
Abstract
Background
The merozoite surface protein 7 (MSP7) is a Plasmodium protein which is involved in parasite invasion; the gene encoding it belongs to a multigene family. It has been proposed that MSP7 paralogues seem to be functionally redundant; however, recent experiments have suggested that they could have different roles.
Results
The msp7 multigene family has been described in newly available Plasmodium genomes; phylogenetic relationships were established in 12 species by using different molecular evolutionary approaches for assessing functional divergence amongst MSP7 members. Gene expansion and contraction rule msp7 family evolution; however, some members could have had concerted evolution. Molecular evolutionary analysis showed that relaxed and/or intensified selection modulated Plasmodium msp7 paralogous evolution. Furthermore, episodic diversifying selection and changes in evolutionary rates suggested that some paralogous proteins have diverged functionally.
Conclusions
Even though msp7 has mainly evolved in line with a birth-and-death evolutionary model, gene conversion has taken place between some paralogous genes allowing them to maintain their functional redundancy. On the other hand, the evolutionary rate of some MSP7 paralogs has become altered, as well as undergoing relaxed or intensified (positive) selection, suggesting functional divergence. This could mean that some MSP7s can form different parasite protein complexes and/or recognise different host receptors during parasite invasion. These results highlight the importance of this gene family in the Plasmodium genus.
Similar content being viewed by others

Background
DNA duplication is an important source of novelty regarding evolution, providing the basis for new molecular activities [1, 2]. The genomes from the three kingdoms of life have been modulated by this mechanism, having multiple copies of genes [2]. Multigene families might evolve in line with a concerted or birth-and-death evolutionary model [3]; paralogous genes keep the same function in the former due to gene conversion whilst paralogous genes could lose or acquire a new function in a birth-and-death model. Since functional importance is highly correlated with evolutionary conservation [4, 5], molecular biologists have used evolutionary approaches to infer functional changes in paralogous genes/proteins using DNA/amino acid sequences [5–8].
Gene duplication seems to be recurrent in Plasmodium genus. These parasites are able to infect several vertebrates such as birds, rodents and primates. More than 200 species have been described to date. Clustering in different lineages occurs according to host (bird/reptile-parasite, rodent-parasite, monkey-parasite and hominid-parasite lineages) [9, 10]. Several genes produced by gene duplication are involved in host-cell invasion [11–14]. MSP7 is a merozoite surface protein encoded by a gene belonging to a multigene family located in chromosome 13 in hominid- and rodent-parasites but in chromosome 12 in monkey-parasites. This family has a different copy number amongst Plasmodium species [15, 16]. These genes are expressed simultaneously but they are independently regulated [17–20]. Functional assays have shown that P. falciparum MSP7 (MSP7I) is proteolytically processed; the resulting 22 kDa C-terminal region fragment is not covalently associated with MSP1 [7] and has cross-reactivity with other MSP7 proteins [17]. Furthermore, this fragment appears be involved in invasion by binding to red blood cells [21]. The C-terminal regions in P. yoelii from different MSP7s seem to be necessary to interact with the 83 kDa MSP1 fragment [17]. The MSP7 knockout reduces the normal growth rate of the mutant parasite in P. berghei; however, it becomes restored a few days later [22].
The msp7 family appears to follow the birth-and-death evolutionary model; some gene copies have been maintained in the genome for a long time and others appear to be more recent [15]. This family has had a complex evolutionary history regarding P. vivax. The C-terminal region is involved in gene conversion [23]. Moreover, this region is highly conserved and under negative selection, suggesting functional/structural constraint [23–25]; by contrast, some P. vivax MSP7 proteins’ central regions have high genetic diversity, maintained by balancing selection, possibly as an immune evasion mechanism [23, 24].
Recent protein-protein interaction assays have shown that MSP7 proteins do not appear to bind to the same host receptor [26]; moreover, these proteins seem to be forming different protein complexes in the parasite [7, 27–30], maybe to perform different parasite-host interactions. Such results flout the functional redundancy hypothesis [15, 18]; functional divergence in MSP7 paralogs thus appears to be probable. This study has analysed data concerning msp7 multigene family evolution, including 13 available Plasmodium genomes by evaluating their phylogenetic relationships and adopting different and new molecular evolutionary approaches for assessing functional divergence amongst MSP7 proteins.
Methods
Sequence data, alignments and phylogenetic tree reconstruction
Genome sequences from 11 Plasmodium species (and one subspecies, GenBank access number: P. reichenowi, GCA_000723685.1; P. falciparum, GCA_000002765.1; P. vivax, GCA_000002415.2; P. cynomolgi, GCA_000321355.1; P. inui, GCA_000524495.1; P. knowlesi, GCA_000006355.1; P. coatneyi, GCA_000725905.1; P. chabaudi, GCA_000003075.2; P. vinckei, GCA_000709005.1 and GCA_000524515.1; P. yoelii, GCA_000003085.2 and P. berghei, GCA_000005395.1) as well as the partial genome sequences from P. gallinaceum (Wellcome Trust Sanger Institute, http://www.sanger.ac.uk/resources/downloads/protozoa/plasmodium-gallinaceum.html) were analysed to obtain msp7 multigene family genomic regions. The msp7 gene copy number for these 13 genomes was established, as reported previously [15, 24].
All gene sequences found were used to deduce amino acid sequences by using Gene Runner software; these sequences were then screened to distinguish the MSP_7C domain using the Pfam server [31] (domain access number: PF12948). All amino acid sequences were then aligned using the MUSCLE algorithm [32] and manually edited by GeneDoc software [33]. The best amino acid substitution model was selected by Akaike’s information criterion using the ProtTest algorithm [34]; the JTT + G + F model was used to infer phylogenetic tress using maximum likelihood (ML) and Bayesian (BY) methods. RAxML was used for ML analysis [35] and topology reliability was evaluated by bootstrap, using 1000 replicates. A Metropolis-coupled Markov chain Monte Carlo (MCMC) algorithm was used for BY analysis [36] with MrBayes [37]. This analysis was run until reaching a standard deviation of split frequencies (ASDSF) value lower than 0.01; sump and sumt commands were used for tabulating posterior probabilities and building a consensus tree; in addition to ASDSF, the PSRF parameter was used for monitoring convergence. Both analyses were performed at CIPRES Science Gateway [38, 39]. A recent evolutionary multigene family model called DLRTS [40] was also performed; this method infers a gene tree by evolving down on a given species tree (with divergence times) by means of duplication, loss and transfer events according to a birth-death-like process [40, 41]. The species tree was inferred with a fragment of cytochrome C oxidase subunit 2 and divergence times were obtained from Pacheco et al. [42]. DLRTS was run using the MCMC algorithm for 10 million generations.
