
Abstract
Background
USP4, USP15 and USP11 are paralogous deubiquitinating enzymes as evidenced by structural organization and sequence similarity. Based on known interactions and substrates it would appear that they have partially redundant roles in pathways vital to cell proliferation, development and innate immunity, and elevated expression of all three has been reported in various human malignancies. The nature and order of duplication events that gave rise to these extant genes has not been determined, nor has their functional redundancy been established experimentally at the organismal level.
Methods
We have employed phylogenetic and syntenic reconstruction methods to determine the chronology of the duplication events that generated the three paralogs and have performed genetic crosses to evaluate redundancy in mice.
Results
Our analyses indicate that USP4 and USP15 arose from whole genome duplication prior to the emergence of jawed vertebrates. Despite having lower sequence identity USP11 was generated later in vertebrate evolution by small-scale duplication of the USP4-encoding region. While USP11 was subsequently lost in many vertebrate species, all available genomes retain a functional copy of either USP4 or USP15, and through genetic crosses of mice with inactivating mutations we have confirmed that viability is contingent on a functional copy of USP4 or USP15. Loss of ubiquitin-exchange regulation, constitutive skipping of the seventh exon and neural-specific expression patterns are derived states of USP11. Post-translational modification sites differ between USP4, USP15 and USP11 throughout evolution.
Conclusions
In isolation sequence alignments can generate erroneous USP gene phylogenies. Through a combination of methodologies the gene duplication events that gave rise to USP4, USP15, and USP11 have been established. Although it operates in the same molecular pathways as the other USPs, the rapid divergence of the more recently generated USP11 enzyme precludes its functional interchangeability with USP4 and USP15. Given their multiplicity of substrates the emergence (and in some cases subsequent loss) of these USP paralogs would be expected to alter the dynamics of the networks in which they are embedded.
Similar content being viewed by others

Background
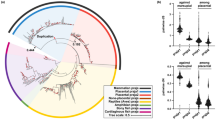
Protein ubiquitin tags are post-translational modifications that serve to either target substrates for proteasomal degradation or modify their interactive capacities [1]. Protein ubiquitination status is determined by the activities of the ubiquitin ligases that conjugate the ubiquitin moieties and the deubiquitinating enzymes (DUBs) that remove them; the balance of these activities thus affects key cellular processes. Among the most extensively networked [2] DUBs are the ubiquitin-specific protease (USP) paralogs USP4 and USP15, which regulate cell growth, embryonic development and innate immunity via their interactions with TGF-β [3, 4], Wnt/β-catenin [5] and NF-κB [6–8] pathway proteins respectively. USP4 and USP15 are also the only catalytic DUBs known to interact with the spliceosome [9–11], with more than eleven splicing factors identified as overlapping substrates [2]. This functional redundancy likely relates to their homology (there is 56.9 % amino acid identity in Homo sapiens as indicated in Fig. 1). One other DUB, USP11, bears considerable, albeit lesser, sequence identity to USP4 (44.5 % identity) and USP15 (43.2 %). The three paralogs share a common domain organization, consisting of a DUSP (domain in USP), two UBL (ubiquitin-like) and a bi-part catalytic domain (Fig. 1).

Comparison of USP4, USP15 and USP11 features. The red, blue and green boxes arranged in a circle represent USP4, USP15 and USP11, respectively. Domain structures are marked as follows: DUSP, domain in USP (N-terminal domain specific to these USPs); UBL, ubiquitin-like domain; D1 & D2, bi-part catalytic domain mediating ubiquitin cleavage. The interior of the circle links amino acid identities among paralogs, where each line represents an identical aligned residue. Links are colored as follows: USP4-USP15 purple; USP11-USP15 teal; USP4-USP11 gold. Alignment links are separated into two outer rings to facilitate viewing. The exterior of the circle features two rings illustrating the following: Inner ring: orthologous protein conservation. The histogram shows site-specific entropy among vertebrate species in black. High entropy reflects high dissimilarity. For comparative measure, the number of species containing the aligned region in question is below the histogram in gray. Low species count indicates amino acid indels. Outer ring: GC content. The heat map indicates relative GC content at the third codon position (GC3), where high GC content is red and low GC content is blue.
Overexpression of these DUBs has been noted in various human cancers, which may be attributable to their collective regulation of oncogenic proteins. For instance, all three paralogs regulate the type I TGF-β receptor while USP15 and USP11 also regulate several of its downstream effectors [4, 12, 13]. Conversely, whereas USP4 and USP15 target p53-inhibiting ligases ARF-BP1 [14] and MDM2 [15], respectively, USP11 stabilizes p53 [16] as well as several other tumor suppressors including PML [17], BRCA2 [18] and Mre11 complex members MRE11 & RAD50 [2]. In sum, though these paralogs are functionally redundant in some capacities, each appears to have undergone substantial subfunctionalization and neofunctionalization. A summary of their known protein interactions is presented in Table 2.
Functional versions of USP4, USP15 and USP11 are detectable in most branches of the vertebrate lineage including human. Of the three, USP4 and USP15 are most similar in terms of sequence identity (Fig. 1) and deubiquitination substrates (Table 2), which is consistent with the (USP11,(USP15,USP4)) branching pattern observed in phylogenetic analyses of these DUBs [19, 20]. This would suggest that the duplication that gave rise to USP4 and USP15 occurred most recently. However, a survey of USP paralogs encoded by metazoan genomes (Fig. 2) contradicts this hypothesis: while functional USP4 and USP15 are present in cartilaginous fish at the emergence of the gnathostome branch, USP11 is not identifiable until bony vertebrates make their appearance. What is more, all single-copy USP sequences have most identity with either USP4 or USP15. It is nevertheless possible that the USP11 duplication occurred earlier though its traces were erased by pseudogenization in deeper-branching species. One phylogeny represents the USP4, USP15 and USP11 relationship as a trifurcation [21], acknowledging its cryptic nature.

Phylogenetic distribution of USP4, USP15 and USP11. Red, blue, green, purple and black boxes represent USP4, USP15, USP11, ancestral (single copy) USP, and pre-USP ancestor sequences, respectively. Sequences are annotated and aligned according to their Reciprocal Best BLAST Hits (RBBHs), where lateral positioning of ancestral sequences indicates relative identity to human USP sequences. Translucent diagonally striped boxes indicate pseudogenes. Orange arrows indicate disruptive LINE1 element insertions in gibbon USP15 and green arrows indicate potentially disruptive insertion of a repetitive sequence of unknown origin in zebrafish USP15. Highlighted vertical bars indicate poly-glutamate sequences in USP15 and USP11
To understand the evolutionary changes in sequence, structure, and function among these paralogs, it is very important to know the temporal sequence of duplication. This enables us to determine which are the ancestral states and which are the derived states that potentially represent adaptation in response to an ancient environment. This motivated us to do phylogenetic studies to characterize the branching pattern and the timing of duplication events. An integrative in silico approach probing these systematic changes in a comparative genomic framework was employed to trace the duplication and subsequent radiation of USP4, USP15 and USP11. We first quantified and characterized USP paralogs in a set of representative metazoan genomes and delineated their divergence times in reference to known whole genome duplication events. We then evaluated ortholog variability to gain insight into the evolutionary processes that gave rise to the three paralogs observable in humans.
