
Abstract
Background
Rhynchocypris oxycephalus is a cold water fish with a wide geographic distribution including the relatively warm temperate regions of southern China. It also occurs in second- and third-step geomorphic areas in China. Previous studies have postulated that high-altitude populations of R. oxycephalus in southern China are Quaternary glacial relics. In this study, we used the mitochondrial gene Cytb and the nuclear gene RAG2 to investigate the species phylogeographical patterns and to test two biogeographic hypotheses: (1) that divergence between lineages supports the three-step model and (2) climatic fluctuations during the Quaternary resulted in the present distribution in southern China.
Results
Phylogenetic analysis detected three major matrilines (A, B, and C); with matrilines B and C being further subdivided into two submatrilines. Based on genetic distances and morphological differences, matriline A potentially represents a cryptic subspecies. The geographic division between matrilines B and C coincided with the division of the second and third geomorphic steps in China, suggesting a historical vicariance event. Pliocene climatic fluctuations might have facilitated the southwards dispersal of R. oxycephalus in matriline C, with the subsequent warming resulting in its split into submatrilines C1 and C2, leaving submatriline C2 as a relic in southern China.
Conclusions
Our study demonstrates that geological events (three steps orogenesis) and climate fluctuations during the Pliocene were important factors in shaping phylogeographical patterns in R. oxycephalus. Notably, no genetic diversity was detected in several populations, all of which possessed unique genotypes. This indicates the uniqueness of local populations and calls for a special conservation plan for the whole species at the population level.
Similar content being viewed by others

Background
Both geological events and climatic fluctuations usually influence genetic variation distribution in populations and species. The uplifting of mountain systems and the development of river systems may form vicariance between populations, resulting in genetic diversification and even speciation [1]-[5]. China has a complex topography; the terrain gradually slopes from west to east forming a flight of three steps [6]. The first step consists of the Tibetan Plateau, with an average elevation of over 4,000 m above sea level. The second step includes the Yun-Gui, Loess, and Inner Mongolia Plateaus, the Tarim, Jungar, and Sichuan Basins, and varies from 1,000–2,000 m in elevation. The third step, dropping to 500–1,000 m in elevation, refers to the vast area in the east of the Great Hinggan Ridge-Taihang-Wushan-Xuefeng Mountains and mainly consists of hills and plains [6].
These steps remain geographically and ecologically dynamic; geomorphic evolution is responsible for the isolation and differentiation of many plant and animal populations [5],[7]. For example, the geographic division of two lineages of Chinese gecko (Gekko swinhonis) coincides with a boundary consisting of the Qinling and Taihang Mountains, suggesting a historical vicariance pattern [8].
Global cyclic fluctuations from the Pliocene and through the Quaternary have resulted in periodic habitat expansions and contractions of species ranges, shaping the species distributions and their genetic patterns [9]-[11]. Species ranges typically shifted southwards during cold glacial periods and northwards during warm interglacial ones [12]. Quaternary climatic fluctuations and associated habitat expansions and contractions have been investigated extensively in extant organisms [10]. In contrast, the impacts of more ancient climatic changes, as far back as the Pliocene, have been largely neglected despite remarkable shifts in global climate and continental ecosystems [13]-[15]. Studies have demonstrated that Quaternary glacial climatic oscillations are too recent to explain many of the deep genetic divergences observed today [15],[16].
Pliocene climatic oscillations have also occurred in China [17]. During cold periods, continental China was mainly affected by cold air masses and glaciation developed in high mountains. In warm periods, the continental climate was dominated by warm humid air masses, causing glaciers in high mountains to retreat. Similar to current global warming projections, warm humid periods had the potential to eliminate cold-adapted organisms [18],[19]. Individual population ranges likely shifted upwards along altitudinal gradients [20],[21]. Nevertheless, our knowledge of the genetic consequences of Pliocene climatic change in China remains sparse.
The Chinese minnow (Rhynchocypris oxycephalus) is a small riverine cyprinid widely distributed in East Asia. They live in cold, running, or still (but well oxygenated) waters, generally inhabiting stream headwaters at high altitude [22],[23]. In China, this species occurs in second- and third-step geomorphic areas. They also occur in the temperate regions of southern China where the average annual temperature is approximately 18°C [24]. Therefore, this is an excellent bioindicator species for low-temperature water and a good model species for studying freshwater fish biogeographic patterns in China because of their typically low dispersal ability and restriction to small mountaintop habitats. It has been suggested that high-altitude populations of R. oxycephalus in southern China are Quaternary glacial relics [25]; however, this claim remains unsubstantiated because no phylogeographic studies have been conducted on this minnow to date.
In the present study, we employed mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) markers to test hypotheses on the drivers of biogeographic processes in R. oxycephalus. Large-scale sampling allowed us to test hypotheses on the evolutionary history and population demographics of R. oxycephalus, to determine: (1) whether or not three steps orogenesis provided different ecological regions that led to high- and low-altitude populations and (2) if Quaternary climatic fluctuations resulted in their present geographic distribution in southern China. Finally, we also assessed the level of genetic diversity for all populations and proposed suitable conservation strategies.
Methods
Ethics statement
The experiments were performed in accordance with the Ethics Committee of the Institute of Hydrobiology, Chinese Academy of Sciences. The policies were enacted according to Chinese Association for Laboratory Animal Sciences, and coordinated with the Institutional Animal Care and Use Committee (IACUC) protocols.
Sample collection
A total of 753 R. oxycephalus individuals were collected from 35 localities across most of its distribution in China (Figure 1 and Additional file 1: Table S1). Samples were obtained using a dip net during 2010–2012. All specimens were preserved in 95% ethanol. We aimed to obtain 20–30 samples per site and the median sample size was 23. However, smaller samples were obtained from some localities (Additional file 1: Table S1). We defined samples from different localities as different populations, because they were distributed in different branches of rivers. Four R. percnurus and R. lagowaskii individuals were used as the outgroup taxa. Detailed information for each specimen used in the present analysis and their localities with GPS records are provided in Additional file 1. All voucher specimens were archived at the Institute of Hydrobiology, Chinese Academy of Sciences.