Gene conversion amongst msp7 members
It has been shown that some msp7 family members (msp7H and msp7I) have evolved by gene conversion, thereby contributing to these members’ homogenisation [23]. msp7 sequences were obtained from 6 P. vivax isolates (Salvador-I, Mauritania-I, India-VII, North Korean, Brazil-I and ctg isolate) [43, 44] and used to assess whether this pattern has also taken place in other msp7 members. Betran’s method was used for gene conversion amongst paralogous genes [45] as well as the GENECONV algorithm [46]. DnaSP software was used for the former method [47] where only conversion tracts larger than 10 nucleotides were considered whilst RDP3 v3.4 software was used for GENECONV [48], considering just conversion tracts having p < 0.01. The same approach was followed for P. falciparum msp7 genes, using the 3D7, FCR3, RO33, 7G8, K1, T9/102 and w2mef isolates.
Identifying episodic diversifying selection on msp7 tree branches
The random effects branch-site model (Branch-site REL) was used for assessing whether msp7 multigene family lineages had been subject to episodic diversifying selection [49]. This method identifies branches where a percentage of sites have evolved under episodic diversifying selection. The MUSCLE algorithm was used for independently aligning each orthologous cluster’s amino acid sequences (Figs. 1 and 2); PAL2NAL software [50] was then used for inferring codon alignments from the aligned amino acid sequences. The best evolutionary models for DNA and protein alignments were inferred by using jModelTest [51] and ProtTest [34], respectively. ML phylogenetic trees were obtained for DNA and protein alignments for each orthologous cluster and used as phylogenetic framework to perform the Branch-site REL method using the HyPhy software package [52]; additionally, the Datamonkey web server [53] was also used to perform this method. The MEME method [54] from Datamonkey server was used to infer which sites were under episodic positive selection in each cluster.

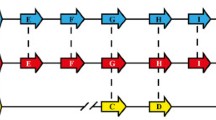
Schematic representation of the chromosomal msp7 loci in 13 Plasmodium genomes. The genes flanking the msp7 chromosome region in Plasmodium species are represented by orange boxes. The coloured boxes within flanking sequences represent msp7 genes in each species whilst black boxes symbolise pseudogenes. The genes are given in alphabetical order from left to right. The dashed lines connect orthologous genes. All genes are represented to scale, but the distance between them is not representative. Question marks refer to what were not clearly orthologous relationships. By contrast with hominid-parasites, species from monkey-parasite and rodent-parasite lineages seem to have similar evolutionary histories regarding msp7 expansion. The grey boxes are lineage-specific genes only found in hominid-parasite species, as they do not belong to the msp7 family (the latter gene representations are not to scale)

msp7 gene family phylogeny inferred by the DLTRS evolutionary model. a Species tree used for generating the MSP7 tree. b MSP7 tree created by evolving down the species tree. Numbers represent different clades whilst numbers on branches are posterior probability values. Nine major clades were identified on the tree. Proteins were clustered in agreement with parasite phylogenetic relationships, clades 1 (red), 2 (yellow) and 5 (purple) being the most ancestral ones. The clades clustering genes from monkey-parasite lineage are depicted in green, proteins from rodent-parasite lineage in blue and hominid-parasite lineage in grey. The P. inui specie-specific duplicate was not considered in this analysis. Due to the family’s complex evolutionary history (which includes gene conversion, intragenic recombination, positive and/or balancing selection) the MCMC analysis did not converge and therefore the duplication/lost rates were not obtained even though a tree reconciliation similar to other topologies was inferred (BY and ML)
Evolutionary analysis for testing functional divergence
Functional redundancy was previously proposed for the MSP7 family by using sequences from seven species [15]. The sequence number in this research was increased and two different phylogeny-based approaches were used to assess functional divergence or redundancy between MSP7 members; one involved using DIVERGE v.3 software [55] to estimate type-I functional divergence [5] which is an indicator of functional changes between members of a multigene family [5, 6, 56–61]. This method is based on (site-specific) shifted evolutionary rates. It assesses whether there has been a significant change in evolution rate after duplication (or speciation) events by calculating the coefficient of divergence (θD) and determining (e.g. by a Likelihood ratio test [5]) whether it is statistically significant for rejecting the null hypothesis (no functional divergence). The software then computes a posterior probability for detecting amino acids responsible for such divergence. Taking into account that new functions in paralogous proteins might emerge after gene duplication whenever selective strength is relaxed or whether positive selection is intensified, a second approach was used with the RELAX method [62]. This method allows partitioning a phylogeny into two subsets of branches to determine whether selective strength was relaxed or intensified in one of these subsets (test branch) relative to the other (reference branch). The Datamonkey web server was used for this analysis.
Results
msp7 chromosomal locus genetic structure in Plasmodium spp
Whole genome sequences from 11 Plasmodium species, 1 subspecies and 1 partial draft genome sequence were screened for describing the msp7 chromosomal locus. The msp7 locus is circumscribed by PVX_082640 and PVX_082715 genes in P. vivax [15, 16, 24]; genes sharing high similarity to them were thus searched in the remaining species. Since MSP7 proteins appear be encoded by a single exon [63], the contigs enclosing flanking genes were analysed using ORFFinder and Gene Runner software to identify open reading frames (ORFs) encoding proteins larger than 200 amino acids. Seventy-nine ORFs (Additional file 1) in 13 genomes having the same transcription orientation were found (Fig. 1). These ORFs had 0.6 to 1.3 kilobases (kb) but P. gallinaceum msp7 had a 3.1 kb length. Like previous studies [15, 16, 24], the copy number was different in Plasmodium spp. P. vivax and P. cynomolgi had the largest copy number (12 ORFs) whilst the lowest copy number was found in P. gallinaceum (just one gene). Shorter fragments (having more than 30% similarity with the identified ORFs) were also found in P. vivax, P. cynomolgi, P. falciparum and P. reichenowi. These ORFs (and small fragments) were named in alphabetical order regarding PVX_082640 and its homologous genes (Fig. 1 and Additional file 1).
Data regarding P. inui, msp7B (pinumsp7B) was incomplete due to gaps in the contig whilst pcynmsp7J, pcynmsp7L, pcoamspB, preimsp7D and pvinvmsp7A had premature stop codons. However, pcynmsp7L could encode a full MSP7 protein since it was shown to have intron donor/acceptor sites by GeneScan [64] screening, as previously shown [24]. Despite GeneScan not showing an intron/exon structure for pvinvmsp7A, it has putative donor/acceptor sites (Additional file 2). The Phobius algorithm was used for determining the presence of signal peptides within ORFs and Pfam for the MSP_7C domain; some genes did not have a signal peptide or the characteristic MSP_7C domain in the C-terminal region (Table 1).