Results
Phylogenetics based on aligned nucleotide and amino acid sequence
Fifty USP4, USP15 and USP11 coding sequences from 23 representative vertebrate and invertebrate genomes (listed in Table 1, Material and Methods) were aligned using MUSCLE [22] with Gblocks cleaning [23], yielding an aligned length of 3981 sites. For phylogenetic reconstruction, we used the maximum likelihood method implemented in DAMBE [24] with the estimated transition/transversion ratio, the F84 model, and Amphimedon queenslandica (sea sponge) as the outgroup. The resulting unrooted tree (Fig. 3) has drastically different evolutionary rates among different lineages, with USP11 evolving particularly faster than other lineages. We performed a likelihood ratio test of the molecular clock hypothesis with the 50 sequences and the TN93 model, and the clock hypothesis is strongly rejected (lnL without clock = −17452.3864, lnL with clock = −17630.0485, likelihood ratio chi-square = 355.3242, DF = 48, p < 0.0001). We have also tested the clock hypothesis by using the third codon positions only, but the clock hypothesis is still strongly rejected (lnL with no clock = −4053.1815, lnL with clock = −4116.3185, Likelihood ratio chi-square = 126.2739, DF = 48, p < 0.0001). Thus, the paralogous sequences are not appropriate for dating. Indeed, age-calibrated phylogenetic dating of the codon sequences generated a tree that placed the divergence of USP15 vertebrate sequences before that of the single-copy ancestral sequences (Additional file 1: Figure S1). This erroneous topology reflects a discord between the substitution model’s assumptions and the nature of the sequences: USP15 orthologs are situated in low-GC regions in vertebrates (human, mouse, chicken, lizard) while USP11 and USP4 are in moderately high-GC isochores. This bias is reflected in their respective GC3 contents (Fig. 1) and thus violates the fundamental assumption of time homogeneity of all practical substitution models. We note that the paralogous genes in vertebrate species are often located in different GC isochores [25, 26]. For this reason, a nucleotide-based or codon-based analysis may bias phylogenetic estimation. To address this problem, we have also analyzed aligned amino acid sequences of the 23 species by the likelihood method. We have adopted the approach recommended by Rodriguez-Ezpeleta et al. [27] by recoding amino acids by size and polarity into four groups: small and polar (SCTND), large and polar (QEKRHY), small and non-polar (PAGV), large and non-polar (ILMFW). This approach not only results in more robust phylogenetic reconstruction, but also dramatically reduces computation time. The resulting tree (Fig. 4) is largely concordant with the maximum likelihood tree topology based on aligned nucleotide sequences (Fig. 3), i.e., USP15 splitting first from USP4/USP11, followed by the USP4 and USP11 split, with the primitive species encompassing a single ortholog clustered close to the root.

Maximum likelihood reconstruction of aligned codon sequences. A maximum likelihood tree of three paralogous genes from representative vertebrate species is represented together with their orthologs from invertebrate species. The unrooted tree was constructed with the F84 model and the maximum likelihood method implemented in DAMBE

Maximum likelihood reconstruction of recoded amino acid sequences. Depicted is a maximum likelihood tree of 50 aligned amino sequences after recoding amino acids by size and polarity into four groups: small and polar (SCTND), large and polar (QEKRHY) small and non-polar (PAGV), large and non-polar (ILMFW). The rooted tree was produced using the ProtML method implemented in DAMBE
The branching pattern of Figs. 3 and 4 enables us to infer an approximate time for gene duplication events. The USP15 lineage splits from (USP4,USP11) during the period between the divergence of vertebrates from primitive chordates (from 581 to 460.6 millions of years ago, or MYA [28]) and the branching of shark from teleost (462.5 to 421.75 MYA [28]), corresponding to the timing of a known whole genome duplication event [29]. A second gene duplication leads to the USP4/USP11 split which occurred in the common ancestor of bony fishes represented by gar, fugu, zebrafish and coelacanth (421.75 to 416 MYA [28]). USP11 is absent in shark. Given that the shark genome has evolved little [30], we may infer that the absence is ancestral instead of secondary loss, i.e., the USP4/USP11 split occurs after the divergence of shark from the ancestor of teleosts.
Synteny of USP11 and USP4 loci supports duplication in a Euteleostome ancestor
Gene homologs often bear not only high sequence identity to their ancestors, but can also retain their genomic context. Synteny, the linear conservation of physically linked gene clusters within or between genomes, can be revelatory of paralogous or orthologous evolutionary relationships. We thus conducted a comparative analysis of the genomic region encompassing USP4 in Callorhincus milii (elephant shark) and the regions surrounding USP4 and the USP11 pseudogene in Lepisosteus oculatus (spotted gar), representing putative pre- and post-duplication loci. We found that several genes adjacent to shark USP4 map physically near to the USP4 orthologs in gar and other higher vertebrates including human and anole (Fig. 5). In fact, the synteny of the region is remarkably well conserved after duplication: in addition to USP11, six other functional paralogs of genes surrounding USP4 in shark and in gar can be identified within 1 Mb of gar pseudo-USP11, while these co-duplicates are absent from the shark genome. Invertebrate genomes likewise encode only a single copy of these genes. In contrast, no USP11 co-duplicates can be identified at the USP15 locus. This supports our inferred branching patterns (Figs. 3 and 4) and altogether suggests that a concerted duplication of the USP4 chromosomal region median to the divergences of gnathostomes and euteleostomes gave rise to USP11.

Shared synteny of USP4 and USP11 loci in Euteleosts. a Illustrated comparison of USP loci for elephant shark (Callorhincus milii), spotted gar (Lepisosteus oculatus), green anole (Anolis carolinensis) and human (Homo sapiens). Genes are represented by arrows, where black outlines indicate paralogous genes and striping indicates pseudogenes. Paralogs shared by USP4 and USP11 are coloured gold, while those shared by USP4 and USP15 are coloured purple. Genomic location of loci is indicated to the right. Upper and lower estimates of divergence times (in millions of years) indicated to the left for the following clades (in ascending order): jawed vertebrates (incl. shark), euteleosts (incl. gar), tetrapods (incl. anole) and mammals (human). Stars indicate inferred divergence times for USP4-15 (purple) and USP4-USP11 (gold). b Schema of paralogous gene collinearity and rearrangement events in (USP4-USP11) and (USP4-USP15) loci
As a consequence of significantly different rates of evolution, Bayesian molecular dating of USP4 and USP11 aligned sequences overestimates their divergence time at 583–885 MYA. Three parallel runs of aligned USP4, USP11 and ancestral USP sequences with six calibration points converged at the rooted tree topology shown in Additional file 2: Figure S2 (note that several of the speciation node patterns and timing are largely inconsistent with known evolutionary relationships). Two sets of identified co-duplicates, RBM5/UBA7 and RBM10/UBA1, are co-localized with USP4 and USP11 respectively throughout vertebrate evolution, and can be used to date the duplication event by proxy. While neither RBM5/RBM10 nor UBA7/UBA1 follow a strict molecular clock, ∆lnL of RBM is greatly reduced compared to that of USP4 and USP11 (∆lnLUSP = 177.6621, ∆lnLRBM = 77.8252; ∆lnLUBA = 412.3984). Fig. 6 presents a phylogenetic reconstruction of RBM5 and RBM10 using a relaxed molecular clock; at 512 MYA, the 95 % credible interval upper bound of the predicted divergence time for these co-duplicates falls nearer the expected range and thus represents a rough estimate for the timing of duplication of the USP4 loci.