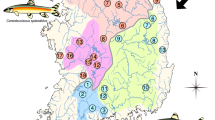
Map of sampling localities for R . oxycephalus populations. The map was downloaded from the Wikimedia Commons on May 10, 2014 and slightly modified [26]. The locality numbers are given in Tables 1 and Additional file 1: Table S1. The matrilines are labeled yellow, blue, and brown for matrilines A, B, and C, respectively.
DNA extraction, PCR amplification, and sequencing
Total genomic DNA was extracted from muscle tissue following the salt-extraction methods of Tang et al. [27]. The mitochondrial cytochrome b gene (Cytb 1,140 bp) was amplified for all samples with primers the GluF (5′-AAC CAC CGT TGT ATT CAA CTA CAA-3′) and ThrR (5′-ACC TCC GAT CTT CGG ATT ACA AGA CCG-3′) [28]. Two Cytb sequences from Japan were also added to the present study [Genbank: AB236730 and AB198971]. The nuclear Recombinase Activating Gene 2 (RAG2) encodes components of the recombinase involved in immunoglobin recombination and T-cell receptor genes and appears as conserved single copies in all vertebrates examined to date [29],[30]. Partial RAG2 sequences (1,127 bp in this study) were obtained from a subset of samples consisting of 127 individuals with the primers RAG2-108f (5′-CCV ARA CGC TCA TGT CCA AC-3′) and RAG2-1324r (5′-TGG ARC AGW AGA TCA TKG C-3′) [31]. Amplifications were performed in a 60-μL reaction volume containing 6 μL of 10 × PCR buffer, 30-50 ng genomic DNA, 1 μL of each primer (each 10 μM), 1.5 μL of dNTPs (each 2.5 mM), and 2.0 U Taq DNA polymerase. Sterile water was added to reach the final volume. Reactions took place in the following program: an initial denaturation at 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 52-58°C for 45 s, extension at 72°C for 1 min, and a final extension at 72°C for 8 min. PCR products were sent to commercial sequencing companies (Shanghai Sangon Biotech Inc.) for purifying and then sequenced in both directions with BigDye™ Terminator Cycle Sequencing Kit on an ABI PRISM® 3730 DNA Analyzer (Applied Biosystems, Life Technologies). The same PCR primers were used for sequencing.
Sequence analysis
A total of 753 Cytb and 127 RAG2 sequences have been newly sequenced in this study. Multiple alignments of sequences were performed using CLUSTAL X [32] and subsequently checked by eye in SeaView [33]. The nucleotide sequence compositions were calculated in PAUP* v. 4.0b10a [34]. Sequence variations were calculated in MEGA 4.0 [35]. Nuclear gene sequences containing more than one ambiguous site were resolved using PHASE version 2.1.1 [36], for which the input files were prepared using SeqPHASE [37]. Recombination tests for detecting the longest non-recombining region for nuclear loci were conducted in IMGC [38]. Identical haplotypes were collapsed in DnaSP 5.10 [39]. Newly determined sequences were submitted to Genbank [accession numbers KM675061-KM675238] (Additional file 1: Table S1).
Phylogenetic analysis
Phylogenetic relationships among mitochondrial haplotypes were reconstructed using Bayesian inference (BI), maximum likelihood (ML), and maximum parsimony (MP) approaches. Selection of the best-fit nucleotide substitution models was based on codon position using the Akaike Information Criterion in jmodeltest 2.0 [40],[41]. For BI and ML analysis, the most appropriate model of nucleotide substitution was (TIM+I+G). BI analysis was carried out in MrBayes v. 3.1.2 [42]. Four simultaneous Markov chains were run for 6 million generations sampling every 100 generations using two independent analyses in parallel runs. The average standard deviation of split frequencies reached a value below 0.01, the potential scale reduction factor (PSRF) value approached 1.0. After the first 6,000 sampled trees were discarded as burn-in, 50% majority-rule consensus trees with posterior probability values for each node were obtained. ML analysis was conducted with the best model of nucleotide substitution (TIM+I+G) in PhyML 3.0 [43]. Nodal support values were estimated from 100 nonparametric bootstrap replicates. MP analysis was implemented in PAUP 4.0b10a [34]. Heuristic searches with tree-bisection-reconnection were executed for 1,000 random addition replicates with all characters treated as unordered and equally weighted. We constructed the BI tree using RAG2 alleles under the same settings as for Cytb, except for model of substitution (GTR+I+R). Furthermore, the Network 4.6.0.0 program [44] was employed to construct median-joining (MJ) networks to visualize relationships among haplotypes, using the MP calculation set to the default parameters.
Divergence time estimates
For divergence time estimation, the molecular clock was evaluated using a two-cluster test in the LINTREE program [45]. Sequences that showed significantly different substitution rates were excluded from further analysis. To estimate the divergence time and ancestral distribution of the extant matrilines, we used a coalescent time estimation method in BEAST v 1.7.5 [46]. Major matriline divergence times were estimated using a Yule speciation tree prior. Because of the absence of fossil or geological data for calibrations, divergence times were estimated under the strict-clock model. Based on the mitochondrial Cytb data, a range of mutation rates (1-2% per my) were adopted, as they have been widely employed for mitochondrial Cytb gene analysis in cyprinid fish [47]-[50]. Two fixed mutation rates of 1% and 2% were conducted and from these an average divergence time was generated. The credibility interval included the lower limit from the 1% clock and the upper limit from the 2% clock. Analyses were performed for 20 million generations while sampling every 1,000th tree, and the first 10% of trees sampled were treated as burn-in. The effective sample size for parameter estimates and convergence was checked with Tracer 1.5 [51]. FigTree v.1.3.1 [52] was used to display summarized and annotated phylogenetic trees with a molecular clock constraint yielded by BEAST.
Population genetic analysis
Diversity estimations for both pooled and individual population datasets were calculated in DnaSP, including the number of haplotypes (n), haplotype diversity (h), and nucleotide diversity (π). The mean divergence among haplotypes and divergence between matrilines were calculated by an uncorrected p-distances model as implemented in MEGA, standard errors were estimated by bootstrapping using 10,000 replicates. Pairwise population differentiation was estimated by computing pairwise Φst for the Cytb data between populations having more than 10 samples in ARLEQUIN 3.5 [53]. To investigate the level of genetic variation between populations, a two-level hierarchical analysis of molecular variance (AMOVA) was performed. The significance was assessed by 1,000 random permutations.