The Plasmodium msp7 family’s phylogenetic relationships
Phylogenetic relationships for this family were identified as previously described [15, 24, 65, 66]. A multiple alignment was performed for deducing 80 msp7 genes’ amino acid sequences (excluding the shorter gene fragments and P. gallinaceum gene). A phylogenetic tree was then inferred by using ML and BY methods with the JTT + G + F model; both topologies gave similar branch patterns, displaying 12 major clades (Additional file 3), with clade 1 clustering sequences from 12 Plasmodium species considered in this study, clade 2 another 10 of them and clade 5 clustering the last gene in the chromosomal region from rodent-parasites, P. vivax and P. cynomolgi. The remaining clades put together sequences according to Plasmodium lineages (i.e. MSP7 sequences from the monkey-parasite lineage were in clades 3, clade 4 clustered the sequences from rodent-parasite lineage and clades 6 clustered sequences from the hominid-parasite lineage). Clades 1, 2, 3a, 3b, 4 and 6b only contained orthologous proteins whilst clades 3c and 3d had orthologous and paralogous proteins from monkey-parasite lineage, the clade 6a clustered sequences from hominid-parasite lineage whilst the sequence in clade 7 appeared to be exclusive to P. inui. In addition to previous studies [15], the DLTRS model was implemented which reconciles the gene tree to the species tree [40, 41]. The fraction of sampled values discarded as burn-in during analysis was 0.25 but the MCMC chain did not converge after more than 10 million generations, therefore, gene duplication, loss or transfer rates were not obtained. However, the reconciliation of the msp7 gene tree to the Plasmodium species tree was obtained. The tree inferred by DLTRS had 9 major clades (Fig. 2), thereby agreeing with the ML and BY clades (Fig. 2 and Additional file 3). In all phylogenies (Fig. 2 and Additional file 3) the sequences without the MSP_7C domain (PvivMSP7D, PcynMSP7D, PknoMSP7B and PcoaMSP7B) appeared to be phylogenetically related to sequences having an MSP_7C domain. However, this relationship was only supported by posterior probabilities but not by bootstrapping (Fig. 2 and Additional file 3). Furthermore, these sequences, like others not containing an MSP_7C domain, had noticeable similarity (>48%) with MSP7 members at the N-terminal end (Additional file 4).
Since PgalMSP7A is larger than other MSP7s, we did not take it into account for the aforementioned analysis; then, only the MSP_7C domain was used for inferring their phylogenetic relationships to determine whether PgalMSP7A was orthologous to MSP7A/H. The branch pattern from this topology (Additional file 5) was similar to the phylogeny obtained by using all sequences (Fig. 2, Additional files 3 and 5). PgalMSP7A clustered with MSP7A; however, posterior probability and bootstrapping were low. PgalMSP7A did not cluster with any other MSP7 in DLTRS tree; instead it appeared as an outgroup.
Gene conversion amongst msp7 genes
An atypical pattern in the clades clustering MSP7H/7I and MSP7B/7E/7G was observed in the phylogenies inferred above (Fig. 2, Additional files 3 and 5). Contrary to what would have been expected, PvivMSP7B was clustered with PvivMSP7E and PvivMSP7G and not with their respective orthologues; likewise, PvivMSP7H and PvivMSP7I seemed to share a common origin. It has previously been reported that gene conversion takes place between PvivMSP7H and 7I [23]. We obtained the pvivmsp7s sequences from 6 P. vivax isolates to assess whether gene conversion takes place in PvivMSP7B, 7E and 7G. Alignments for pvivmsp7C, 7H and 7I and another for pvivmsp7B, 7E and 7G were performed; Betran’s method and the GENECONV algorithm were then used, displaying recombination tracks between isolates but also between paralogous genes. The Betran algorithm found 2 conversion tracks between pvivmsp7C and 7I, whilst there were 3 conversion tracks between pvivmsp7B and 7E and another two between pvivmsp7E and 7G. Figure 3 shows the conversion tracks found by GENECONV. The same approach was followed for P. falciparum by using different reference isolates. By contrast with P. vivax, pfmsp7 members did not seem to be affected by gene conversion since no conversion tracks were found amongst them (data not shown).

Schematic representations of gene conversion tracks identified by the GENECONV method. Each gene is represented by a colour bar (pvivmsp7C (blue), pvivmsp7H (red), pvivmsp7I (green), pvivmsp7B (purple), pvivmsp7E (fuchsia), and pvivmsp7G (orange)); a different coloured rectangle is a graphical representation of sequence fragments potentially originating from gene conversion. Conversion tracks were mainly located in the 3′-ends. The % value refers to the similarity value for the sequence region involved in gene conversion (or intragenic recombination)
Episodic positive selection on msp7 branches
Previous studies have shown ancestral positive selection regarding the msp1 gene in the monkey-parasite lineage [67, 68]. Such episodic positive selection could have occurred in order to adapt to newly-appeared macaque species [67]. Since MSP1 and MSP7 form a protein complex [7, 18, 29] involved in parasite invasion, both proteins should have similar selective pressures. Since this has been assessed just for msp7E and 7L [24], the Branch-site REL method was used here for assessing whether other lineages are subject to episodic diversifying selection in msp7 evolutionary history; evidence was found of strong episodic diversifying selection in a few internal branches and in several external branches (Fig. 4 and Additional file 6). Regarding rodent-parasites, the lineages leading to MSP7A and 7B in P. vinckei vinckei; P. vinckei petteri, P. chabaudi (Fig. 4a) and P. berghei (Fig. 4i) were under selection. Just one internal branch (the P. berghei/P. yoelii MSP7C ancestor, Fig. 4g) displayed episodic selection. Concerning monkey-parasites, a percentage of sites regarding the lineages leading to MSP7A in P. vivax, P. cynomolgi and P. inui as well as the P. inui/P. knowlesi/P. coatneyi and P. knowlesi/P. coatneyi lineage ancestors (Fig. 4a) were under very strong episodic positive selection (ω > 33). Likewise, the lineages that gave rise to P. cynomolgi MSP7B, 7E, P. vivax MSP7B, 7E (Fig. 4b); P. inui 7B (Fig. 4c); P. vivax 7F (Fig. 4d); P. knowlesi 7C, P. cynomolgi 7G, P. inui 7D (Fig. 4e); P. coatneyi 7D, P. knowlesi 7D, P. vivax 7H, P. inui 7E, P. cynomolgi 7I (Fig. 4f) and P. vivax 7L (Fig. 4h) also were under positive selection. Very strong episodic positive selection was observed in the P. knowlesi/P. coatneyi 7C, P. inui 7D/P. vivax 7G, P. coatneyi/P. knowlesi 7D, P. inui 7E/P. cynomolgi 7I/P. vivax 7I and P. cynomolgi/P. vivax MSP7I ancestral branches (Fig. 4 and Additional file 6).

