
Abstract
Background
In the field of drug repositioning, it is assumed that similar drugs may treat similar diseases, therefore many existing computational methods need to compute the similarities of drugs and diseases. However, the calculation of similarity depends on the adopted measure and the available features, which may lead that the similarity scores vary dramatically from one to another, and it will not work when facing the incomplete data. Besides, supervised learning based methods usually need both positive and negative samples to train the prediction models, whereas in drug-disease pairs data there are only some verified interactions (positive samples) and a lot of unlabeled pairs. To train the models, many methods simply treat the unlabeled samples as negative ones, which may introduce artificial noises. Herein, we propose a method to predict drug-disease associations without the need of similarity information, and select more likely negative samples.
Results
In the proposed EMP-SVD (Ensemble Meta Paths and Singular Value Decomposition), we introduce five meta paths corresponding to different kinds of interaction data, and for each meta path we generate a commuting matrix. Every matrix is factorized into two low rank matrices by SVD which are used for the latent features of drugs and diseases respectively. The features are combined to represent drug-disease pairs. We build a base classifier via Random Forest for each meta path and five base classifiers are combined as the final ensemble classifier. In order to train out a more reliable prediction model, we select more likely negative ones from unlabeled samples under the assumption that non-associated drug and disease pair have no common interacted proteins. The experiments have shown that the proposed EMP-SVD method outperforms several state-of-the-art approaches. Case studies by literature investigation have found that the proposed EMP-SVD can mine out many drug-disease associations, which implies the practicality of EMP-SVD.
Conclusions
The proposed EMP-SVD can integrate the interaction data among drugs, proteins and diseases, and predict the drug-disease associations without the need of similarity information. At the same time, the strategy of selecting more reliable negative samples will benefit the prediction.
Similar content being viewed by others

Background
De novo drug discovery is a complex systematic project which is expensive, time-consuming and with high failure risks. As reported, it will take 0.8–1.5 billion dollars and about 10–17 years to bring a small molecule drug into market, and during the development stage, almost 90% of the small molecules can not pass the Phase I clinical trial and finally be eliminated [1, 2]. For the approved drugs in market, their pharmacological and toxicological properties are clear and the drug safeties are often guaranteed, but only some of their indications are found. For example, there are 2589 approved small molecule drugs in DrugBank [3], and more than 25000 diseases in UMLS medical database [4], resulting in over 60 millions of drug-disease pairs. However, only less than 5% of the drug-disease pairs were identified to have therapeutic relationships, and most of the drug-disease relationships are unknown [5]. Therefore, to discover the new indications of approved drugs, known as drug repositioning, can greatly save money and time, especially can improve the success rate, has become a promising alternative for de novo drug development.
Historically, finding a new indiction of a drug is likely to be an accidental event with a bit of luck. For example, Minoxidil, originally for the treatment of hypertension, was found by chance to have the treatment efficacy for hair loss [6]; Sildenafil (trade name: Viagra), originally for the treatment of angina, was occasionally found to have the potential to treat erectile dysfunction [7]. Such occasional findings of the drugs’ new indictions suggest a new methodology of drug development. However, the “pot-luck” approach can not promise drug repositioning effectively and efficiently. It is necessary to develop a computational method that helps to redirect approved drugs. Fortunately, with the accumulation of multiple omics data and the development of machine learning methods, it is possible to mine the drugs’ potential indications in silico. Up to now, many computational methods have been proposed to find new indictions of drugs by predicting potential treatment relationships of drug-disease pairs.
Based on the hypothesis that the gene expression signature of a particular drug is opposite to the gene expression signature of a disease, some gene expression based methods [8, 9] have been proposed. Noticing that such kind of methods may fail to consider the different roles of genes and their dependencies at the system level, system-level based approach that integrates the gene expressions and related network has recently been proposed [10].
Recently, along with the increase of drugs and diseases related multi-omics data, many methods have been proposed to integrate multiple sources of data to predict the drug-disease interactions based on machine learning techniques. Gottlieb et al. proposed a method (PREDICT) to predict new associations between drugs and diseases by integrating five drug-drug similarities and two disease-disease similarities data [11]. Wang et al. proposed a computational framework based on a three-layer heterogeneous network model (TL-HGBI) by integrating similarities and interactions among diseases, drugs and drug targets [12]. Luo et al. utilized some comprehensive similarities about drugs and diseases, and proposed a Bi-Random walk algorithm (MBiRW) to predict potential drug-disease interactions [13]. Martinez et al. developed a method named DrugNet for drug-disease and disease-drug priorization by integrating heterogeneous data [14]. Wu et al. integrated comprehensive drug-drug and disease-disease similarities from chemical/phenotype layer, gene layer and treatment network layer, and proposed a semi-supervised graph cut method (SSGC) to predict the drug-disease associations [15]. Moghadam et al. adopted the kernel fusion technique to combine different drug features and disease features, and then built SVM models to predict novel drug indications [16]. Liang et al. integrated drug chemical information, target domain information and gene ontology annotation information, and proposed a Laplacian regularized sparse subspace learning method (LRSSL) to predict drug-disease associations [17]. Zhang et al. introduced a linear neighborhood similarity [18] and a network topological similarity [19], then proposed a similarity constrained matrix factorization method (SCMFDD) to predict drug-disease associations by making use of known drug-disease associations, drug features and disease semantic information [20].