The plausibility of an isolation-by-distance scenario was explicitly tested by Mantel's tests in ARLEQUIN to analyze the relationships between genetic (mean number of pairwise nucleotide differences, Φst) and geographical (straight line differences) distances between sampling localities.
Demographic history
Three methods were used with the Cytb data to trace the demographic history of matrilines B1, B2, C1, and C2. Matriline A was excluded form demographic analysis because of the small sample size. First, Tajima's D[54] and Fu’s Fs[55] were used to check for neutral evolution of the mtDNA in ARLEQUIN. In the neutrality test, we assessed significance by generating null distributions from 10,000 coalescent simulations; significantly negative values indicated population expansion. Second, mismatch distributions [56] were calculated in ARLEQUIN to test for signals of demographic expansion. For a population undergoing exponential growth, its mismatch distribution should fit a smooth unimodal curve [57]. The significance of sum of squared deviations (SSD) and raggedness indices were carried out by bootstrap resampling (10,000 replicates). Third, Extended Bayesian Skyline Plot EBSP; [58] were implemented in the program BEAST to describe demographic history by assessing the time variation of effective population size. This analysis was performed using the GTR+I+R substitution model and no partition into codon positions. The coalescent tree prior was specified as the Extended Bayesian Skyline Plot. The strict-clock model with mutation rates of 1% per million years was used. The analysis was performed for 20 million generations. The remaining settings were in default mode.
Morphological differentiation analysis
The ratio of caudal peduncle depth to head length (CPD/HL) and caudal peduncle length to depth (CPL/CPD) are key morphometric variables for discrimination between R. oxycephalus and R. lagowaskii[59],[60]. We measured these two variables for 28 populations (Figure 1: localities 3-12, 14-24, and 27-33) of R. oxycephalus and the Xinyang (locality 35) population using digital calipers (IP54 produced by NSCING in Nanjing, China; accurate to the nearest 0.01 mm). Variation of these key morphometric characteristics among the three matrilines (A, B, and C) was subsequently assessed by ANOVA. Tukey’s Honest Significant Difference was employed for post-hoc pairwise comparisons. The sample sizes for morphological analysis were 30, 170, and 433 for matrilines A, B, and C, respectively.
Results
Sequence data
For Cytb, we obtained 755 R. oxycephalus sequences from 36 populations, including two sequences from GenBank. The sequence alignment provided a data matrix of 1,097 bp, of which 289 bp (26.3%) were parsimony informative. Base frequencies were unequal (A =25.9%, T =30.4%, C = 27.1%, G =16.6%). Strong compositional biases against G existed at the second and especislly at the third position (just 10.1%). A total of 124 haplotypes were identified among the 755 R. oxycephalus sequences (Table 1 and Additional file 1: Table S1). The haplotypes exhibited restricted geographical distributions: 96.7% of the haplotypes were unique for a single location and there were no broadly distributed ones.
For RAG2, we sequenced 127 individuals from a subset of samples selected on the basis of the Cytb matrilines (Additional file 1: Table S1). Fourteen individuals were heterozygous. The 1,127-bp fragment included 89 polymorphic sites. All fragments consisted of 62 alleles.
Matrilineal genealogy and nDNA allele tree
Based on the mtDNA data, three methods (BI, ML, and MP) yielded congruent topologies and they consistently supported three matrilines (the BI tree is shown in Figure 2). Matriline A, at the base of the tree, corresponded to two individuals from Japan and samples from Xinyang (locality 35) belonging to the Huaihe River. It exhibited high divergence from the other matrilines (divergence = 6.9-7.7%). Matriline B, sister to matriline C, contained two submatrilines, one on the east of the Middle Yellow River (B1) and the other on the west of the same river and the Upper Yangtze River tributaries (B2). Matriline C was also subdivided into two submatrilines but with poor node support. Submatriline C1 was composed of individuals from Liaohe River and Haihe River streams in northern China, while submatriline C2 was widely distributed, mainly in the Middle and Lower Yangtze River streams and in Zhejiang Province, southern China.

R . oxycephalus maternal genealogy from Bayesian inference analysis. Numbers above branches are Bayesian posterior probabilities and bootstrap values from the maximum likelihood analysis, which are only shown for major and minor matrilines. The three major matrilines are identified by different colors (yellow, matriline A; blue, matriline B; and brown, matriline C).
For the RAG2 data, both the BI tree and MJ network showed strong historical patterning: three well-supported groups of alleles, I, II, and III, were identified (Figure 3a and b). Group I contained all Xinyang individuals (Figure 1: locality 35) corresponding to matriline A (Figure 2) from the Huaihe River. Group II comprised matriline B individuals (Figure 2) (Figure 1: localities 27-34) from the Middle Yellow and Upper Yangtze Rivers. Interestingly, Group II clustered with some individuals from matriline C1. Group III consisted of samples from matriline C2 (Figure 2) (Figure 1: localities 1-21) from the Middle and Lower Yangtze River basin and Zhejiang Province. The relationships of the individuals in matriline C1 were unresolved.

Allele tree and MJ network for R . oxycephalus based on the nuclear gene RAG2 . The colors correspond to matrilines in Figure 2 (yellow, matriline A; blue, matriline B; and brown, matriline C). The small black rhombus represents each mutational step. (a) A Bayesian tree detected three allelic groups I, II, and III. Numbers above the nodes are Bayesian posterior probabilities. (b) MJ network. Three clusters that correspond to the allelic groups in (a) are also identified.
Divergence time estimate
The two-cluster test showed that three sequences (haplotypes 22-24, locality 8) had significantly different substitution rates compared with the other sequences. Therefore, these three sequences were excluded from further analysis. A strict molecular clock was conducted for the remaining samples (Figure 4). Time since the most recent common ancestor of the entire ingroup was estimated to be 6.43 mya (4.30-8.61 mya). Matriline A diverged 3.93 mya (2.62-5.25 mya) and matrilines B and C diverged 3.68 mya (2.45-4.92 mya). Submatrilines C1 and C2 diverged 2.98 mya (1.98-3.99 mya).

Divergence time tree for R . oxycephalus. Branch lengths are proportional to divergence times. Bars on the nodes are 95% confidence intervals. Matrilines A, B1, B2, and C1, C2 correspond to those in Figure 2.