Phylogenies analysed for episodic selection. Each orthologous cluster was analysed by the Branch-site REL method. The shade of each colour on branches indicates strength of selection (red shows ω >13, blue ω ≤1 and grey ω = 1). The size of each colour represents the percentage of sites in the corresponding class found by Branch-site REL. Branches have been classified as undergoing episodic diversifying selection by the p-value corrected for multiple testing using the Holm-Bonferroni method at p < 0.05. a. clade 1; b. pviv/pcynmsp7B and 7E; c. pviv/pcynmsp7C and pinumsp7B; d. pviv/pcynmsp7F and pinumsp7C; e. pviv/pcynmsp7G, pkno/pcoamsp7C and pinumsp7D; f. pviv/pcynmsp7H/7I, pkno/pcoamsp7D and pinumsp7E; g. clade 2; h. pviv/pcynmsp7L and pinumsp7H and i. clade 4. At the bottom of each phylogeny there is a scale representation of msp7s. The blue boxes represent the encoded N-terminal region, the light brown ones symbolise the central region and the purple boxes the MSP_7C domain. Numbers within boxes represent the number of codons under positive selection inferred by MEME, SLAC, FEL, REL and FUBAR methods using the Datamonkey web server
Evolutionary analysis for testing functional divergence
Gu’s type-I functional divergence and RELAX methods were used to identify functional divergence amongst MSP7 proteins. Pairwise comparisons between paralogous proteins (e.g. from different clades) as well as between orthologues (e.g. proteins from different parasite lineage but within the same clade), showed high coefficient of divergence (θD) values (Table 2); between 10 and 20% of the sites had a significant change in their evolutionary rate (Additional file 7). Likewise, we found either a relaxed selection or an intensified selection amongst MSP7 paralogues (Table 2 and Additional file 8).
Discussion
It has been suggested that DNA duplication is the main source of evolutionary innovation [1, 2] since duplicate DNA fragments might evolve to new functions. However, acquiring new functions (neofunctionalisation) is not always the duplicate’s outcome [69]. There are other fates for paralogous fragments such as non-functionalisation (or pseudogenisation), subfunctionalisation [69] or even functional redundancy. Encoded-protein gene duplication in parasitic organisms involved in host recognition could provide an advantage, regardless of whether such duplicates increase host recognition ability. Plasmodium genomes have a lot of genes as multigene families [11–14, 44], some of them are functionally equivalent whilst others have functionally diverged (they recognise different host receptors) [70–73].
The msp7 family has been previously described in eight species [15, 16, 24], displaying different expansion. The present study has analysed 4 new species and completed analysis for P. reichenowi. An unequal copy gene number was found in Plasmodium species, suggesting lineage or specie-specific duplications or deletions. We also found genes which have become pseudogenes, thereby confirming the birth-and-death model of evolution for this family (Fig. 1). Then again, phylogenetic analysis was used for establishing phylogenetic relationships amongst msp7 paralogues. According to the phylogenetic trees (Fig. 2 and Additional file 3), MSP7A/H (clade 1) was the most ancestral gene, followed by clade 2 which is shared by monkey- and rodent-parasites. Clade 5 was also an old clade since it is shared amongst monkey- and rodent-parasites; however, not all monkey-parasites had this copy and it thus became lost in P. knowlesi, P. coatneyi and P. inui. Moreover, these proteins in monkey- and rodent-parasites did not have the MSP_7C domain. The remaining clades were clustered in agreement with Plasmodium species’ relationships (e.g. P. vivax genes clustered with P. cynomolgi genes) and they were syntenic, suggesting they are orthologues. These expansions reproduced the genus phylogenetic relationships. Species belonging to monkey-parasite lineage had the highest copy number. P. vivax and P. cynomolgi (sister taxa) shared the whole msp7 repertory. P. inui is the phylogenetically closest species to the aforementioned ones, having 7 orthologues followed by P. knowlesi and P. coatneyi having 5 orthologues. The latter species are sister taxa sharing a common ancestor as well as the whole msp7 repertory. The monkey-parasite lineage is a sister taxon to rodent-parasite lineage. At least two orthologous genes were found within these two lineages whilst only one gene was shared between these and the hominid-parasite lineage (Fig. 1). The most ancient Plasmodium lineage is the bird/reptile-parasite. We analysed P. gallinaceum and found just one large msp7 gene. According to the MSP7 C-terminal phylogenetic tree (Additional file 5), it is still unclear whether this large gene is orthologous to the most ancestral gene (msp7A/H). As we did not find any more msp7 genes in the P. gallinaceum partial genome, gene expansion should have taken place after mammal-parasite radiation 40 million year ago [42].
We found 83 sequences between bordering genes in the 13 genomes (Fig. 1 and Additional file 1); however, 11 protein sequences did not have the MSP_7C domain at the C-terminal end, though some of them did cluster with MSP7 proteins (those containing the MSP_7C domain). Proteins lacking such domain had high similarity with MSP7s at the N-terminal end (Additional file 4). This suggested that proteins lacking an MSP_7C domain (MSP7-like) are incomplete duplicates or have lost the domain throughout Plasmodium evolutionary history.
On the other hand, groups clustering paralogous proteins (3d clade) displayed an unusual pattern (Fig. 2 and Additional file 3). Proteins such as Pviv/PcynMSP7E and Pviv/PcynMSP7B seemed to be more similar within species than between species. A similar branch pattern was observed in the MSP_7C phylogenetic tree (Additional file 5), where PvivMSP7B was more similar to PvivMSP7E than PcynMSP7B. Likewise, PvivMSP7H was more similar to PvivMSP7I whilst PcynMSP7H clustered together with PcynMSP7I. A previous study has shown that gene conversion takes place in pvivmsp7H and 7I genes [23]; such branching pattern is therefore a consequence of gene conversion. We also observed gene conversion tracks amongst P. vivax reference isolates in the aforementioned genes, suggesting that this mechanism also occurs in pvivmsp7B, 7E and 7G; however, such genes are not near each other (Fig. 1) and there was no complete gene homogenisation. Therefore, it is not clear whether this mechanism is taking place at present or they are ancient gene conversion events.
Parasite invasion involves several protein-protein interactions between parasite and host. MSP1 is the protein mediating initial interaction, this protein and MSP7 form a complex involved in parasite-host interaction [7, 18, 21, 29]. MSP1 has shown an episodic positive selection signal throughout its evolutionary history [67, 68]; such positive selection is shown at ancestral branches and is likely the result of adapting to newly-appeared macaque species 3.7–5.1 million years ago [67] or during human switching [74]. Since MSP7 is in a complex with MSP1 [7, 18, 29], the former should have similar selective pressures and therefore similar behaviour. We have found several lineages under strong diversifying selection (ω > 10 [49], Additional file 6). As in MSP1 [67, 68], few internal branches were under episodic selection (Fig. 4a, e, f and g); this pattern could be the outcome of adaptation to new hosts during Plasmodium sympatric speciation, as has been suggested for other antigens [67, 68]. On the other hand, several external lineages (branches) were under selection. This could be the outcome of changes in evolutionary rates throughout msp7 paralogous evolution which have been favoured by selection since they may have promoted adaptation to a new host or acquiring new molecular activities.