Bayesian dating of aligned co-duplicate codon sequences. Phylogenetic reconstruction and fossil-calibrated dating of aligned codon sequences for RBM5 and RBM10 was generated using BEAST v. 1.8. 95 % credible intervals are indicated. Calibration points were obtained from TimeTree. The gold star indicates the inferred RBM5-RBM10 divergence time. Red stars indicate major deviations from true topology
We believe our analyses provide overwhelming evidence in favor of a (USP15,(USP4,USP11)) branching pattern as opposed to the (USP11(USP4,USP15)) pattern that would be inferred based on sequenced relatedness [19, 20]. We posit that USP11 experienced greater coding sequence drift immediately following its duplication, resulting in complete pseudogenization in some species (e.g. gar) while in others a fast-evolving (Figs. 3 and 4), subfunctionalized (Table 2) protein emerged that is less similar than its well-conserved ancestors, USP4 and USP15. Adopting this novel understanding of their evolutionary relationship, we next examined the variability among USP homologs.
Signature features of USP paralogs
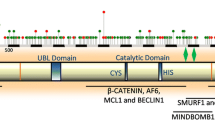
Four key molecular traits are thought to engender paralog functional radiation: structure-function innovations, distinctive spliced isoforms, altered cellular regulation (via post-translational modification), and specific spatiotemporal expression patterns [31]. While the defining domain architecture presented in Fig. 1 is pervasive in all USP4, USP11 and USP15 as well as ancestral (single copy) homologs, divergence among the structured domains and the unstructured linking regions is observed, which has been reported to confer differential enzymatic properties [20, 32]. We herein derive the constitutive evolutionary differences, or molecular signatures, that have defined USP4, USP15 and USP11 from their inception using branching pattern knowledge and ancestral state reconstruction. Filtering paralog-defining features is more informative than monospecies sequence alignment, which contains intraparalog (species-specific) variations likely to be especially pronounced in the fast-evolving USP11. These conserved molecular signatures, explained below, are summarized in Fig. 7.

Summary of signature features of USP4, USP15 and USP1. a Alignment of signature sequences for USP4 (red), USP15 (blue) and USP11 (green) with the single-copy sequence (purple). The DUSP-UBL compound domain is shown and coloured as in Fig. 1. b Schematic illustration of signature phosphorylation sites and loss of alternatively spliced exon in USP11. Vertical bars traverse sequences with shared phosphorylation sites
Structure-function innovations
First let us consider the molecular signatures of structured regions in USP4, USP15, and USP11. The “domain in USP” (DUSP) and “ubiquitin-like” (UBL) structured regions form the N-terminal domain that distinguishes this subgroup of USPs. DUSP-UBL domains mediate some enzyme-substrate interactions [9, 33–35] and confer intrinsic regulatory capacities that have been structurally modeled for mammalian USP4, USP15 and USP11 [20, 32, 35, 36]. For instance, USP4 dimerization occurs in equilibrium through this domain, while neither USP15 nor USP11 are expected to dimerize in vivo [20]. The DUSP-UBL domain of USP4 also regulates ubiquitin active- site binding dynamics through its association with the unstructured insert region [32], though the absence of key residues impedes this regulatory function in human USP11 [32, 35]. The enzyme kinetics of USP15 are more similar to that of USP4 [32]. Given our derivation of their duplication chronology, it seems likely that the loss of ubiquitin-exchange regulation in USP11 is a derived and not ancestral state, though structural information is available only for mammalian proteins. Fig. 7a presents an alignment of the DUSP-UBL domains of an ancestral USP with the signature sequences of USP4, USP15 and USP11. Lancelet was selected as the ancestral species because it is the closest single-copy relative (Fig. 2). In addition, the domain sequence is identical in Branchiostoma floridae and the newly sequenced B. belcheri, two lancelets that have experienced a high degree of protein evolution [37], suggesting that it is an accurate depiction of a pre-duplication USP. USP signature sequences indicate residues that are conserved in a majority of members from each phylogenetic clade (elimination of species-specific substitutions). While the key residues are largely conserved in USP4, USP15, and the ancestral USP, disruption of the hydrophobic pocket and shortening of DUSP-UBL linker [32, 35] are signatures of USP11. This derived state implies that USP11 has had a different mode of action throughout time.
The two parts of the structured catalytic domain of these USPs, D1 and D2, are the most highly conserved regions among paralogs and orthologs (Fig. 1). Both are required for catalytic activity, and their conservation extends beyond the USPs under current consideration to the entire USP subfamily of deubiquitinating enzymes.
Distinctive spliced isoforms
Whereas the seventh exon (E7) is alternatively spliced in USP4 and USP15, a corresponding exon is absent in USP11. The flexible linker region separating the DUSP-UBL and catalytic domains is roughly 20 residues long in USP11, its length in USP4 and USP15 short isoforms and the minimal length required for the aforementioned domain interaction [32]. Shark USP4 and the lancelet single-copy USP, ancestral to USP11, contain E7; what is more, both long and short isoforms have been reported in chondrichthyes. Thus, the “permanent skipping” of E7 in all USP11 represents a derived state. Alterations in the stoichiometry of USP4 isoforms have been reported for a rare bone disease [38], though the functional consequences of E7 alternative splicing have not been studied. In all species, the polypeptides encoded by USP4 and USP15 E7 are serine-rich, and many serve as putative post-translational modification (PTM) sites as identified in large-scale studies on human proteins [39]. In sum, the loss of E7 is a signature derived state of USP11 with potential functional or regulatory implications.
Altered cellular regulation
Post-translational modification (PTM) regulation can differ among gene duplicates. Some PTM sites are well conserved while others stably differentiate the USP paralogs in question. For one, Ser445 (a known Akt phosphorylation site [13]), there is conservation in all USP4, USP11, USP15 and ancestral homologs. There are, on the other hand, multiple cases wherein a putative phosphorylation site has been lost or gained in USP11 relative to its ancestor, USP4. Within the insert region, USP4 Ser675 and Ser680 (identified phosphorylation substrates in multiple studies [40–48]) are conserved in USP15 but absent from USP11. Similarly, in an alignment of all USPs, putative phosphorylation site USP4 Tyr539 [39] is conserved in USP4 and USP15 while it is substituted by Phe in USP11. Slightly downstream, USP11 has Tyr551 (a reported phosphorylation site [39]) and Tyr554 whereas His and Phe, respectively, are universally present in USP4 and USP15. Still within the insert region, at positions 607 and 608, there exists in USP11 a pair of tyrosines that have been identified as phosphorylation sites in several large-scale studies [39]. The region in question aligns poorly with other paralogs, though there are two reported, albeit low confidence, serine phosphorylation sites in USP4 and none in USP15. As previously mentioned, the alternatively spliced exon, lost in USP11, contains several reported phosphorylated serines in USP4 and USP15 [39].