Molecular diversity and genetic structure
The overall Cytb haplotype (h) and nucleotide (π) diversities in all samples were 0.9770 ± 0.0010 and 0.0523 ± 0.0006, respectively. The number of haplotypes (n), h, and π varied among populations (Table 1). The Gaizhou population (Figure 1: locality 22), which exhibited the highest h value, contained haplotypes from submatriline C1. The Mianchi population (Figure 1: locality 32) had the highest π value, which corresponded to the co-occurrence of haplotypes from submatrilines B1 and B2. In contrast, 12 populations (Daqinggu, Jinyun, Shangrao, Huangshan, Yuexi, Jinzhai, Wuyishan, Wuyuan, Laishui, Lichuan, Baoji, and Xinyang, corresponding to localities 9, 14-16, 18-21, 25, 27, 31, and 35) each contained only a single haplotype. The two-level AMOVA suggested that R. oxycephalus was highly geographically structured: 96.75% of the genetic variation was attributed to differentiation among populations and this was highly significant (P <0.001). All pairwise Φst values were statistically significant, varying from 0.099 to 1.000 (P <0.001 to P = 0.027). Genetic divergence varied from 6.5 to 7.4% among matrilines A, B, and C, and from 5.5 to 5.6% between submatrilines B1-B2 and C1-C2 (Table 2).
A significant positive correlation was found between geographical distance and genetic divergence among populations (for Cytb: r = 0.30, P <0.01; for RAG2: r = 0.25, P <0.01). However, some populations (e.g., Wuyuan and Huangshan, Wuyishan and Shangrao, Baoji, and Jialingjiang, corresponding to localities 21 and 16, 20 and 15, 28 and 31, respectively) were genetically distinct despite their geographical proximity. These populations are distributed on opposite sides of the same mountains.
Demographic history
For submatriline B1, Tajima’s D and Fu’s Fs were negative but not significant (Table 3). However, values of SSD and raggedness index did not reject the hypothesis of sudden expansion and the B1 mismatch distribution was unimodal (Additional file 2: Figure S1a). Extended Bayesian Skyline Plots (EBSP) suggested that the effective population size was slowly increasing (Additional file 3: Figure S2a). For submatrilines B2, C1, and C2, population expansions were not supported in neutrality tests (Table 3) and the mismatch analyses significantly rejected the expansion hypothesis. However, the EBSP suggested a decline followed by a steep increase in effective population size for submatrilines C1 and C2 (Additional file 3: Figure S2b and d) and a steep decline for submatriline B2 (Additional file 3: Figure S2c). Therefore, in all matrilines there was ambiguity in the demographic scenarios suggested by all three methods.
Morphological differentiation
The ANOVA analysis showed that two key taxonomic characteristics varied significantly among the three matrilines (CPD/HL: df = 2, 630, P = 0.000; CPL/CPD: df = 2, 630, P = 0.001). For CPD/HL, there was no significant difference between matrilines A and B (Tukey’s post-hoc tests, P = 0.607). However, significant pair-wise distinctions were detected between matrilines A and C and between matrilines B and C (Tukey’s post-hoc tests, P = 0.017 and P = 0.001, respectively). For CPL/CPD, matriline A values differed significantly from those of B and C (Tukey’s post-hoc tests, P = 0.027 and P = 0.001, respectively), but there was no significant difference between matrilines B and C (Tukey’s post-hoc tests, P = 0.202).
Discussion
Deep matriline divergence and implication of cryptic subspecies
Phylogenetic analysis based on Cytb revealed three matrilines (Figure 2): these are highly consistent with the geographic structure of the R. oxycephalus range. Matriline A was first to split from the other two and exhibits high divergence from them. The Taihang-Wushan Mountains seem to act as dividing lines between matrilines B and C. Further divergence occurred between populations in matriline B (B1 and B2) and C (C1 and C2). The nDNA phylogenetic and network analysis revealed a deep genetic divergence analogous to mtDNA (Figure 3).
There is a deep genetic divergence between matriline A and the other matrilines for Cytb (6.9-7.7%, Table 2). Compared with other cyprinid fishes, this amount of divergence has been attributed to subspecies-level differences [61]. The RAG2 tree results were consistent with those from the Cytb tree, matriline A is basal in the BI tree and the MJ network, indicating that matriline A is most distant from the others (Figure 3). We also sequenced some Cytb and RAG2 sequences from other closed Rhynchocypris species (e.g., R. percnurus, R. lagowaskii, and R. czekanowskii). The results revealed that matriline A is a distinct monophyletic group and forms a sister relationship with R. oxycephalus. Furthermore, samples from Xinyang (locality 35), representing matriline A in our samples, are morphologically distinct from the other populations in taxonomically relevant ratios. Therefore, we suggest that matriline A represents a cryptic subspecies, which may be geographically restricted to Japan and parts of China including the Huaihe River. Although Japan is currently separated from eastern China, studies have proposed that these regions most likely had the same freshwater ichthyofauna during the Miocene and Pliocene [62].
The Cytb sequences analysis carried out in our study revealed three distinct evolutionary lineages in R. oxycephalus. Do these major lineages correspond to two or three distinct species? Compared with the general definitions of five different groups of candidate species [63], all uncorrected p-distances between the R. oxycephalus lineages reached a value associated with subspecies divergences (including matrilines B and C). Additionally, morphological differences (CPD/HL) were also observed between the major lineages (matrilines A and C, matrilines B and C). However, the relationship between matrilines B and C is not completely supported by the RAG2 gene data, but matriline A is indeed distinct from the others (as discussed in the previous paragraph). Therefore, the hypothesis of matrilines B and C being distinct subspecies or even species requires further research.