Previous work did not recognise codons under positive selection [15]; however, here we identified codons under selection by using improved and/or newly developed methods (Fig. 4). The greatest amount of codons under positive selection was located in central msp7 regions and a few at 5′ or 3′-ends. Population genetics studies have shown the central region to be the most polymorphic and it seems to be involved in immune evasion [23, 24]. The positive selected sites found amongst species in central regions could thus be the consequence of host adaptation to avoid host immune responses. On the other hand, positive selected sites at the encoding C-terminal region could be the outcome of coevolution between host receptor and parasite MSP7 ligands and also the result of the acquisition of new roles (Additional file 8).
Despite functional redundancy having been suggested for the MSP7 family [15, 18], some groups have shown that MSP7 proteins appear not to bind to the same host receptor [26]. Likewise, some PvivMSP7 proteins seem to be forming different protein complexes in the parasite [7, 27–30] which might allow different parasite-host interactions. We could not find evidence of functional divergence in MSP7 paralogues when comparing different clades (e.g. clade 1 against clade 2) in a previous in silico study [15]. This could have been because orthologous proteins might use different regions to interact with a host or with their own parasite proteins. The P. falciparum (hominid-parasite lineage) MSP1 region involved in host recognition is the 19 kDa fragment [75]; nevertheless, the 33 kDa fragment in P. vivax (monkey-parasite lineage) facilitates parasite-host interaction [76]. Therefore, even though they are orthologous, both proteins have differences in their evolutionary rates within functional regions [77]. Whether this behaviour also took place in MSP7, DIVERGE gave false negatives regarding functional divergence. We thus analysed MSP7 proteins from other Plasmodium species to assess functional divergence amongst MSP7 clades (paralogous) but also within clades (orthologous). Unlike a previous study [15], just the MSP7 C-terminal region was analysed here, taking into account that this region has the domain (MSP_7C) defining members of this family, it is the only MSP7 region in the protein complex [7], in P. vivax MSP7s C-terminal regions are highly conserved and under negative selection whilst other regions are highly polymorphic [23–25] and most regions binding to red blood cells are in the MSP7 C-terminal region [21]. We have found changes in evolutionary rates amongst paralogous proteins in this region. The coefficients of divergence (θD) were statistically significantly larger than 0 between some clades (Table 2 and Additional file 7), suggesting that some MSP7s have diverged functionally. Such divergence could mean that different MSP7 proteins could form different parasite protein complexes or that MPS7s could interact with different host receptors. We also found changes in evolutionary rates within clades (between orthologues) leading to large θD values (e.g. between monkey-parasite MSP7A and rodent-parasite MSP7A). This could have been due to functional divergence or different protein regions carrying out the function (as previously shown for MSP1 [75, 76]).
It has been demonstrated that duplicated genes experience a brief period of relaxed selection early in their history [69]. This relaxation could have led to pseudogenisation or, rarely, evolving to new functions [69]. Moreover, positive selection in duplicates could also lead to them acquiring new molecular activities [62]; relaxed selection or intensification of positive selection must thus be identified in MSP7s having functional divergence. Our DIVERGE results were consistent with RELAX results (Table 2). Proteins showing functional divergence also displayed functional constraint relaxation or intensification of positive selection. However, some clades having high θD did not show relaxation or intensification. This could have been due to episodic positive selection. This kind of selection acts very quickly and involves a switch from negative selection to positive selection and back to negative selection [62]. Episodic selection was found in all msp7 paralogous clusters by two different approaches (Fig. 4, Additional files 6 and 7); consequently, episodic positive selection allowing functional divergence could not have been detected by RELAX.
On the other hand, some paralogous proteins had no statistical θD values; they also displayed intensification of negative selection, thereby suggesting that they are functionally equivalent. Furthermore, functional redundancy in MSP7H, 7I and/or 7B and 7E could be favoured by gene conversion; some MSP7 members could therefore evolve by “partial gene conversion” affecting some but not all MSP7 paralogous proteins.
Conclusion
We have described the msp7 family in different Plasmodium species, using different phylogenetic and molecular evolution analyses. Although msp7 evolved mainly in line with a birth-and-death evolutionary model, some members have evolved in a concerted way. Gene conversion has taken place between some paralogous genes allowing gene sequence homogenisation, these paralogous genes consequently keeping the same function. However, some gene conversion tracks could be ancient and thus the homogenisation has been lost. In addition, some paralogous proteins did not show changes in their evolutionary rates; thus, MSP7A, 7B, 7C, 7E, 7H and 7I in monkey-parasites seem to be functionally equivalent copies. Other MSP7 members showed alteration in their evolutionary rate as well as relaxed or intensified (positive) selection; functional divergence may thus have occurred in them. Such functional divergence could enable MSP7F, 7G, 7K and 7L (from monkey-parasites) and MSP7A, 7B and 7C (from rodent-parasites) to form different parasite protein complexes and/or recognise different host receptors during invasion. In fact, protein-protein assays have shown that PvivMSP7A interacts with PvivTRAg56.2 [30] whilst PvivMSP7L has been found forming a complex with a member of the MSP3 family [27]. Moreover, PvivMSP7G (but not 7C or 7L) is able to bind to human P-selectin whilst PberMSP7C binds better to mouse P-selectin than PberMSP7A [26]. The results described here highlight this family’s importance in the Plasmodium genus. Further functional assays should be performed based on these results to gain a deeper understanding of the biology of Plasmodium invasion.
Abbreviations
- ASDSF:
-
A standard deviation of split frequencies
- Branch-site REL:
-
Random effects branch-site method
- BY:
-
Bayesian method
- DLTRS:
-
Duplications, losses, transfers, rates & sequence evolution
- HyPhy:
-
Hypothesis testing using phylogenies
- Kb:
-
Kilobases
- MCMC:
-
Metropolis-coupled Markov chain Monte Carlo
- MEME:
-
Mixed effects model of evolution
- ML:
-
Maximum likelihood
- MSP3:
-
Merozoite surface protein 3
- MSP7:
-
Merozoite surface protein 7
- ORFs:
-
Open reading frames
- PvivTRAg56.2:
-
P. vivax tryptophan-rich antigen 56.2
- RDP:
-
Recombination detection program
- θD :
-
Coefficient of divergence
- ω:
-
Omega rate (dN/dS)
References
Ohno S. Evolution by gene duplication. London, New York: Allen & Unwin; Springer-Verlag; 1970.
Zhang J. Evolution by gene duplication: an update. Trends Ecol Evol. 2003;18(6):292–8.
Nei M, Rooney AP. Concerted and birth-and-death evolution of multigene families. Annu Rev Genet. 2005;39:121–52.
Kimura M. The neutral theory of molecular evolution. Cambridge Cambridgeshire, New York: Cambridge University Press; 1983.
Gu X. Statistical methods for testing functional divergence after gene duplication. Mol Biol Evol. 1999;16(12):1664–74.
Gaucher EA, Gu X, Miyamoto MM, Benner SA. Predicting functional divergence in protein evolution by site-specific rate shifts. Trends Biochem Sci. 2002;27(6):315–21.