The N and C-termini are remarkably different among USP4, USP11 and USP15. The N-terminus of USP11 is longer, more disordered and more hydrophobic (rich in alanine). In addition, the C-termini of all gnathostome USP15 present a segment rich in aspartic acid, glutamatic acid and asparagine (e.g. human: 962-DEDSNDNDNDIENEN-976; shark: 978-DEDCNENDVENEN-990), except those of teleost fish, which instead have C-terminal segment(s) exceptionally rich in glutamate (e.g. zebrafish: 775-EKEEEEEDEDEEDVNDSEQEED-795; tongue sole: 966-DEEDEEEEEEEEGEVEEEDEEEEEGRE-981, 1015-NEREDEEEEEEEEEEEEEEEQE-1035). A poly-E repetitive sequence is also found in USP11 of various organisms, including teleost fish, some reptiles, the opossum and the Chinese hamster. These regions are schematically highlighted in Fig. 2. Aspartic acid and asparagine residues can be hydroxylated [49], though it remains to be seen whether any hydroxylation of such residues occurs within the acidic domains of the USPs. In addition, the D- & N- rich C-terminus of non-teleost fish USP15 presents two validated serine phosphorylation sites [39, 50–55], absent from USP4, whereas human USP11 has seven of these sites [39, 50, 55–57] within its final 20 residues that are conserved among mammals. While many of these conserved and differential phosphorylation sites remain to be functionally characterized, most are all located within unstructured regions, namely the insert, linker, and C-terminal regions. This is consistent with reports that disordered region often serve as PTM substrates [58–61] and changes in PTM regulation contributes to the functional divergence of paralogs [31]. In addition to phosphorylation and hydroxylation the disordered regions of the USPs may be subject to a number of other modifications including acetylation, methylation, and/or the addition of peptide moieties such as ubiquitin or SUMO. The contribution of this growing repertoire of PTMs to USP4, USP15, and USP11 regulation has yet to be established.
In sum, each paralog has distinctive signature features that represent common evolutionary categories namely structure-function innovations, distinctive spliced isoforms and altered cellular regulation. The fourth common differentiating trait, different spatiotemporal expression patterns, will be discussed in a later section.
Variable mechanisms of USP11 loss
As depicted in Fig. 2, USP11 has been lost multiple times throughout vertebrate evolution. In select fish, reptile and mammalian genomes, the syntenic loci where USP11 habitually resides hosts USP11 pseudogenes in lieu of functional genes. For instance, among reference primate genomes, USP11 is uniquely pseudogenized in Gorilla gorilla. The phylogenetic dispersal of pseudogenization suggests that these events occurred independently. In birds such as chicken, however, the entire syntenic region containing USP11 has been deleted while USP4, USP15 and their respective neighbouring genes are conserved, as illustrated in Fig. 8. A chromosomal rearrangement event in the avian ancestor may be responsible for the deletion of the segment containing USP11.

Synteny of USP4, USP15, and USP11 in anole compared to chicken and human. USP11 and surrounding region is absent in chicken. Asterisks represent the positions of anole USP genes (green indicates USP11, red is USP4, and teal is USP15)
In vivo demonstration of a minimal requirement for USP4 or USP15
Whereas the variable retention or loss of USP11 suggests that it is dispensable, all species contain either or both USP4 and USP15. It is reasonable to speculate that one functional copy from this gene pair is essential for viability, and we tested this hypothesis using mouse strains in which the Usp4 or Usp15 gene had been inactivated by the insertion of a retroviral provirus. The TF2497 and TF2834 strains have gene-trap proviruses in the Usp4 and Usp15 genes respectively; in both cases the insertion disrupts the gene near the 5′ end and precludes expression of a functional gene product (indeed no transcript can be detected by the sensitive method of reverse-transcription/polymerase chain reaction). Both strains are viable when homozygous for the inactivating insertion, and we have found no evidence of reduced fertility or obvious phenotypic effects. The lack of phenotypic consequences could be explained by functional redundancy between the USP4 and USP15 enzymes, but to determine if this is the case we conducted genetic crosses between mice heterozygous for the two genes (approval for these experiments was provided by the Animal Care Committee, University of Ottawa). As reported in Table 3, of 166 pups born we were unable to identify any progeny that had inactivating mutations in all four alleles, though all other expected genotypes were detected. Given that one of sixteen pups would be expected with the compound null genotype the lethality of this genotype can be asserted with a high level of confidence (from a binomial analysis p = (15/16)166 = 0.000022). We therefore conclude that USP4 and USP15 have sufficient functional redundancy to rescue inactivating mutations in a reciprocal fashion. The presence of functional USP11 genes is insufficient to rescue pups that are null for both USP4 and USP15. Mice are viable with one functional allele from the USP4/USP15 gene pair, though some apparent deviation from Mendelian ratios suggests that there may be phenotypic consequences of this haploinsufficient state. The nature of these consequences will be explored in future studies.
Discussion
In the present work we have established the duplication chronology of a subgroup of highly networked ubiquitin-specific proteases, USP4, USP15 and USP11, and have characterized their subsequent radiation. According to the widely accepted 2R theory [29], vertebrate genomes have undergone two rounds of whole genome duplication (WGD). While it was conventionally assumed that these WGD events predated the divergence of jawless and jawed vertebrates [62], recent analysis of the elephant shark genome [30] placed at least one WGD event median to cyclostome and gnathostome divergence. In fact, subsequent studies have suggested that the 2R events occurred independently in cyclostomes and gnathostomes [63, 64], and that the former expansion was further shaped by an additional, lamprey-specific WGD. Thus, the USP15-like duplicate in lampreys is likely not orthologous to gnathostome USP4; rather, USP4 and USP15 appear to be ohnologs derived from WGD in a jawed vertebrate ancestor. In addition, the USP15-likeness of the lamprey version suggests that this paralog is the ancestral sequence, though there is no consensus among invertebrates as to whether their single copy most resembles USP4 or USP15 (Fig. 2). The issue cannot be settled by genomic synteny reconstruction, which is considerably more difficult in earlier species due to increased divergence time and the present lack of chromosomal assembly data for many species. In contrast, well-conserved intragenomic synteny points to the emergence of USP11 as the result of a more recent duplication event that does not coincide with any reported WGD event; it is likely the product of a small-scale duplication (SSD). The characterization of USP4/15 and USP4/11 duplications as WGD and SSD, respectively, corroborates well with reported trends for these phenomena: SSD-derived paralog sequences tend to evolve faster and are more functionally divergent [65].