However, most of the existed methods are facing two main problems: one is that most of them are based on the hypothesis that similar drugs treat similar diseases, thus they need the similarity information between drugs, proteins, diseases, and so on. However, the similarity data can be not easily obtained. People often need to customize a program to collect data and to calculate the similarities so as to satisfy their own needs. Moreover, the calculation of similarity scores depends on the adopted measures, which may lead that the similarity score of a pair varies dramatically from one method to another. For example, two proteins are similar according to their structures, while they may be dissimilar according to their sequences. Even worse, some features required for calculating the similarities may be unknown or unavailable, resulting that these methods fail to work [21]. The other problem is that supervised learning based methods usually need both positive and negative samples to train the prediction models, whereas the drug-disease pair data, like other biological data, is lack of experimental validated negative samples. To train the models, most of the existing methods randomly select some unlabeled samples as the negative ones. Obviously, such strategy is very rough, for we are not sure whether there are some positive samples uncovered in the unlabeled data.
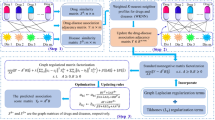
In this paper, we propose a method, called EMP-SVD (Ensemble Meta Paths and Singular Value Decomposition), to detect drug-disease treatment relations by using drug-disease, drug-protein and disease-protein interaction data. Unlike other methods, EMP-SVD needs no similarity information at all. In order to integrate different kinds of interaction data and consider different dependencies, we introduce five meta paths. For each meta path, we first generate a commuting matrix based on the corresponding interaction data, and then get latent features of drugs and diseases by using SVD (Singular Value Decomposition). All drug-disease pairs can be represented by the features. Finally, we train a base classifier by using the Random Forest algorithm. Five base classifiers are combined as an ensemble model to predict the drug-disease interactions. The framework of our method is shown in Fig. 1. In order to train out a more reliable prediction model, we select more likely negative ones from unlabeled samples under the assumption that non-associated drug and disease pair have no common interacted proteins, which is different from other methods. To evaluate our proposed method, we will compare it with the state-of-the-art methods, and also do case studies by literature investigation.

The framework of our proposed EMP-SVD
Materials and methods
Data sets
In this paper, we mainly made use of the interaction data of drug-disease, drug-protein and disease-protein to build the prediction model. We collected such data from DrugBank [3, 22, 23], OMIM [24] and Gottlieb’s data set [11]. Concretely, we collected 4642 drug-protein interaction data from DrugBank, involving 1186 drugs and 1147 proteins; 1365 disease-protein interactions from OMIM, involving 449 diseases and 1147 proteins; and 1827 drug-disease interactions from Gottlieb’s data set, involving 302 disease, 551 drugs. Obviously, the heterogenous network composed of drugs, proteins, diseases and the known interactions is sparse. The statistic of the data is shown in Table 1.
Although our method does not need the similarity information, most of other machine learning based methods do need. For the convenience of comparison, we still collected the chemical structure of drugs and the sequence data of proteins from DrugBank. We computed the drug-drug chemical similarities according to their SMILES strings [25] via Openbabel tool [26], and the protein-protein similarities according to the sequence data by Smith-Waterman algorithm [27]. Moreover, we directly downloaded the disease-disease similarities from MimMiner [28].
Definitions and notations
In this section, we will give the formal definitions and notations used in this paper.
Definition 1
(Heterogeneous drug-protein-disease network schema). For a given heterogenous drug-protein-disease network G=(V,E), where V=D∪P∪S, D, P and S are the sets of drug, protein, disease nodes in the network respectively, while E=Ed,p∪Ep,d∪Ep,s∪Es,p∪Ed,s∪Es,d are the sets of heterogeneous links in G, which include the “binds to” link between drugs and proteins, “causes/caused by” link between proteins and diseases, “treats/treated by” link between drugs and diseases. The schema of G can be defined as \(M_{G} = (\mathcal {T,R})\), where \(\mathcal {T}=\{Drug, Protein, Disease\}\), \(\mathcal {R}=\{binds~to, cuases, caused~by, treats, treated~by\}\), \(\mathcal {T}\) and \(\mathcal {R}\) are the sets of node types and link types in G, respectively.
The network schema MG severs as a template of a network G. For a drug-protein-disease heterogenous network, the network schema is shown in Fig. 2.

Schema of drug-protein-disease heterogeneous network
Definition 2
(Heterogenous network meta path) Based on a given heterogenous network schema \(M_{G} = (\mathcal {T,R})\), \(\mathcal {P} = T_{1} \xrightarrow {R_{1}} T_{2}\xrightarrow {R_{2}}...\xrightarrow {R_{k-1}} T_{k} \) is defined to be a heterogenous network meta path in network G, where \(T_{i} \in \mathcal {T}\), i∈{1,2,...,k} and \(R_{i} \in \mathcal {R}\), i∈{1,2,...,k−1} and if (T1,T2,...,Tk are not all the same) ∨ (R1,R2,...,Rk−1 are not all the same).
