA, Heled J, Xie D, Drummond AJ. Computational performance and statistical accuracy of∗BEAST and comparisons with other methods. Syst Biol. 2016;65:381–96.
Edwards SV, Potter S, Schmitt CJ, Bragg JG, Moritz C. Reticulation, divergence, and the phylogeography–phylogenetics continuum. Proc Natl Acad Sci. 2016;113:8025–32 Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1601066113.
Mirarab S, Warnow T. ASTRAL-II: Coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics. 2015:44–52.
Zhang C, Rabiee M, Sayyari E, Mirarab S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics BMC Bioinformatics. 2018;19:15–30 Available from: https://doi.org/10.1186/s12859-018-2129-y.
Ogilvie HA, Bouckaert RR, Drummond AJ. StarBEAST2 brings faster species tree inference and accurate estimates of substitution rates. Mol Biol Evol. 2017;34:2101–14.
Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10:e1003537.
Matzke NJ. BEASTmasteR: R tools for automated conversion of NEXUS data to BEAST2 XML format, for fossil tip-dating and other uses.". 2015 [cited 2017 May 3]. Available from: http://phylo.wikidot.com/beastmaster.
Gamble T, Greenbaum E, Jackman TR, Bauer AM. Into the light: diurnality has evolved multiple times in geckos. Biol J Linn Soc. 2015;115:896–910 Available from: https://academic.oup.com/biolinnean/article/115/4/896/2440166.
Oliver PM, Hugall AF. Phylogenetic evidence for mid-Cenozoic turnover of a diverse continental biota. Nat Ecol Evol. 2017;1:1896 Available from: http://www.nature.com/articles/s41559-017-0355-8.
Esri. ArcGIS Desktop: Release 10. Redlands. 2011. p. CA, USA.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25.
Ramos-Onsins SE, Rozas J. Statistical properties of new neutrality tests against population growth. Mol Biol Evol. 2002;19:2092–100.
Harvey MG, Seeholzer GF, Tilston B, Rabosky DL, Cuervo AM. Positive association between population genetic differentiation and speciation rates in New World birds. Proc Nat Acad Sci. 2017;114:6328–33.
Orme CDL, Freckleton RP, Thomas GH, Petzoldt T, Fritz SA, Isaac NJB. CAPER: comparative analyses of Phylogenetics and evolution in R. Methods Ecol Evol. 2012;3:145–51.
Matzke NJ. Probabilistic historical biogeography: new models for founder-event speciation, imperfect detection, and fossils allow improved accuracy and model-testing. Front Biogeogr. 2013;5(4):242–8.
Ree RH, Smith SA. Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst Biol. 2008;57:4–14.
Ronquist F. Dispersal-vicariance analysis: a new approach to the quantification of historical biogeography. Syst Biol. 1997;46:195–203.
Landis MJ, Matzke NJ, Moore BR, Huelsenbeck JP. Bayesian analysis of biogeography when the number of areas is large. Syst Biol. 2013;62:789–804.
Lewis PO. A likelihood approach to estimating phylogeny from discrete morphological character data. Syst Biol. 2001;50:913–25.
Burnham KP, Anderson DR. Model selection and multimodel inference: a practical information-theoretic approach (2nd ed). Ecol Model. 2002; Available from: http://cds.cern.ch/record/1608735/files/9780387953649_TOC.pdf.
Doughty P, Palmer R, Sistrom MJ, Bauer AM, Donnellan SC. Two new species of Gehyra (Squamata: Gekkonidae) geckos from the north-West Kimberley region of Western Australia. Rec West Aust Museum. 2012;27:117–34 Available from: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.453.9878&rep=rep1&type=pdf.
Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, et al. Picante: R tools for integrating phylogenies and ecology. Bioinformatics. 2010;26:1463–4.
Revell LJ. Phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol Evol. 2012;3:217–23.
Pennell MW, Eastman JM, Slater GJ, Brown JW, Uyeda JC, Fitzjohn RG, et al. Geiger v2.0: An expanded suite of methods for fitting macroevolutionary models to phylogenetic trees. Bioinformatics. 2014;30:2216–8.
Pinheiro J, Bates D, DebRoy S, Sarkar D. nlme: Linear and Nonlinear Mixed Effects Models. R Packag. version. 2016:1–86.
Uyeda JC, Caetano DS, Pennell MW. Comparative analysis of principal components can be misleading. Syst Biol. 2015;64:677–89.
Ingram T, Mahler DL. SURFACE: detecting convergent evolution from comparative data by fitting Ornstein-Uhlenbeck models with stepwise Akaike information criterion. Methods Ecol Evol. 2013;4:416–25.
Khabbazian M, Kriebel R, Rohe K, Ané C. Fast and accurate detection of evolutionary shifts in Ornstein–Uhlenbeck models. Methods Ecol Evol. 2016;7:811–24.
Harvey MS, Rix MG, Framenau VW, Hamilton ZR, Johnson MS, Teale RJ, et al. Protecting the innocent: studying short-range endemic taxa enhances conservation outcomes. Invertebr Syst. 2011;25:1–10.