Several notable differences in USP composition exist between and within clades. Primate species appear to have inconsistent USP repertoires: USP15 was inactivated by insertion of a LINE1 element in the gibbon, while erasure of USP11 and reduction of unstructured USP4 domains can both be observed in the gorilla. USP11 was also lost in the avian ancestor, inferred by the consistent absence of its genomic locus in all bird genomes (Fig. 8). Curiously, avian USP4 presents notable deviations from the signatures of this paralog: of the six bird genomes surveyed, all bear mutations in crucial residues for the ubiquitin-exchange mechanism [32], i.e. Arg40 and/or Met24 mutated in all, disruption of DUSP-UBL linker residues (a.a. 88–92) in chicken, QQD box region deleted in duck, and so on. In fact, USP4 is also more divergent in other species where USP11 was lost, which lead for example to the consistently incorrect branching patterns for frog and gorilla USP4 (Figs. 3 and 4; Additional file 1: Figure S1 and Additional file 2: Figure S2). What is more, avian USP4 adopts some USP11 signatures: these have collectively lost the Ser675 and Ser680 phosphorylation sites, while USP4 of the pigeon and zebra finch have also lost E7. The loss of these features that define all other USP4 (and USP15) is a derived state of USP11 and may thus represent a homoplastic convergence of avian USP4 toward the USP11 sequence.
Most of the signature features distinguish USP11 from USP4 and USP15, though the divergence of these last two is of practical interest due to their high protein sequence identity (Fig. 1), functional overlap (Table 2), and capacity for reciprocal rescue at the organismal level (Table 3). USP4 and USP15 differ in their codon usage: USP15, located in GC-poor isochores, employs more AT-ending codons than USP4. Low GC content is common in germ-line specific genes [25]. USP15 is in fact expressed at notably elevated levels in mature oocytes [66] (oocytes being the cell type for which its expression is the highest in mice [67]) while USP4 is at low abundance throughout oocyte maturation [68]. USP4 is predominantly expressed in somatic cells, particularly those of the immune system [67]. The distinct spatiotemporal expression patterns of USP4 and USP15 could explain why these redundant proteins have been maintained: vertebrate genomes could optimally encode two versions of an ancestral protein to accommodate its important roles in germ and somatic cells. While we show that one functional copy of USP4 or USP15 is a minimum requirement for viability (Table 3), the observed departure from Mendelian ratios may arise from a functional deficiency in oocytes haploinsufficient for USP15. Planned experiments (including in vitro culture of early embryos) should be informative in this regard. The expansion of TGF-β pathway substrates in USP15 may reflect an enhanced role in the regulation of oocyte development [69–71], while USP4 may have become the USP of greater importance in innate immunity pathways, as reflected by an increased number of substrates (Table 2). Further, an inserted in-frame zebrafish-specific repetitive element has modified the USP15 catalytic domain coding sequence of this species. While it remains to be seen whether the enzymatic activity of USP15 has been altered or inactivated by this insertion, we anticipate that perturbation of USP15 will provide insights into DUB network rewiring in the zebrafish. As a model system that is amenable to the testing of hypotheses through genome manipulation, the zebrafish should be ideal for future investigations of the respective roles and expression patterns of USP4 and USP15. The expression pattern of USP11 is notably distinct: without exception in human, mouse, rat, and pig its expression is predominantly neuronal [67]. In contrast to its paralogs [4], USP11 exerts a protective effect in glioma [17] as it stabilizes many tumor suppressors (Table 2).
While all organisms minimally retain USP4 or USP15 and some have in addition USP11, none have more than these three closely related USPs (including teleost fish and lamprey, which have experienced a third whole genome duplication). Genomes coding for USP4, USP15 and USP11 may thus represent the optimal system, where USP11 is an optional descendant whose functional contributions remain largely unexplored. Prior to the advent of whole genome sequencing it would have been reasonable to predict that with increasing organismal complexity there would be increasing complexity in molecular systems essential for development and tissue homeostasis, and the machinery relating to ubiquitin conjugation and removal would be high on the list of molecular systems expected to become more elaborate. In the case of deubiquitinating enzymes such a prediction would have been validated by whole genome sequencing: whereas vertebrate genomes encode more than 50 USP enzymes, roughly half this number are encoded by the genome of the fruit fly, and roughly half again by the genome of the budding yeast. In the evolution of complex molecular pathways the additional USP genes generated by WGD or SSD events could have provided substrate material for neofunctionalization or subfunctionalization. One can easily imagine how innovation within an augmented USP repertoire could facilitate innovation in complex signaling cascades (as exemplified by the NF-κB pathway central to innate immunity, or the TGF-β pathway). While we have restricted our focus to USP4, USP15, and USP11 we believe our analysis of the evolutionary history of this subset of deubiquitinating enzymes has been instructive in a broader sense. It demonstrates, for example, that BLAST alignments, while intuitive, can be misleading in the construction of an accurate USP phylogeny. Sequence similarity alone would not predict the branching pattern summarized in Fig. 9, which arose from extensive phylogenetic reconstruction of the DUSP-containing USP family incorporating aligned nucleotide and amino acid sequences, taxonomic distribution and patterns of synteny. We are hopeful that our approach can serve as a template for future studies of USP gene evolution, and will ultimate lead to a better understanding of the origins of this important gene family.

Phylogeny of USP genes. Percentages refer to amino acid identity between indicated USPs after global alignment. The pink asterisk denotes a whole genome duplication event, and the yellow asterisk denotes a small-scale duplication event involving USP4 and surrounding genes
Methods
Sequence retrieval
Identification and proper annotation of homologs in an array of species is a first essential step in studying the evolution of duplicated genes. Coding sequences were retrieved from GenBank [72] and from genome project databases [62, 73, 74] using the well-annotated human sequences for USP4, USP15 and USP11 as tBLASTn queries [75]. Reciprocal Best BLAST Hit (RBBH) annotation transfer was applied to unannotated genomes. Accession IDs for all sequences are below.
Sequence entropy
The site-wise Shannon entropy of aligned vertebrate USP amino acid sequences was calculated using DAMBE [24]. The results were plotted as a histogram using Circos [76].
GC content analysis
The seqinr package in R was employed to generate plots for the GC content of the third codon positions (GC3) of the Homo sapiens USP4, USP15 and USP11 coding sequences using a sliding window of width 10. A heatmap of GC3 content was generated using Circos [76].
Species tree reconstruction
A taxonomic phylogeny was generated using PhyloT [77]. Paralog affiliations were attributed as per their RBBH (described in Sequence retrieval). Putative non-processed pseudogene loci were confirmed using GenScan [78]. The SynMap function in CoGe [79] enabled comparison of the synteny of USP neighbouring regions in Anolis carolinensis, Homo sapiens, and Gallus gallus, which was visualized using Circos.
Phylogenetic analysis
USP4, USP15 and USP11 codon sequences were aligned using MUSCLE [22]. The maximum-likelihood method using estimated transition/transversion ratio and F84 model, as implemented in DAMBE [24], was used to derive a phylogeny rooted on Amphimedon queenslandica. Molecular clock analyses were also conducted using DAMBE [24].