Geologically driven divergence between matrilines B and C
The deep divergence observed between matrilines B and C likely reflects long-term isolation and ecological differences between high- and low-elevation regions, which is probably associated with China’s complex geological history. As indicated in Figure 1, matriline B populations are mainly distributed in high-altitude areas; while matriline C occurs in the lowland regions. The Taihang-Wushan Mountains, part of the demarcation line for the second- and third-step geomorphic areas in China [15], separate matrilines B and C. Therefore, R. oxycephalus divergence is likely associated with the orogenesis of China’s three steps. Geological evidence indicates that recent large-scale intense uplift of the Qinghai-Tibetan Plateau took place during the Pliocene 3.6 mya [64], contributing to the formation of three steps land features [65]. During this period, new habitats and different ecological regions may have been created leading to upper and lower populations. These ecological differences may have resulted in high levels of divergence between matrilines B and C, fostering the accumulation of autapomorphic mutations. The mean estimate of divergence between matrilines B and C was 3.68 mya (2.45-4.92 mya in the Pliocene), suggesting that the speciation event occurred at approximately the same time as the geological event. Similar effects have been reported for several species of plants [3] and animals [66],[67]. Based on the Cytb gene sequences, He and Chen [68] suggested that the highly specialized schizothoracine fishes may have originated in the early Pliocene and the major cladogenetic events in this species are closely correlated with marked environmental changes caused by the violent upheaval of the plateau. The geographic division between two Chinese gecko (Gekko swinhonis) lineages also coincides with the Qinling and Taihang Mountains boundary, suggesting a historical vicariance pattern. The orogeny of the Qinling Mountains may have launched the independent lineage divergence [8]. Thus, barriers to gene flow produced by the complex geological history appear to be responsible for driving the high level of species diversity in the mountains of China.
Pliocene climate fluctuation driven split between submatrilines C1 and C2
There is a 5.5% mtDNA difference between submatrilines C1 and C2 (Table 2). This high level of divergence likely reflects long-term isolation between the two submatrilines. Our molecular clock analysis estimated that submatriline C1 split from C2 approximately 2.98 mya (1.98-3.99 mya), corresponding to the Pliocene warming (2.6-5.0 mya) [69]-[72]. This warming period was preceded by an extensive cooling (5.5-5.2 mya) [14],[71]. During the colder period, matriline C may have extended its range southwards to southern China, resulting in a widespread distribution of the species. The subsequent warming then resulted in the split of matriline C into submatrilines C1 and C2; submatriline C1 is now distributed in cold northern China, while C2 remains in southern China. Similar effects have been reported in speckled dace (R. osculus) in western North America [73]. In Asian salamanders (Pachytriton), all major intraspecific lineages originated before the Quaternary period and pre-Quaternary climate change played an important role in the formation of regional biodiversity [15].
After the split of submatrilines C1 and C2, intraspecific divergence may have been intensified through cyclical climatic fluctuations in the Pleistocene, which also facilitated the relatively wide distribution of R. oxycephalus in southern China. Similar processes have been described in terrestrial montane organisms. Thus, our conclusion is inconsistent with the hypothesis that R. oxycephalus populations in southern China are Quaternary glacial relics in the high-altitude mountains of this region [25].
Species-level genetic diversity and structure
Chinese R. oxycephalus populations are highly genetically diverse. For the Cytb gene in all samples, there were 289 (26.3%) parsimony informative sites and the most variable sites occurred in different haplotypes. AMOVA analysis revealed that 96.75% of the genetic variation was attributed to differentiation among populations. In contrast, genetic diversity within populations was very low and 12 populations possessed a single haplotype despite typically analyzing over 20 fish per population. These populations may have had extremely small founding populations. A similar phenomenon has been reported in Quasipaa boulengeri, a montane stream-dwelling frog, where 85% of the haplotypes only occurred in a single location and 16 populations out of 45 had zero haplotype and nucleotide diversity [74]. High levels of genetic diversity among populations and low levels within populations may commonly occur in montane streams [75]. In these species, local extinctions may considerably impoverish species genetic diversity via the loss of unique haplotypes.
The genetic structure of a species is greatly influenced by the palaeogeographical history of the region that comprises its geographical range. In the present study, mtDNA analysis suggested that R. oxycephalus was geographically restricted to local montane areas. Approximately 97% of the haplotypes are unique for their respective populations. The majority of genetic variance was seen at the population level and was correlated with geographic distance; more distant sites within the hydrological network were more genetically differentiated. In addition to complex topological changes, this pattern may have resulted from the isolation of populations because of specific habitat requirements. Rhynchocypris oxycephalus mainly occurs in the clear cold permanent headwaters of montane streams. This specific ecological requirement results in small population sizes and restricts gene flow among populations (each population possesses unique haplotypes). Therefore, these populations most likely survive in isolated habitats and differentiate through genetic drift and selection.
Implications for conservation
The Chinese minnow currently has a wide geographical distribution in East Asia and to date is ranked as `not threatened’. However, individual populations are small because they are restricted to stream headwaters and may be sensitive to climate change and anthropogenic threats. Using ecological niche modeling, Yu et al. [24] predicted that climate change poses a severe threat to R. oxycephalus. The geographical distribution of this species is expected to contract with progressing climate change and become severely limited, particularly in south-eastern China. Anthropogenic threats, such as deforestation, illegal fishing, and habitat destruction (e.g., road construction and hydro-electric power station) may represent major threats to this species. These factors place R. oxycephalus at risk for extinction.
Genetic variability is widely recognized as an important component of natural biodiversity. The maintenance of high genetic diversity could prevent the loss of a species evolutionary potential [76]. The distinct genetic differentiation in R. oxycephalus has important implications for the conservation of this species. However, many R. oxycephalus populations inhabit areas that are not protected. Our study may serve as a guide for monitoring and developing a conservation strategy for this species. Within this framework, our analyses suggest that the three matrilineal ranges (B, C1, and C2) be considered individual Management Units. All of these require protection because of their genetic uniqueness, which includes exclusive mtDNA haplotypes and private nuclear alleles.
Additionally, the southern populations including those from the Yangtze River, Zhejiang Province, and the Min River demand special attention. This species is geographically restricted to local streams and many of these populations possess negligible genetic diversity. Overall genetic diversity in R. oxycephalus will be reduced if these populations become extinct. Climate change projections indicate severe threats to R. oxycephalus and to these southern populations in particular [24]. Consequently, we recommend that conservation efforts should pay particular attention to areas with unique genetic information. Long-term monitoring programs should also be established, irrespective of whether local population genetic diversity is high or low.
Conclusions
This study assembles a large dataset to infer R. oxycephalus phylogeographic patterns in China. The results support the hypothesis that geological events (three steps orogenesis) have driven divergence between matrilines B and C. Based on the molecular clock, it is unlikely that climate fluctuations during the Quaternary facilitated the southwards dispersal of R. oxycephalus in matriline C and established relics in south China as postulated by Zhang and Chen [25]. Pliocene cooling and warming might have resulted in the split of submatrilines C1 and C2. Notably, no genetic diversity was detected in several populations and all of these populations possessed unique genotypes. This indicates the uniqueness of local populations and calls for a special conservation concern for this species at the population level. These conclusions are based on Cytb and RAG2 gene data; analysis with additional markers would greatly enhance our understanding of this species.