Simó-Riudalbas M, Metallinou M, de Pous P, Els J, Jayasinghe S, Péntek-Zakar E, et al. Cryptic diversity in Ptyodactylus (Reptilia: Gekkonidae) from the northern Hajar Mountains of Oman and the United Arab Emirates uncovered by an integrative taxonomic approach. PLoS One. 2017;12:e0180397 Available from: http://dx.plos.org/10.1371/journal.pone.0180397.
Grismer LL, Wood PL, Thura MK, Zin T, Quah ES, Murdoch ML, et al. Twelve new species of Cyrtodactylus gray (Squamata: Gekkonidae) from isolated limestone habitats in east-central and southern Myanmar demonstrate high localized diversity and unprecedented microendemism. Zool J Linnean Soc. 2017;182:862–959.
Meiri S, Bauer AM, Allison A, Castro-Herrera F, Chirio L, Colli G, et al. Extinct, obscure or imaginary: the lizard species with the smallest ranges. Divers Distrib. 2018;24:262–73.
Byrne M, Yeates DK, Joseph L, Kearney M, Bowler J, Williams M, et al. Birth of a biome: insights into the assembly and maintenance of the Australian arid zone biota. Mol Ecol. 2008;17:4398–417.
Byrne M, Steane DA, Joseph L, Yeates DK, Jordan GJ, Crayn D, et al. Decline of a biome: evolution, contraction, fragmentation, extinction and invasion of the Australian Mesic zone biota. J Biogeogr. 2011;38:1635–56.
Potter S, Eldridge MDB, Taggart DA, Cooper SJB. Multiple biogeographical barriers identified across the monsoon tropics of northern Australia: Phylogeographic analysis of the brachyotis group of rock-wallabies. Mol Ecol. 2012;21:2254–69.
Moritz C, Fujita MK, Rosauer D, Agudo R, Bourke G, Doughty P, et al. Multilocus phylogeography reveals nested endemism in a gecko across the monsoonal tropics of Australia. Mol Ecol. 2016:1354–66 Available from: http://doi.wiley.com/10.1111/mec.13511.
Ladiges PY, Udovicic F, Nelson G. Australian biogeographical connections and the phylogeny of large genera in the plant family Myrtaceae. J Biogeogr. 2003;30:989–98 Available from: http://doi.wiley.com/10.1046/j.1365-2699.2003.00881.x.
Oliver PM, Laver RJ, Smith KL, Bauer AM. Long-term persistence and vicariance within the Australian monsoonal tropics: the case of the giant cave and tree geckos (Pseudothecadactylus). Aust J Zool. 2014:462–8.
Fujioka T, Chappell J, Fifield LK, Rhodes EJ. Australian desert dune fields initiated with Pliocene - Pleistocene global climatic shift. Geology. 2009;37:51–4.
Glaw F, Kohler J, Townsend TM, Vences M. Rivaling the world’s smallest reptiles: discovery of miniaturized and microendemic new species of leaf chameleons (Brookesia) from northern Madagascar. PLoS One. 2012;7:e31314.
Scheffers BR, Edwards DP, Diesmos A, Williams SE, Evans TA. Microhabitats reduce animal’s exposure to climate extremes. Glob Chang Biol. 2014;20:495–503.
Rosauer DF, Byrne M, Blom MPK, Coates DJ, Donnellan S, Doughty P, et al. Real-world conservation planning for evolutionary diversity in the Kimberley, Australia, sidesteps uncertain taxonomy. Conserv Lett. 2018;11:e12438.
Heinicke MP, Jackman TR, Bauer AM. The measure of success: geographic isolation promotes diversification in Pachydactylus geckos. BMC Evol Biol. 2017;17:9 Available from: http://bmcevolbiol.biomedcentral.com/articles/10.1186/s12862-016-0846-2.
Lim JY, Marshall CR. The true tempo of evolutionary radiation and decline revealed on the Hawaiian archipelago. Nature. 2017;543:710–3 Available from: https://doi.org/10.1038/nature21675.
Revell LJ, Johnson MA, Schulte JA, Kolbe JJ, Losos JB. A phylogenetic test for adaptive convergence in rock-dwelling lizards. Evolution. 2007;61:2898–912.
Goodman BA, Isaac JL. Convergent body flattening in a clade of tropical rock-using lizards (Scincidae: Lygosominae). Biol J Linn Soc. 2008;94:399–411.
Hipsley CA, Miles DB, Müller J. Morphological disparity opposes latitudinal diversity gradient in lacertid lizards. Biol Lett. 2014;10:20140101 Available from: https://royalsocietypublishing.org/doi/full/10.1098/rsbl.2014.0101.
Pincheira-Donoso D, Harvey LP, Ruta M. What defines an adaptive radiation? Macroevolutionary diversification dynamics of an exceptionally species-rich continental lizard radiation. BMC Evol Biol BMC Evolutionary Biology. 2015;15 Available from: http://www.biomedcentral.com/1471-2148/15/153.
Schoener TW. Resource partitioning in ecological communities. Science . 1974;185:27–39. Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.185.4145.27
Rabosky DL, Reid J, Cowan MA, Foulkes J. Overdispersion of body size in Australian desert lizard communities at local scales only: no evidence for the Narcissus effect. Oecologia. 2007;154:561–70.