Divergence dating
Codon alignments were produced by the MUSCLE algorithm using default GBlocks parameters as implemented in TranslatorX [22]. AIC and LRT nucleotide substitution model tests in DAMBE [24] designated the Generalized Time Reversible (GTR) as most appropriate for all alignments. BEAST v.1.8.2 [80], a Bayesian Markov chain Monte Carlo (MCMC)-based phylogenetic dating program, was employed to quantify USP age-calibrated divergence times. All analyses used a log normal relaxed molecular clock. The USP4/USP11 analysis used six calibration points obtained from TimeTree [28] (in millions of years): Dog-Cow[USP4,USP11]: 60, Human-Opossum[USP4]: 112, Human-Anole [USP4, USP11]: 320 & Gar-Human [USP4]: 418. The RBM5/RBM10 analysis employed four pairs of calibration points: Human-Gorilla [RBM5,RBM10]: 8, Mouse-Rat[RBM5,RBM10]: 10.4, Human-Anole[RBM5,RBM10]: 320 & Human-Zebrafish [RBM5,RBM10]: 425. Tracer was used to verify similar convergence after 20 million steps for 3 runs in each case.
Mouse genetic crosses
TF2497 and TF2834 strains were purchased from Taconic Laboratories (Germantown, New York, USA), and were housed in a barrier facility at the University of Ottawa under protocol ME-305, approved by the Animal Care Committee, University of Ottawa. The strains were crossed to obtain mice heterozygous for proviral insertions in both the Usp4 and Usp15 genes. Eight pairs of compound heterozygous mice were mated under standard conditions, and progeny were obtained for genotyping. Genotyping was performed at 3–4 weeks of age, using tissue from ear punches. DNA was prepared using the REDExtract-N-Amp™ Tissue PCR Kit (Sigma-Aldrich Canada, Oakville, Ontario) and polymerase chain reaction was performed using the kit reagents. For genotyping of USP4 the forward primer used was derived from the third exon (upstream of the proviral insertion site): 5′- CCAGCAGCCTATTGTCAGAA -3′, where reverse primers were derived from the third intron (downstream of the proviral insertion site): 5′- TCAGTACTTAGGGATCTCTGA -3′ or from the neomycin phosphotransferase gene within the provirus: 5′- AACCTGCGTGCAATCCATCT -3′. Amplification conditions for USP4 were as follows: initial denaturation at 95C for 3 min followed by 30 cycles of 95C for 30 s, 57C for 30 s and 72C for 60 s and a final cycle at 72C for 5 min. A PCR product of approximately 250 base pairs was generated from the wild type gene, whereas the disrupted gene generated a product of approximately 1000 bp as detected by ethidium bromide staining of 1 % agarose gels. For USP15 a similar strategy was adopted using the forward primer: 5′ – GGTTTGAAGGATAACGTAGGC -3′, and reverse primers 5′ – ATAAACCCTCTTGCAGTTGCATC -3′ and 5′- GAGTACCTAACAGGCACTTGAGACG -3′. USP15 PCR conditions were similar except that annealing was done at 55C for 30 s and elongation at 72C was reduced to 45 s.
References
Grabbe C, Husnjak K, Dikic I. The spatial and temporal organization of ubiquitin networks. Nat Rev Mol Cell Biol. 2011;12:295–307.
Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403.
Aggarwal K, Massagué J. Ubiquitin removal in the TGF-β pathway. Nat Cell Biol. 2012;14:656–7.
Eichhorn PJA, Rodón L, Gonzàlez-Juncà A, Dirac A, Gili M, Martínez-Sáez E, et al. USP15 stabilizes TGF-β receptor I and promotes oncogenesis through the activation of TGF-β signaling in glioblastoma. Nat Med. 2012;18:429–35.
Zhao B, Schlesiger C, Masucci MG, Lindsten K. The ubiquitin specific protease 4 (USP4) is a new player in the Wnt signalling pathway. J Cell Mol Med. 2009;13:1886–95.
Fan Y-H, Yu Y, Mao R-F, Tan X-J, Xu G-F, Zhang H, et al. USP4 targets TAK1 to downregulate TNFα-induced NF-κB activation. Cell Death Differ. 2011;18:1547–60.
Xiao N, Li H, Luo J, Wang R, Chen H, Chen J, et al. Ubiquitin-specific protease 4 (USP4) targets TRAF2 and TRAF6 for deubiquitination and inhibits TNFα-induced cancer cell migration. Biochem J. 2012;441:979–86.
Schweitzer K, Bozko PM, Dubiel W, Naumann M. CSN controls NF‐κB by deubiquitinylation of IκBα. EMBO J. 2007;26:1532–41.
Song EJ, Werner SL, Neubauer J, Stegmeier F, Aspden J, Rio D, et al. The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev. 2010;24:1434–47.
Long L, Thelen JP, Furgason M, Haj-Yahya M, Brik A, Cheng D, et al. The U4/U6 recycling factor SART3 has histone chaperone activity and associates with USP15 to regulate H2B deubiquitination. J Biol Chem. 2014;289:8916–30.
Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, et al. InnateDB: systems biology of innate immunity and beyond—recent updates and continuing curation. Nucleic Acids Res. 2013;41:D1228–33.
Al-Salihi MA, Herhaus L, Macartney T, Sapkota GP. USP11 augments TGFβ signalling by deubiquitylating ALK5. Open Biol. 2012;2:120063.
Zhang L, Zhou F, Drabsch Y, Snaar-Jagalska E, Mickanin C, Huang H, et al. USP4 is regulated by Akt phosphorylation and deubiquitylates TGF-beta type I receptor. Nat Cell Biol. 2012;14:717–26.
Zhang X, Berger FG, Yang J, Lu X. USP4 inhibits p53 through deubiquitinating and stabilizing ARF‐BP1. EMBO J. 2011;30:2177–89.
Zou Q, Jin J, Hu H, Li HS, Romano S, Xiao Y, et al. USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat Immunol. 2014;15:562–70.
Ke J, Dai C, Wu W, Gao J, Xia A, Liu G, et al. USP11 regulates p53 stability by deubiquitinating p53. J Zhejiang Univ Sci B. 2014;15:1032–8.
Wu H-C, Lin Y-C, Liu C-H, Chung H-C, Wang Y-T, Lin Y-W, et al. USP11 regulates PML stability to control Notch-induced malignancy in brain tumours. Nat Commun. 2014;5:3214.
Schoenfeld AR, Apgar S, Dolios G, Wang R, Aaronson SA. BRCA2 is ubiquitinated in vivo and interacts with USP11, a deubiquitinating enzyme that exhibits prosurvival function in the cellular response to DNA damage. Mol Cell Biol. 2004;24:7444–55.
Clague MJ, Barsukov I, Coulson JM, Liu H, Rigden DJ, Urbé S. Deubiquitylases from genes to organism. Physiol Rev. 2013;93:1289–315.
Elliott PR, Liu H, Pastok MW, Grossmann GJ, Rigden DJ, Clague MJ, et al. Structural variability of the ubiquitin specific protease DUSP-UBL double domains. FEBS Lett. 2011;585:3385–90.
Jacq X, Kemp M, Martin NMB, Jackson SP. Deubiquitylating enzymes and DNA damage response pathways. Cell Biochem Biophys. 2013;67:25–43.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–52.