Additional files
References
Mayr E: Systematics and the Origin of Species. 1942, Columbia University Press, New York
Rundle HD, Nosil P: Ecological speciation. Ecol Lett. 2005, 8: 336-352. 10.1111/j.1461-0248.2004.00715.x.
Liu JQ, Wang YJ, Wang AL, Hideaki O, Abbott RJ: Radiation and diversification within the Ligularia-Cremanthodium-Parasenecio complex (Asteraceae) triggered by uplift of the Qinghai-Tibetan Plateau. Mol Phylogenet Evol. 2006, 38: 31-49. 10.1016/j.ympev.2005.09.010.
Che J, Zhou WW, Hu JS, Yan F, Papenfuss TJ, Wake DB, Zhang YP: Spiny frogs (Paini) illuminate the history of the Himalayan region and Southeast Asia. Proc Natl Acad Sci U S A. 2010, 107: 13765-13770. 10.1073/pnas.1008415107.
Li ZJ, Yu GH, Rao DQ, Yang JX: Phylogeography and demographic history of Babina pleuraden (Anura, Ranidae) in southwestern China. PLoS ONE. 2012, 7: e34013-10.1371/journal.pone.0034013.
Jiang F, Wu X: Fundamental characteristics of the stepped landform in China continent. Mar Geol & Quat Ge. 1993, 13: 15-24.
Yuan QJ, Zhang ZY, Peng H, Ge S: Chloroplast phylogeography of Dipentodon (Dipentodontaceae) in Southwest China and northern Vietnam. Mol Ecol. 2008, 17: 1054-1065. 10.1111/j.1365-294X.2007.03628.x.
Yan J, Wang QX, Chang Q, Ji X, Zhou K: The divergence of two independent lineages of an endemic Chinese gecko, Gekko swinhonis, launched by the Qinling orogenic belt. Mol Ecol. 2010, 19: 2490-2500. 10.1111/j.1365-294X.2010.04660.x.
Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF: Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol. 1998, 7: 453-464. 10.1046/j.1365-294x.1998.00289.x.
Hewitt GM: Genetic consequences of climatic oscillations in the Quaternary. Philos T Roy Soc B. 2004, 359: 183-195. 10.1098/rstb.2003.1388.
Soltis DE, Morris AB, McLachlan JS, Manos PS, Soltis PS: Comparative phylogeography of unglaciated eastern North America. Mol Ecol. 2006, 15: 4261-4293. 10.1111/j.1365-294X.2006.03061.x.
Hewitt G: The genetic legacy of the Quaternary ice ages. Nature. 2000, 405: 907-913. 10.1038/35016000.
Cerling TE, Harris JM, MacFadden BJ, Leakey MG, Quade J, Eisenmann V, Ehleringer JR: Global vegetation change through the Miocene/Pliocene boundary. Nature. 1997, 389: 153-158. 10.1038/38229.
Zachos J, Pagani M, Sloan L, Thomas E, Billups K: Trends, rhythms, and aberrations in global climate 65 Ma to present. Science. 2001, 292: 686-693. 10.1126/science.1059412.
Wu Y, Wang Y, Jiang K, Hanken J: Significance of pre-Quaternary climate change for montane species diversity: insights from Asian salamanders (Salamandridae: Pachytriton). Mol Phylogenet Evol. 2013, 66: 380-390. 10.1016/j.ympev.2012.10.011.
Miralles A, Carranza S: Systematics and biogeography of the Neotropical genus Mabuya, with special emphasis on the Amazonian skink Mabuya nigropunctata (Reptilia, Scincidae). Mol Phylogenet Evol. 2010, 54: 857-869. 10.1016/j.ympev.2009.10.016.
Huang PH: Quaternary climatic changes in china and problem of Lushan glaciation remnants. J Glaciol Geocryol. 1982, 4: 1-14.
Pounds JA, Bustamante MR, Coloma LA, Consuegra JA, Fogden MPL, Foster PN, La Marca E, Masters KL, Merino-Viteri A, Puschendorf R, Ron SR, Sanchez-Azofeifa GA, Still CJ, Young BE: Widespread amphibian extinctions from epidemic disease driven by global warming. Nature. 2006, 439: 161-167. 10.1038/nature04246.
Milanovich JR, Peterman WE, Nibbelink NP, Maerz JC: Projected loss of a salamander diversity hotspot as a consequence of projected global climate change. Plos ONE. 2010, 5: 10-10.1371/journal.pone.0012189.
Thomas CD, Cameron A, Green RE, Bakkenes M, Beaumont LJ, Collingham YC, Erasmus BFN, Siqueira MF, Grainger A, Hannah L, Hughes L, Huntley B, Jaarsveld AS, Midgley GF, Miles L, Ortega-Huerta MA, Peterson AT, Phillips OL, Williams SE: Extinction risk from climate change. Nature. 2004, 427: 145-148. 10.1038/nature02121.
Wake DB: Facing extinction in real time. Science. 2012, 335: 1052-1053. 10.1126/science.1218364.
Bogutskaya NG, Naseka AM, Shedko SV, Vasil’eva ED, Chereshnev IA: The fishes of the Amur River: updated check-list and zoogeography. Ichthyol Explor Fres. 2008, 19: 301-366.
Jang MH, Kim JG, Park SB, Jeong KS, Cho G, Joo G: The current status of the distribution of introduced fish in large river systems of South Korea. Int Rev Hydrobiol. 2002, 87: 319-328. 10.1002/1522-2632(200205)87:2/3<319::AID-IROH319>3.0.CO;2-N.
Yu D, Chen M, Zhou ZC, Eric R, Tang QY, Liu HZ: Global climate change will severely decrease potential distribution of the East Asian coldwater fish Rhynchocypris oxycephalus (Actinopterygii, Cyprinidae). Hydrobiologia. 2013, 700: 23-32. 10.1007/s10750-012-1213-y.