Pachebat JA, Ling IT, Grainger M, Trucco C, Howell S, Fernandez-Reyes D, Gunaratne R, Holder AA. The 22 kDa component of the protein complex on the surface of Plasmodium falciparum merozoites is derived from a larger precursor, merozoite surface protein 7. Mol Biochem Parasitol. 2001;117(1):83–9.
Kondrashov FA, Rogozin IB, Wolf YI, Koonin EV. Selection in the evolution of gene duplications. Genome Biol. 2002;3(2):RESEARCH0008.
Duval L, Fourment M, Nerrienet E, Rousset D, Sadeuh SA, Goodman SM, Andriaholinirina NV, Randrianarivelojosia M, Paul RE, Robert V, et al. African apes as reservoirs of Plasmodium falciparum and the origin and diversification of the Laverania subgenus. Proc Natl Acad Sci U S A. 2010;107(23):10561–6.
Hall N. Genomic insights into the other malaria. Nat Genet. 2012;44(9):962–3.
Tachibana S, Sullivan SA, Kawai S, Nakamura S, Kim HR, Goto N, Arisue N, Palacpac NM, Honma H, Yagi M, et al. Plasmodium cynomolgi genome sequences provide insight into Plasmodium vivax and the monkey malaria clade. Nat Genet. 2012;44(9):1051–5.
Singh V, Gupta P, Pande V. Revisiting the multigene families: Plasmodium var and vir genes. J Vector Borne Dis. 2014;51(2):75–81.
Sundararaman SA, Plenderleith LJ, Liu W, Loy DE, Learn GH, Li Y, Shaw KS, Ayouba A, Peeters M, Speede S, et al. Genomes of cryptic chimpanzee Plasmodium species reveal key evolutionary events leading to human malaria. Nat Commun. 2016;7:11078.
Gupta A, Thiruvengadam G, Desai SA. The conserved clag multigene family of malaria parasites: essential roles in host-pathogen interaction. Drug Resist Updat. 2015;18:47–54.
Garzon-Ospina D, Cadavid LF, Patarroyo MA. Differential expansion of the merozoite surface protein (msp)-7 gene family in Plasmodium species under a birth-and-death model of evolution. Mol Phylogenet Evol. 2010;55(2):399–408.
Kadekoppala M, Holder AA. Merozoite surface proteins of the malaria parasite: the MSP1 complex and the MSP7 family. Int J Parasitol. 2010;40(10):1155–61.
Mello K, Daly TM, Long CA, Burns JM, Bergman LW. Members of the merozoite surface protein 7 family with similar expression patterns differ in ability to protect against Plasmodium yoelii malaria. Infect Immun. 2004;72(2):1010–8.
Mello K, Daly TM, Morrisey J, Vaidya AB, Long CA, Bergman LW. A multigene family that interacts with the amino terminus of plasmodium MSP-1 identified using the yeast two-hybrid system. Eukaryot Cell. 2002;1(6):915–25.
Bozdech Z, Mok S, Hu G, Imwong M, Jaidee A, Russell B, Ginsburg H, Nosten F, Day NP, White NJ, et al. The transcriptome of Plasmodium vivax reveals divergence and diversity of transcriptional regulation in malaria parasites. Proc Natl Acad Sci U S A. 2008;105(42):16290–5.
Zhu L, Mok S, Imwong M, Jaidee A, Russell B, Nosten F, Day NP, White NJ, Preiser PR, Bozdech Z. New insights into the Plasmodium vivax transcriptome using RNA-Seq. Sci Rep. 2016;6:20498.
Garcia Y, Puentes A, Curtidor H, Cifuentes G, Reyes C, Barreto J, Moreno A, Patarroyo ME. Identifying merozoite surface protein 4 and merozoite surface protein 7 Plasmodium falciparum protein family members specifically binding to human erythrocytes suggests a new malarial parasite-redundant survival mechanism. J Med Chem. 2007;50(23):5665–75.
Tewari R, Ogun SA, Gunaratne RS, Crisanti A, Holder AA. Disruption of Plasmodium berghei merozoite surface protein 7 gene modulates parasite growth in vivo. Blood. 2005;105(1):394–6.
Garzon-Ospina D, Lopez C, Forero-Rodriguez J, Patarroyo MA. Genetic diversity and selection in three Plasmodium vivax merozoite surface protein 7 (Pvmsp-7) genes in a Colombian population. PLoS One. 2012;7(9):e45962.
Garzon-Ospina D, Forero-Rodriguez J, Patarroyo MA. Heterogeneous genetic diversity pattern in Plasmodium vivax genes encoding merozoite surface proteins (MSP) -7E, −7 F and -7 L. Malar J. 2014;13:495.
Garzon-Ospina D, Romero-Murillo L, Tobon LF, Patarroyo MA. Low genetic polymorphism of merozoite surface proteins 7 and 10 in Colombian Plasmodium vivax isolates. Infect Genet Evol. 2011;11(2):528–31.
Perrin AJ, Bartholdson SJ, Wright GJ. P-selectin is a host receptor for Plasmodium MSP7 ligands. Malar J. 2015;14:238.
Hostetler JB, Sharma S, Bartholdson SJ, Wright GJ, Fairhurst RM, Rayner JC. A Library of Plasmodium vivax Recombinant Merozoite Proteins Reveals New Vaccine Candidates and Protein-Protein Interactions. PLoS Negl Trop Dis. 2015;9(12):e0004264.
Lin CS, Uboldi AD, Epp C, Bujard H, Tsuboi T, Czabotar PE, Cowman AF. Multiple Plasmodium falciparum Merozoite Surface Protein 1 Complexes Mediate Merozoite Binding to Human Erythrocytes. J Biol Chem. 2016;291(14):7703–15.
Kauth CW, Woehlbier U, Kern M, Mekonnen Z, Lutz R, Mucke N, Langowski J, Bujard H. Interactions between merozoite surface proteins 1, 6, and 7 of the malaria parasite Plasmodium falciparum. J Biol Chem. 2006;281(42):31517–27.
Tyagi K, Hossain ME, Thakur V, Aggarwal P, Malhotra P, Mohmmed A, Sharma YD. Plasmodium vivax Tryptophan Rich Antigen PvTRAg36.6 Interacts with PvETRAMP and PvTRAg56.6 Interacts with PvMSP7 during Erythrocytic Stages of the Parasite. PLoS One. 2016;11(3):e0151065.
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44(D1):D279–85.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Nicholas KB, Nicholas HBJ. GeneDoc: A tool for editing and annotating multiple sequence alignments. 1997.
Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21(9):2104–5.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3.
Altekar G, Dwarkadas S, Huelsenbeck JP, Ronquist F. Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics. 2004;20(3):407–15.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42.
Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE). New Orleans, LA; 2010. p. 1–8. http://www.phylo.org/sub_sections/portal/sc2010_paper.pdf.