Xia X. DAMBE5: a comprehensive software package for data analysis in. Mol Biol Evol. 2013;30:1720–8.
Vinogradov AE. Isochores and tissue-specificity. Nucleic Acids Res. 2003;31:5212–20.
Rodin SN, Parkhomchuk DV. Position-associated GC asymmetry of gene duplicates. J Mol Evol. 2004;59:372–84.
Rodríguez-Ezpeleta N, Brinkmann H, Roure B, Lartillot N, Lang BF, Philippe H. Detecting and overcoming systematic errors in genome-scale phylogenies. Syst Biol. 2007;56:389–99.
Hedges SB, Dudley J, Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22:2971–2.
Dehal P, Boore JL. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 2005;3:e314.
Venkatesh B, Lee AP, Ravi V, Maurya AK, Lian MM, Swann JB, et al. Elephant shark genome provides unique insights into gnathostome evolution. Nature. 2014;505:174–9.
Nguyen Ba AN, Strome B, Hua JJ, Desmond J, Gagnon-Arsenault I, Weiss EL, et al. Detecting functional divergence after gene duplication through evolutionary changes in posttranslational regulatory sequences. PLoS Comput Biol. 2014;10:e1003977.
Clerici M, Luna-Vargas MPA, Faesen AC, Sixma TK. The DUSP-Ubl domain of USP4 enhances its catalytic efficiency by promoting ubiquitin exchange. Nat Commun. 2014;5:5399.
Zhao B, Velasco K, Sompallae R, Pfirrmann T, Masucci MG, Lindsten K. The ubiquitin specific protease-4 (USP4) interacts with the S9/Rpn6 subunit of the proteasome. Biochem Biophys Res Commun. 2012;427:490–6.
Hayes SD, Liu H, MacDonald E, Sanderson CM, Coulson JM, Clague MJ, et al. Direct and indirect control of mitogen-activated protein kinase pathway-associated components, BRAP/IMP E3 ubiquitin ligase and CRAF/RAF1 kinase, by the deubiquitylating enzyme USP15. J Biol Chem. 2012;287:43007–18.
Harper S, Gratton HE, Cornaciu I, Oberer M, Scott DJ, Emsley J, et al. Structure and catalytic regulatory function of ubiquitin specific protease 11 N-terminal and ubiquitin-like domains. Biochemistry (Mosc). 2014;53:2966–78.
Harper S, Besong TMD, Emsley J, Scott DJ, Dreveny I. Structure of the USP15 N-terminal domains: a β-hairpin mediates close association between the DUSP and UBL domains. Biochemistry (Mosc). 2011;50:7995–8004.
Huang S, Chen Z, Yan X, Yu T, Huang G, Yan Q, et al. Decelerated genome evolution in modern vertebrates revealed by analysis of multiple lancelet genomes. Nat Commun. 2014;5:5896.
Klinck R, Laberge G, Bisson M, McManus S, Michou L, Brown JP, et al. Alternative splicing in osteoclasts and Paget’s disease of bone. BMC Med Genet. 2014;15:98.
Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40 (Database issue):D261–70.
Zanivan S, Gnad F, Wickström SA, Geiger T, Macek B, Cox J, et al. Solid tumor proteome and phosphoproteome analysis by high resolution mass spectrometry. J Proteome Res. 2008;7:5314–26.
Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–89.
Yu Y, Yoon S-O, Poulogiannis G, Yang Q, Ma XM, Villén J, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332:1322–6.
Gnad F, Young A, Zhou W, Lyle K, Ong CC, Stokes MP, et al. Systems-wide analysis of K-Ras, Cdc42, and PAK4 signaling by quantitative phosphoproteomics. Mol Cell Proteomics. 2013;12:2070–80.
Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, et al. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat Commun. 2012;3:876.
Nishi H, Fong JH, Chang C, Teichmann SA, Panchenko AR. Regulation of protein-protein binding by coupling between phosphorylation and intrinsic disorder: analysis of human protein complexes. Mol Biosyst. 2013;9:1620–6.
Trost M, Sauvageau M, Hérault O, Deleris P, Pomiès C, Chagraoui J, et al. Posttranslational regulation of self-renewal capacity: insights from proteome and phosphoproteome analyses of stem cell leukemia. Blood. 2012;120:e17–27.
Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJR, et al. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res. 2013;12:260–71.
Sharma K, D’Souza RCJ, Tyanova S, Schaab C, Wiśniewski JR, Cox J, et al. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014;8:1583–94.
Iyuke FO, Green JR, Willmore WG. Active Learning for the Prediction of Asparagine/Aspartate Hydroxylation Sites on Proteins. Calgary: ACTAPRESS; 2011.
Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, et al. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105:10762–7.
Gauci S, Helbig AO, Slijper M, Krijgsveld J, Heck AJR, Mohammed S. Lys-N and trypsin cover complementary parts of the phosphoproteome in a refined SCX-based approach. Anal Chem. 2009;81:4493–501.
Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3:ra3.
Christensen GL, Kelstrup CD, Lyngsø C, Sarwar U, Bøgebo R, Sheikh SP, et al. Quantitative phosphoproteomics dissection of seven-transmembrane receptor signaling using full and biased agonists. Mol Cell Proteomics. 2010;9:1540–53.
Phanstiel DH, Brumbaugh J, Wenger CD, Tian S, Probasco MD, Bailey DJ, et al. Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat Methods. 2011;8:821–7.
Bian Y, Song C, Cheng K, Dong M, Wang F, Huang J, et al. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J Proteomics. 2014;96:253–62.
Van Hoof D, Muñoz J, Braam SR, Pinkse MWH, Linding R, Heck AJR, et al. Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell Stem Cell. 2009;5:214–26.
Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci Signal. 2011;4:rs5.
Kurotani A, Tokmakov AA, Kuroda Y, Fukami Y, Shinozaki K, Sakurai T. Correlations between predicted protein disorder and post-translational modifications in plants. Bioinformatics. 2014;30:1095–103.
Vuzman D, Hoffman Y, Levy Y: Modulating protein-DNA interactions by post-translational modifications at disordered regions. Pac Symp Biocomput 2012:188–199.
Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208.
Ma B, Nussinov R. Regulating highly dynamic unstructured proteins and their coding mRNAs. Genome Biol. 2009;10:204.
Smith JJ, Kuraku S, Holt C, Sauka-Spengler T, Jiang N, Campbell MS, et al. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat Genet. 2013;45:415–21. 421e1–2.
Mehta TK, Ravi V, Yamasaki S, Lee AP, Lian MM, Tay B-H, et al. Evidence for at least six Hox clusters in the Japanese lamprey (Lethenteron japonicum). Proc Natl Acad Sci. 2013;110:16044–9.
Nah GSS, Tay B-H, Brenner S, Osato M, Venkatesh B. Characterization of the Runx gene family in a jawless vertebrate, the Japanese lamprey (lethenteron japonicum). PLoS One. 2014;9, e113445.
Fares MA, Keane OM, Toft C, Carretero-Paulet L, Jones GW. The roles of whole-genome and small-scale duplications in the functional specialization of saccharomyces cerevisiae genes. PLoS Genet. 2013;9, e1003176.