Hutchinson GE. Homage to Santa-Rosalia or why are there so many kinds of animals. Am Nat. 1959;93:145–59.
Weber MG, Wagner CE, Best RJ, Harmon LJ, Matthews B. Evolution in a community context: on integrating ecological interactions and macroevolution. Trends Ecol Evol. 2017;32:291–304 Available from: https://doi.org/10.1016/j.tree.2017.01.003.
Acknowledgments
We thank the following people for providing access to specimens and samples in their care: Andrew Amey (Queensland Museum), Gavin Dally (Northern Territory Museum), Paul Doughty, Ryan Ellis (Western Australian Museum), Conrad Hoskin (James Cook University), Eric Rittmeyer (Rutgers University), Chris Austin (Lousiana State University), and Katie Date and Jane Melville (Museums Victoria). Damien Esquerre and Nick Matzke provided analytical advice. We also thank landholders, Parks staff, and Indigenous Rangers and Traditional Owners for access to their lands and for participating in fieldwork.
Funding
This work was funded by Australian Research Council (ARC) Linkage Grants to Paul Oliver with Mike Lee and Paul Doughty, and to Craig Moritz with Scott Keogh; an ARC Early Career Researcher Fellowship to Paul Oliver; an ARC Laureate Fellowship to Craig Moritz; and additional funding provided by the Australian National University. Collection of outgroup material from New Guinea was in part funded by National Science Foundation grant DEB 1146033 to Chris Austin.
Availability of data and materials
The genetic data alignments and morphological measurement datasets supporting the results of this article are available in the Dryad repository (https://doi.org/10.5061/dryad.d78b42h).
Author information
Authors and Affiliations
Contributions
PMO and CCM designed, conceived and funded the study and fieldwork. RCP, RJL, and LGA generated data. SB, LGA, PMO, and LGT analysed the data, and PMO wrote the paper with contributions from all authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Fieldwork and collecting were undertaken under Australian National University ethics permit number A2013/08.
Consent for publication
All authors give consent for their work to be published in this paper.
Competing interests
The authors declare they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Distribution of sampling (in kmz file format) for saxicoline taxa in the G. australis group in this paper. (KMZ 6 kb)
Additional file 2:
Distribution of sampling (in kmz file format) for generalist taxa in the G. australis group in this paper. (KMZ 8 kb)
Additional file 3:
Appendix 1. Full list of samples included in genetic and morphological analyses inlcuding GenBank accession details. Samples which were assigned to genetic lineages based on locality (no DNA) are indicated with an asterisk. (XLSX 135 kb)
Additional file 4:
Table S1. Net Tamura-Nei corrected genetic divergences amongst major mtDNA lineages. Table S2. Summary species delimitation results using ad hoc mtDNA divergences, bGMYC on mtDNA data, and tr2 on exon capture data. Table S3. Estimates of range-size (km2) for lineages in the Gehyra australis group based on minimum convex hulls for genetically-typed samples. Table S4. Relative likelihood and AIC scores for competing models of ecological trait evolution, as implemented in BioGeoBEARS. Table S5. Summary of population genetics statistics and expansion tests for major lineages (estimated from mtDNA). Table S6. Comparison of mean diversification rate statistic estimates for the Gehyra australis group stratified by ecology. Table S7. Loadings, variance and percentage of data variance explained from the principal components analyses (PCA) on all log-transformed morphological traits (“sPCA”), and on the residuals of body shape traits against SVL (“rPCA”). Table S8. Morphological differentiation between habitat types in the Gehyra australis group using multivariate data and ANOVA (phylogenetic or non-phylogenetic, based on tests for phylogenetic signal). Table S9. Morphological differentiation between habitat types in the Gehyra australis group using univariate data and Phylogenetic Generalized Least Squares (PGLS) regression and most likely model of trait evolution (BM = Brownian Motion, OU = Ornstein-Uhlenbeck). (DOCX 64 kb)
Additional file 5:
Figure S1. Geographic distribution and lineage allocation for samples from the Gehyra australis group included in exon capture. Figure S2. Topology and support values estimated by ASTRAL-II from 556 IQ-TREE gene trees for the Gehyra australis group. Figure S3. ASTRAL-III quartet support mapped onto species tree for the Gehyra australis group. Figure S4. Mean and ranges of values for morphological characters that showed evidence of significant variation across saxicoline and generalist taxa (according to PGLS) in the Gehyra australis group. Figure S5. Morphological regime shifts within the Gehyra australis group inferred by SURFACE based on an analysis including SVL (log-transformed) and 11 other size-corrected morphological characters. Figure S6. Morphological regime shifts within the Gehyra australis group inferred by l1OU based on an analysis including SVL (log-transformed) and 11 other size-corrected morphological characters. (DOC 999 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Oliver, P.M., Ashman, L.G., Bank, S. et al. On and off the rocks: persistence and ecological diversification in a tropical Australian lizard radiation. BMC Evol Biol 19, 81 (2019). https://doi.org/10.1186/s12862-019-1408-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-019-1408-1