Miller MA, Schwartz T, Pickett BE, He S, Klem EB, Scheuermann RH, Passarotti M, Kaufman S, O’Leary MA. A RESTful API for Access to Phylogenetic Tools via the CIPRES Science Gateway. Evol Bioinformatics Online. 2015;11:43–8.
Sjostrand J, Tofigh A, Daubin V, Arvestad L, Sennblad B, Lagergren J. A Bayesian method for analyzing lateral gene transfer. Syst Biol. 2014;63(3):409–20.
Sjostrand J, Sennblad B, Arvestad L, Lagergren J. DLRS: gene tree evolution in light of a species tree. Bioinformatics. 2012;28(22):2994–5.
Pacheco MA, Battistuzzi FU, Junge RE, Cornejo OE, Williams CV, Landau I, Rabetafika L, Snounou G, Jones-Engel L, Escalante AA. Timing the origin of human malarias: the lemur puzzle. BMC Evol Biol. 2011;11:299.
Carlton JM, Adams JH, Silva JC, Bidwell SL, Lorenzi H, Caler E, Crabtree J, Angiuoli SV, Merino EF, Amedeo P, et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008;455(7214):757–63.
Neafsey DE, Galinsky K, Jiang RH, Young L, Sykes SM, Saif S, Gujja S, Goldberg JM, Young S, Zeng Q, et al. The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat Genet. 2012;44(9):1046–50.
Betran E, Rozas J, Navarro A, Barbadilla A. The estimation of the number and the length distribution of gene conversion tracts from population DNA sequence data. Genetics. 1997;146(1):89–99.
Sawyer S. Statistical tests for detecting gene conversion. Mol Biol Evol. 1989;6(5):526–38.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2.
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26(19):2462–3.
Kosakovsky Pond SL, Murrell B, Fourment M, Frost SD, Delport W, Scheffler K. A random effects branch-site model for detecting episodic diversifying selection. Mol Biol Evol. 2011;28(11):3033–43.
Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34(Web Server issue):W609–12.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772.
Pond SL, Frost SD, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21(5):676–9.
Delport W, Poon AF, Frost SD, Kosakovsky Pond SL. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;26(19):2455–7.
Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012;8(7):e1002764.
Gu X, Zou Y, Su Z, Huang W, Zhou Z, Arendsee Z, Zeng Y. An update of DIVERGE software for functional divergence analysis of protein family. Mol Biol Evol. 2013;30(7):1713–9.
Yan J, Cai Z. Molecular evolution and functional divergence of the cytochrome P450 3 (CYP3) Family in Actinopterygii (ray-finned fish). PLoS One. 2010;5(12):e14276.
Zhao Z, Liu H, Luo Y, Zhou S, An L, Wang C, Jin Q, Zhou M, Xu JR. Molecular evolution and functional divergence of tubulin superfamily in the fungal tree of life. Sci Rep. 2014;4:6746.
Wang Y, Gu X. Functional divergence in the caspase gene family and altered functional constraints: statistical analysis and prediction. Genetics. 2001;158(3):1311–20.
Zhou H, Gu J, Lamont SJ, Gu X. Evolutionary analysis for functional divergence of the toll-like receptor gene family and altered functional constraints. J Mol Evol. 2007;65(2):119–23.
McNally D, Fares MA. In silico identification of functional divergence between the multiple groEL gene paralogs in Chlamydiae. BMC Evol Biol. 2007;7:81.
Song W, Qin Y, Zhu Y, Yin G, Wu N, Li Y, Hu Y. Delineation of plant caleosin residues critical for functional divergence, positive selection and coevolution. BMC Evol Biol. 2014;14:124.
Wertheim JO, Murrell B, Smith MD, Kosakovsky Pond SL, Scheffler K. RELAX: detecting relaxed selection in a phylogenetic framework. Mol Biol Evol. 2015;32(3):820–32.
Mongui A, Perez-Leal O, Soto SC, Cortes J, Patarroyo MA. Cloning, expression, and characterisation of a Plasmodium vivax MSP7 family merozoite surface protein. Biochem Biophys Res Commun. 2006;351(3):639–44.
Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268(1):78–94.
Arisue N, Kawai S, Hirai M, Palacpac NM, Jia M, Kaneko A, Tanabe K, Horii T. Clues to evolution of the SERA multigene family in 18 Plasmodium species. PLoS One. 2011;6(3):e17775.
Rice BL, Acosta MM, Pacheco MA, Carlton JM, Barnwell JW, Escalante AA. The origin and diversification of the merozoite surface protein 3 (msp3) multi-gene family in Plasmodium vivax and related parasites. Mol Phylogenet Evol. 2014;78:172–84.
Sawai H, Otani H, Arisue N, Palacpac N, de Oliveira Martins L, Pathirana S, Handunnetti S, Kawai S, Kishino H, Horii T, et al. Lineage-specific positive selection at the merozoite surface protein 1 (msp1) locus of Plasmodium vivax and related simian malaria parasites. BMC Evol Biol. 2010;10:52.
Muehlenbein MP, Pacheco MA, Taylor JE, Prall SP, Ambu L, Nathan S, Alsisto S, Ramirez D, Escalante AA. Accelerated diversification of nonhuman primate malarias in Southeast Asia: adaptive radiation or geographic speciation? Mol Biol Evol. 2015;32(2):422–39.
Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–5.
Orlandi PA, Klotz FW, Haynes JD. A malaria invasion receptor, the 175-kilodalton erythrocyte binding antigen of Plasmodium falciparum recognizes the terminal Neu5Ac(alpha 2–3)Gal- sequences of glycophorin A. J Cell Biol. 1992;116(4):901–9.
Maier AG, Duraisingh MT, Reeder JC, Patel SS, Kazura JW, Zimmerman PA, Cowman AF. Plasmodium falciparum erythrocyte invasion through glycophorin C and selection for Gerbich negativity in human populations. Nat Med. 2003;9(1):87–92.
Triglia T, Duraisingh MT, Good RT, Cowman AF. Reticulocyte-binding protein homologue 1 is required for sialic acid-dependent invasion into human erythrocytes by Plasmodium falciparum. Mol Microbiol. 2005;55(1):162–74.
Stubbs J, Simpson KM, Triglia T, Plouffe D, Tonkin CJ, Duraisingh MT, Maier AG, Winzeler EA, Cowman AF. Molecular mechanism for switching of P. falciparum invasion pathways into human erythrocytes. Science. 2005;309(5739):1384–7.
Mu J, Joy DA, Duan J, Huang Y, Carlton J, Walker J, Barnwell J, Beerli P, Charleston MA, Pybus OG, et al. Host switch leads to emergence of Plasmodium vivax malaria in humans. Mol Biol Evol. 2005;22(8):1686–93.
Urquiza M, Rodriguez LE, Suarez JE, Guzman F, Ocampo M, Curtidor H, Segura C, Trujillo E, Patarroyo ME. Identification of Plasmodium falciparum MSP-1 peptides able to bind to human red blood cells. Parasite Immunol. 1996;18(10):515–26.