Kocabas AM, Crosby J, Ross PJ, Otu HH, Beyhan Z, Can H, et al. The transcriptome of human oocytes. Proc Natl Acad Sci. 2006;103:14027–32.
Zimmermann P, Hennig L, Gruissem W. Gene-expression analysis and network discovery using genevestigator. Trends Plant Sci. 2005;10:407–9.
Chen J, Torcia S, Xie F, Lin C-J, Cakmak H, Franciosi F, et al. Somatic cells regulate maternal mRNA translation and developmental competence of mouse oocytes. Nat Cell Biol. 2013;15:1415–23.
Dragovic RA, Ritter LJ, Schulz SJ, Amato F, Thompson JG, Armstrong DT, et al. Oocyte-secreted factor activation of SMAD 2/3 signaling enables initiation of mouse cumulus cell expansion. Biol Reprod. 2007;76:848–57.
Elvin JA, Yan C, Matzuk MM. Oocyte-expressed TGF-beta superfamily members in female fertility. Mol Cell Endocrinol. 2000;159:1–5.
Jackowska M, Kempisty B, Woźna M, Piotrowska H, Antosik P, Zawierucha P, et al. Differential expression of GDF9, TGFB1, TGFB2 and TGFB3 in porcine oocytes isolated from follicles of different size before and after culture in vitro. Acta Vet Hung. 2013;61:99–115.
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, et al. GenBank. Nucleic Acids Res. 2013;41(Database issue):D36–42.
Simakov O, Marletaz F, Cho S-J, Edsinger-Gonzales E, Havlak P, Hellsten U, et al. Insights into bilaterian evolution from three spiralian genomes. Nature. 2013;493:526–31.
Wyffels JL, King B, Vincent J, Chen C, Wu CH, Polson SW. SkateBase, an elasmobranch genome project and collection of molecular resources for chondrichthyan fishes. F1000Res. 2014;3:191.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Krzywinski MI, Schein JE, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45.
Letunic I, Bork P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39 suppl 2:W475–8.
Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94.
Lyons E, Pedersen B, Kane J, Freeling M. The value of nonmodel genomes and an example using SynMap within CoGe to dissect the hexaploidy that predates the Rosids. Trop Plant Biol. 2008;1:181–90.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73.
Herhaus L, Al-Salihi MA, Dingwell KS, Cummins TD, Wasmus L, Vogt J, et al. USP15 targets ALK3/BMPR1A for deubiquitylation to enhance bone morphogenetic protein signalling. Open Biol. 2014;4:140065.
Blanchette P, Gilchrist CA, Baker RT, Gray DA. Association of UNP, a ubiquitin-specific protease, with the pocket proteins pRb, p107 and p130. Oncogene. 2001;20:5533–7.
Villeneuve NF, Tian W, Wu T, Sun Z, Lau A, Chapman E, et al. USP15 negatively regulates Nrf2 through deubiquitination of Keap1. Mol Cell. 2013;51:68–79.
Wang L, Zhao W, Zhang M, Wang P, Zhao K, Zhao X, et al. USP4 positively regulates RIG-I-mediated antiviral response through deubiquitination and stabilization of RIG-I. J Virol. 2013;87:4507–15.
Pauli E-K, Chan YK, Davis ME, Gableske S, Wang MK, Feister KF, et al. The ubiquitin-specific protease USP15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci Signal. 2014;7:ra3.
Di Donato F, Chan EK, Askanase AD, Miranda-Carus M, Buyon JP. Interaction between 52 kDa SSA/Ro and deubiquitinating enzyme UnpEL: a clue to function. Int J Biochem Cell Biol. 2001;33:924–34.
Hou X, Wang L, Zhang L, Pan X, Zhao W. Ubiquitin-specific protease 4 promotes TNF-α-induced apoptosis by deubiquitination of RIP1 in head and neck squamous cell carcinoma. FEBS Lett. 2013;587:311–6.
Yamaguchi T, Kimura J, Miki Y, Yoshida K. The deubiquitinating enzyme USP11 controls an IkappaB kinase alpha (IKKalpha)-p53 signaling pathway in response to tumor necrosis factor alpha (TNFalpha). J Biol Chem. 2007;282:33943–8.
Sun W, Tan X, Shi Y, Xu G, Mao R, Gu X, et al. USP11 negatively regulates TNFalpha-induced NF-kappaB activation by targeting on IkappaBalpha. Cell Signal. 2010;22:386–94.
Huang X, Langelotz C, Hetfeld-Pechoc BKJ, Schwenk W, Dubiel W. The COP9 signalosome mediates beta-catenin degradation by deneddylation and blocks adenomatous polyposis coli destruction via USP15. J Mol Biol. 2009;391:691–702.
Milojevic T, Reiterer V, Stefan E, Korkhov VM, Dorostkar MM, Ducza E, et al. The ubiquitin-specific protease Usp4 regulates the cell surface level of the A2A receptor. Mol Pharmacol. 2006;69:1083–94.
Maertens GN, El Messaoudi-Aubert S, Elderkin S, Hiom K, Peters G. Ubiquitin-specific proteases 7 and 11 modulate Polycomb regulation of the INK4a tumour suppressor. EMBO J. 2010;29:2553–65.
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada through grant RGPIN-2015-05879 (awarded to DAG). Caitlyn Vlasschaert was the recipient of a Frederick Banting and Charles Best Scholarship from the Canadian Institutes of Health Research. We thank Dr. Stéphane Aris-Brosou for his time and guidance in troubleshooting the BEAST phylogenetic analyses. We are grateful to David Cook for his help with Circos, and acknowledge the kind assistance of Tim Ramsay in the statistical analysis of mouse crosses.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CV contributed to the design of the study, carried out all computational analyses, and wrote the initial draft of the manuscript. XX contributed to the design of the study and advised on computational methodologies and the interpretation of data. JC performed mouse genetic crosses and contributed to the manuscript. DAG conceived of the study, participated in its design, and wrote sections of the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Figure S1. Bayesian dating of aligned codon sequences. Phylogenetic reconstruction and fossil-calibrated dating of aligned codon sequences for USP4, USP15, USP11 and ancestral USP sequences was generated using BEAST v. 1.8. 95 % credible intervals are indicated. Calibration points were obtained from TimeTree. (PDF 64 kb)
Additional file 2:
Figure S2. Bayesian dating of aligned USP4 and USP11 codon sequences. Phylogenetic reconstruction and fossil-calibrated dating of aligned codon sequences for USP4 and USP11 was generated using BEAST v. 1.8. 95 % credible intervals are indicated. Calibration points were obtained from TimeTree. The gold star indicates the inferred USP4-USP11 divergence time. Red stars indicate major deviations from true topology. (PDF 52 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Vlasschaert, C., Xia, X., Coulombe, J. et al. Evolution of the highly networked deubiquitinating enzymes USP4, USP15, and USP11. BMC Evol Biol 15, 230 (2015). https://doi.org/10.1186/s12862-015-0511-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-015-0511-1