Zhang E, Chen YY: Fish Fauna in Northeastern Jiangxi Province with a discussion on the zoogeographical division of East China. Acta Hydrobiologica Sinca. 1997, 3: 254-261. (in Chinese)
Wikimedia Commons. [], [http://commons.wikimedia.org/wiki/File:East_Asia_topographic_map.png]
Tang Q, Freyhof J, Xiong B, Liu H: Multiple invasions of Europe by East Asian cobitid loaches (Teleostei: Cobitidae). Hydrobiologia. 2008, 605: 17-28. 10.1007/s10750-008-9296-1.
Doadrio I, Carmona JA, Machordom A: Haplotype diversity and phylogenetic relationships among the Iberian Barbels (Barbus, Cyprinidae) reveal two evolutionary lineages. J Hered. 2002, 93: 140-147. 10.1093/jhered/93.2.140.
Hansen JD, Kaattari SL: The recombination activating gene 2 (RAG2) of the rainbow trout Oncorhynchus mykiss. Immunogenetics. 1996, 44: 203-211. 10.1007/BF02602586.
Willett CE, Cherry JJ, Steiner LA: Characterization and expression of the recombination activating genes (rag1 and rag2) of zebrafish. Immunogenetics. 1997, 45: 394-404. 10.1007/s002510050221.
Kim KY, Ko MH, Liu H, Tang QY, Chen XL, Miyazaki J, Bang I: Phylogenetic relationships of Pseudorasbora, Pseudopungtungia, and Pungtungia (Teleostei; Cypriniformes; Gobioninae) inferred from multiple nuclear gene sequences. BioMed Res Int. 2013, 2013: Article ID 347242, 6 pages-
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25: 4876-4882. 10.1093/nar/25.24.4876.
Galtier N, Gouy M, Gautier C: SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Bioinformatics. 1996, 12: 543-548. 10.1093/bioinformatics/12.6.543.
Swofford DL: PAUP*: Phylogenetic Analysis using Parsimony (* and other methods), version 4. 2002, Sinauer Associates, Sunderland, Massachusetts
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24: 1596-1599. 10.1093/molbev/msm092.
Stephens M, Smith NJ, Donnelly P: A new statistical method for haplotype reconstruction from population Data. Am J Hum Genet. 2001, 68: 978-989. 10.1086/319501.
Flot JF: SeqPHASE: a web tool for interconverting phase input/output files and FASTA sequence alignments. Mol Ecol Resour. 2010, 10: 162-166. 10.1111/j.1755-0998.2009.02732.x.
Woerner AE, Cox MP, Hammer MF: Recombination-filtered genomic datasets by information maximization. Bioinformatics. 2007, 23: 1851-1853. 10.1093/bioinformatics/btm253.
Librado P, Rozas J: Dnasp v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009, 25: 1451-1452. 10.1093/bioinformatics/btp187.
Darriba D, Taboada GL, Doallo R, Posada D: jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012, 9: 772-10.1038/nmeth.2109.
Guindon S, Gascuel O: A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Systematic Biol. 2003, 52: 696-704. 10.1080/10635150390235520.
Ronquist F, Huelsenbeck JP: MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19: 1572-1574. 10.1093/bioinformatics/btg180.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O: New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Sys Biol. 2010, 59: 307-321. 10.1093/sysbio/syq010.
Bandelt HJ, Forster P, Rohl A: Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999, 16: 37-48. 10.1093/oxfordjournals.molbev.a026036.
Takezaki N, Rzhetsky A, Nei M: Phylogenetic tests of the molecular clock and linearized trees. Mol Biol Evol. 1995, 12: 823-833.
Drummond AJ, Rambaut A: BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007, 7: 214-10.1186/1471-2148-7-214.
Durand JD, Tsigenopoulos CS, Unlü E, Berrebi P: Phylogeny and biogeography of the family Cyprinidae in the Middle East inferred from Cytochrome b DNA-evolutionary significance of this region. Mol Phylogenet Evol. 2002, 22: 91-100. 10.1006/mpev.2001.1040.
Ketmaier V, Bianco PG, Cobolli M, Krivokapic M, Caniglia R, De Matthaeis E: Molecular phylogeny of two lineages of Leuciscinae cyprinids (Telestes and Scardinius) from the peri-Mediterranean area based on Cytochrome b data. Mol Phylogenet Evol. 2004, 32: 1061-1071. 10.1016/j.ympev.2004.04.008.
Tang Q, Liu S, Yu D, Liu H, Patrick D: Mitochondrial capture and incomplete lineage sorting in the diversification of balitorine loaches (Cypriniformes, Balitoridae) revealed by mitochondrial and nuclear genes. Zool Scr. 2012, 41: 233-247. 10.1111/j.1463-6409.2011.00530.x.
Meyer A: Evolution of Mitochondrial DNA in Fishes. Biochemistry and Molecular Biology of Fishes, Volume 2. Edited by: Hochachka PW, Mommsen TP. 1993, Elsevier, Hague, 1-38.
Rambaut A, Drummond AJ: Tracer v1.4. [], [http://beast.bio.ed.ac.uk/Tracer]
Rambaut A: FigTree. [], [http://tree.bio.ed.ac.uk/software/figtree/]
Excoffier L, Laval G, Schneider S: Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol Bioinform. 2005, 1: 47-50.
Tajima F: Statistical methods to test for nucleotide mutation hypothesis by DNA polymorphism. Genetics. 1989, 123: 585-595.
Fu YX: Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997, 147: 915-925.
Slatkin M, Hudson RR: Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics. 1991, 1991 (129): 555-562.
Rogers AR, Harpending H: Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992, 9: 552-569.
Heled J, Drummond AJ: Bayesian inference of population size history from multiple loci. BMC Evol Biol. 2008, 8: 289-10.1186/1471-2148-8-289.
Chen YY: Fauna Sinica. Osteichthyes. Cypriniformes II. 1998, Sciences Press, Beijing
Hosoya K: Cyprinidae. Fishes of Japan with Pictorial Keys to the Species. Edited by: Nakabo T. 2000, Tokai University Press, Tokyo, 2
McPhail JD, Taylor EB: Morphological and genetic variation in northwestern suckers, Castosomus Catostomus: The salish sucker problem. Copeia. 1999, 1999: 884-892. 10.2307/1447964.
Chang MM, Chen YY, Tong HW: A new Miocene Xenocyprinine (Cyprinidae) from Heilongjiang Province, Northeast China and succession of late Cenozoic fish faunas of East Asia. Vertebrata Palasiatica. 1996, 34: 165-183.