Rodriguez LE, Urquiza M, Ocampo M, Curtidor H, Suarez J, Garcia J, Vera R, Puentes A, Lopez R, Pinto M, et al. Plasmodium vivax MSP-1 peptides have high specific binding activity to human reticulocytes. Vaccine. 2002;20(9–10):1331–9.
Parobek CM, Bailey JA, Hathaway NJ, Socheat D, Rogers WO, Juliano JJ. Differing patterns of selection and geospatial genetic diversity within two leading Plasmodium vivax candidate vaccine antigens. PLoS Negl Trop Dis. 2014;8(4):e2796.
Garzón-Ospina D, Forero-Rodríguez JA. PM: Data from: Evidence of functional divergence in MSP7 paralogous proteins: a molecular-evolutionary and phylogenetic analysis. Dryad Digital Repository 2016, http://dx.doi.org/10.5061/dryad.1q26f.
Acknowledgements
We would like to thank Jason Garry for translating and reviewing the manuscript.
Funding
This work was financed by the Departamento Administrativo de Ciencia, Tecnología e Innovación (COLCIENCIAS) through grant RC # 0309-2013.
Availability of data and materials
The datasets supporting this article’s results are available in the Dryad Digital Repository (http://dx.doi.org/10.5061/dryad.3qk84) [78].
Authors’ contributions
DG-O devised and designed the study, performed the molecular evolutionary analysis and wrote the manuscript. JF-R participated in designing the study, the molecular evolutionary analysis and writing the manuscript. MAP coordinated the study and helped to write the manuscript. All the authors have read and approved the final version of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Eighty-tree sequences from MSP7 family found in 13 Plasmodium genomes. The PlasmoDB accession numbers for msp7 genes from eight species are shown. (TXT 88 kb)
Additional file 2:
Putative donor/acceptor sites in P. vinckei vinckei msp7A. (PDF 42 kb)
Additional file 3:
msp7 gene family phylogeny inferred by the maximum likelihood method. Numbers represent different clades (based on Fig. 2 numbers from main text) whilst numbers on branches are bootstrap and posterior probability values. Thirteen major clades were identified on the tree; however, clades 1 and 6 were not clearly supported by bootstrap values. Proteins were clustered in agreement with parasite phylogenetic relationships, clades 1 (red) and 2 (yellow) being the most ancestral ones; clade 5 could also be an old cluster. The clades clustering genes from monkey-parasite lineage are depicted in green, proteins from rodent-parasite lineage in blue and hominid-parasite lineage in grey. The protein cluster in brown is a P. inui specie-specific duplicate. Both ML and BY trees showed similar topologies, but only the ML tree is shown. Some internal branches were inconsistent between BY and ML trees. Since msp7 family has a complex evolutionary history (having gene conversion, intragenic recombination and/or natural selection) the tree inference is affected. However, external branches were consistent in both topologies as well as in Fig 2’s tree. In BY analysis, the settings for prior and likelihood were: prset aamodel = fixed (JONES); prset statefreqpr = fixed (empirical); lset rates = gamma (JTT + G + F). More than 11 million generations were required for reaching an ASDSF value lower than 0.01. The PSRF parameter was also used for monitoring convergence (PSRF: 1.000). (TIF 3836 kb)
Additional file 4:
Similarity values in the N-terminal region between MSP7 proteins and sequences lacking an MSP_7C domain. (PDF 24 kb)
Additional file 5:
Phylogenetic tree inferred with the C-terminal (MSP_7C) domain. Mammal-parasite MSP7 sequences containing the MSP_7C domain and the PgalMSP7 C-terminal domain were aligned and phylogenetic trees were then inferred. PgalMSP7 clustered with the most ancestral MSP7 protein (clade 1, see Fig. 2 in the main text) though this group was not supported by bootstrap and/or posterior probability. The remaining sequences clustered according to host-parasite lineages. Numbers on branches show bootstrap and posterior probabilities values. Clades shown in red and yellow were the most ancestral ones. The clades clustering genes from monkey-parasite lineage are depicted in green, proteins from rodent-parasite lineage in blue and hominid-parasite lineage in grey. Both ML and BY trees showed similar topologies but only the ML tree is shown. Numbers outside the clades represent the number of clades in Fig. 2 from the main text. (TIF 3038 kb)
Additional file 6:
Episodic positive selection on MSP7 branches. ω + values reflect the maximum likelihood estimate rate of positive selection. p-value obtained after Holm-Bonferroni multiple testing correction. The Branch-site REL method was performed by HyPhy software using both amino acid and DNA phylogenies. The Datamonkey web server was also used for calculating this method. The number of sites under episodic positive selection was identified by MEME using Datamonkey. The letters in the first panel correspond to the letters in Fig. 4 from the main text. (PDF 292 kb)
Additional file 7:
Putative sites involved in functional divergence. DIVERGE computed a posterior probability for detecting putative amino acids responsible for functional divergence in pairwise comparisons having statistical significant θD values. Such putative sites were highlighted in green and sites in the C-terminal region (MSP_7C domain) under positive selection found by the MEME method were tagged with a red plus symbol (+). Since functional divergence could involve relaxation of functional constraint or could be due to positive selection, a perfect correlation between putative amino acids responsible for divergence and positive selection would not have been expected. Positive selection might be involved in the acquisition of a new role but could also be the outcome of adaptation to a new host during Plasmodium’s evolutionary history. Positive selected sites were inferred using the clade (e.g. sequences within clade 1) but not using the sequences from the comparison (e.g. Clade 1 Primate-parasites vs Clade 2 Primate-parasites). (PDF 478 kb)
Additional file 8:
Selection intensity (functional constraint relaxation or selection intensification) throughout msp7 paralogous genes using the partitioned descriptive model. Three ω parameters (positive, negative and neutral) and the relative proportion of sites are plotted for test (blue) and reference (red) branches. The grey vertical dashed line at ω = 1 represents neutral evolution. The numbers in parenthesis indicate the comparison number in Table 2 from the main text. Comparisons 2, 4 and 15 show that the percentage of positive selected sites decreased on test branches; they also showed an intensification of negative selected sites (on test branches), having non-significant θD. These results suggested functional redundancy. Comparisons 11, 19, 20 and 28 displayed an increased percentage of positive selected sites having an intensification of this kind of selection on the test branches whilst the percentage of negative selected sites remained equivalent or decreased. The θD values in these comparisons were statistically significant, suggesting functional divergence. Comparison 24 showed a statistically significant θD and relaxed selection, indicating functional divergence. (TIF 6617 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Garzón-Ospina, D., Forero-Rodríguez, J. & Patarroyo, M.A. Evidence of functional divergence in MSP7 paralogous proteins: a molecular-evolutionary and phylogenetic analysis. BMC Evol Biol 16, 256 (2016). https://doi.org/10.1186/s12862-016-0830-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-016-0830-x