Kartavtsev YP: Divergence at Cyt-b and Co-1 mtDNA genes on different taxonomic levels and genetics of speciation in animals. Mitochondrial DNA. 2011, 22: 55-65. 10.3109/19401736.2011.588215.
Zhang LS: Palaeogeography of China. 2012, Science Press, Beijing
Zhang DF, Feng QL, Jian MB: Eco-environmental effects of the Qinghai-Tibet Plateau uplift during the Quaternary in China. Environ Geol. 2000, 39: 1352-1357. 10.1007/s002540000174.
He SP, Cao WX, Chen YY: The uplift of Qinghai-Xizang (Tibet) Plateau and the vicariance speciation of glyptosternoid fishes (Siluriformes: Sisoridae). Sci China C Life Sci. 2001, 44: 644-651. 10.1007/BF02879359.
Zhang DR, Chen MY, Murphy RW, Che J, Pang JF, Hu JS, Luo J, Wu SJ, Ye H, Zhang YP: Genealogy and palaeodrainage basins in Yunnan Province: phylogeography of the Yunnan spiny frog, Nanorana yunnanensis (Dicroglossidae). Mol Ecol. 2010, 19: 3406-3420. 10.1111/j.1365-294X.2010.04747.x.
He DK, Chen YF: Molecular phylogeny and biogeography of the highly specialized grade schizothoracine fishes (Teleostei: Cyprinidae) inferred from cytochrome b sequences. Chinese Sci Bull. 2007, 52: 777-788. 10.1007/s11434-007-0123-2.
Ravelo AC, Andreasen DH, Lyle M, Lyle AO, Wara MW: Regional climate shifts caused by gradual global cooling in the Pliocene epoch. Nature. 2004, 429: 263-267. 10.1038/nature02567.
Salzmann U, Williams M, Haywood AM, Johnson ALA, Kender S, Zalasiewicz J: Climate and environment of a Pliocene warm world. Palaeogeogr Palaeocl. 2011, 309: 1-8. 10.1016/j.palaeo.2011.05.044.
Hodell DA, Elmstrom KM, Kennett JP: Latest Miocene benthic δ18O changes, global ice volume, sea level and the `Messinian salinity crisis’. Nature. 1986, 320: 411-414. 10.1038/320411a0.
Salzmann U, Dolan A, Haywood A, Chan WL, Voss J, Hill D, Abe-Ouchi A, Bragg F, Chandler M, Contoux C, Dowsett H, Jost A, Kamae Y, Lohmann G, Lunt D, Pickering S, Pound M, Ramstein G, Sohl L, Stepanek S, Ueda H, Zhang Z: Challenges in quantifying Pliocene terrestrial warming revealed by data-model discord. Nat Clim Change. 2013, 3: 969-974. 10.1038/nclimate2008.
Pfrender ME, Hicks J, Lynch M: Biogeographic patterns and current distribution of molecular-genetic variation among populations of speckled dace, Rhinichthys osculus (Girard). Mol Phylogenet Evol. 2004, 30: 490-502. 10.1016/S1055-7903(03)00242-2.
Yan F, Zhou W, Zhao H, Yuan Z, Wang Y, Jiang K, Jin J, Murphy RW, Che J, Zhang Y: Geological events play a larger role than Pleistocene climatic fluctuations in driving the genetic structure of Quasipaa boulengeri (Anura: Dicroglossidae). Mol Ecol. 2013, 22: 1120-1133. 10.1111/mec.12153.
Chung MY, Lopez-Pujol J, Maki M, Kim KJ, Chung JM, Sun BY, Chung MG: Genetic diversity in the common terrestrial orchid Oreorchis patens and its rare congener Oreorchis coreana: inference of species evolutionary history and implications for conservation. J Hered. 2012, 103: 692-702. 10.1093/jhered/ess032.
Frankham R, Ballou JD, Briscoe DA: Introduction to Conservation Genetics. 2002, Cambridge University Press, Cambridge
Acknowledgments
We thank Zhuocheng Zhou, Jun Wang, Yuyu Xiong, and Shaorong Yang for their help in the field and with specimen collection. Thanks also to Rao Cui for assistance with map analysis and Martin Reichard for his comments on the paper. This research is funded by the Innovation Projects of the Chinese Academy of Sciences (KSCX2-YW-Z1023) and the National Natural Science Foundation of China (31061160185).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HZL designed the research and provided financial support. DY wrote the manuscript. DY and MC collected materials and performed laboratory work. QYT contributed to the data analysis. XJL participated in laboratory work. All authors read and approved the final manuscript.
Electronic supplementary material
12862_2014_225_MOESM1_ESM.pdf
Additional file 1: Table S1.: Summary of sample localities for Rhynchocypris oxycephalus and outgroups. The locality numbers correspond to Figure 1. Site, coordinates (longitude/latitude), sample size, voucher ID number, Cytb haplotype and identified lineages, nuclear clade, and GenBank accession number for Cytb and RAG2 are presented. (PDF 893 KB)
12862_2014_225_MOESM2_ESM.pdf
Additional file 2: Figure S1.: Mismatch distribution for each R. oxycephalus matriline. The abscissa indicates the number of pairwise differences between compared sequences. The ordinate is the frequency for each value. Histograms are the observed frequencies of pairwise divergences among sequences and the line refers to the expectation under the model of population expansion. (a-d) Mismatch distributions for the matrilines B1, B2, C1, and C2, respectively. (PDF 189 KB)
12862_2014_225_MOESM3_ESM.pdf
Additional file 3: Figure S2.: Bayesian skyline plot estimated by BEAST for each R. oxycephalus matriline. X-axis, time in millions of years; y-axis, effective population size (units = Neτ, the product of effective population size and generation length in millions of years). The mean estimate and both 95% HPD limits are indicated. (a-d) Bayesian skyline plots for matrilines B1, B2, C1, and C2, respectively. (PDF 197 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Yu, D., Chen, M., Tang, Q. et al. Geological events and Pliocene climate fluctuations explain the phylogeographical pattern of the cold water fish Rhynchocypris oxycephalus (Cypriniformes: Cyprinidae) in China. BMC Evol Biol 14, 225 (2014). https://doi.org/10.1186/s12862-014-0225-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-014-0225-9