For simplicity, we also omit the link types in denoting the meta path if there is no multiple links between the two types, for examples, \(\mathcal {P} = T_{1} \xrightarrow {} T_{2}\xrightarrow {}...\xrightarrow {} T_{k} \) denotes the meta path \(\mathcal {P} = T_{1} \xrightarrow {R_{1}} T_{2}\xrightarrow {R_{2}}...\xrightarrow {R_{k-1}} T_{k} \). The length of \(\mathcal {P}\) is the number of links in \(\mathcal {P}\).
Definition 3
(Commuting matrix [29]) Given a network G=(V,E) and its network schema MG, a commuting matrix for a meta path \(\mathcal {P} = T_{1} \xrightarrow {} T_{2}\xrightarrow {}...\xrightarrow {} T_{k} \) is defined as \(X = A_{T_{1} T_{2}} A_{T_{2} T_{3}}...A_{T_{k-1} T_{k}}\), where \(A_{T_{i} T_{j}}\) is the adjacency (interaction) matrix between type Ti and type Tj. X(i,j) represents the number of path instances between object ui∈T1 and object vj∈Tk under meta path \(\mathcal {P}\).
Since we want to detect the interactions between the drugs and the diseases, we only consider the cases of T1=Drug and Tk=Disease.
Now that there are only three kinds of nodes (drug, protein and disease) in the heterogenous network, we think the meta path with length greater than three may be too long to contribute to the prediction. Sun’s work also has shown that short meta paths are good enough, and long meta paths may even reduce the quality [29]. Therefore, in this work, we only selected meta paths with length no longer than three. As a result, we select five meta paths described below.
Let Ads be the drug-disease interaction matrix, Adp be the drug-protein interaction matrix, and Asp be the disease-protein interaction matrix, we can get the commuting matrices of the five meta paths as follows:
Meta-path-1: Drug \(\xrightarrow {{treats}}\) Disease. The commuting matrix of it, denoted as X1, can be obtained by:
Meta-path-2: Drug \(\xrightarrow {{binds\ to}}\) Protein \(\xrightarrow {{causes}}\) Disease. The commuting matrix of it, denoted as X2, can be obtained by :
By using meta-path-2, we can integrate the drug-protein interaction information and the disease-protein interaction information, that is to say, we easily take the protein related information into account.
Meta-path-3: Drug \(\xrightarrow {{binds\ to}}\) Protein \(\xrightarrow {{binds\ to}}\) Drug \(\xrightarrow {treats}\) Disease. The commuting matrix of it, denoted as X3, can be obtained by:
By using meta-path-3, we can integrate drug-protein interaction and drug-disease interaction information. What’s more, meta-path-3 also indicates that if two drugs share some common proteins, they may have similar indications.
Meta-path-4: Drug \(\xrightarrow {{treats}}\) Disease \(\xrightarrow {{treated\ by}}\) Drug \(\xrightarrow {{treats}}\) Disease. The commuting matrix of it, denoted as X4, can be obtained by :
By using meta-path-4, we can integrate the drug-disease interaction information. Besides, meta-path-4 also indicates that if two drugs share some common indications, then the indication of one drug may also be the potential indication of another drug.
Meta-path-5: Drug \(\xrightarrow {treats}\) Disease \(\xrightarrow {caused\ by}\) Protein \(\xrightarrow {causes}\) Disease. The commuting matrix of it, denoted as X5, can be obtained by :
By using meta-path-5, we can integrate the drug-disease interaction and the disease-protein interaction information. What’s more, meta-path-5 also indicates that if two disease share some common proteins, the drug for treating one disease may also be the potential therapeutical drug for another disease.
As the definition, the element X(i,j) of the commuting matrix X denotes the number of path instances from drug di to disease sj under the corresponding meta path. We show an example in Fig. 3. There are two path instances from drug d3 to disease s2 under Meta-path-2, \(d_{3} \xrightarrow {} p_{3} \xrightarrow {} s_{2}\) and \(d_{3} \xrightarrow {} p_{5} \xrightarrow {} s_{2}\), thus we have X2(3,2)=2 in commuting matrix X2.

An example of the meaning of commuting matrix
Feature extraction with singular value decomposition
Now that element X(i,j) in a commuting matrix X denotes the number of path instances from the drug di to disease sj, then row i in the commuting matrix can be used as features of drug di, and column j can be used as features of disease sj. And we can use the concatenation of them to represent the drug-disease pair. Suppose there are m drugs and n diseases, we will have m+n (In this work, m=1186,n=449) features to represent the drug-disease pair. By contrast, the number of drug-disease pairs is small (We only have 1827 known interactions in this work). Obviously, the feature dimension is relatively high, which is not proper to construct a robust prediction model. Now that the singular value decomposition (SVD) has been successfully used to reduce the dimension in many researches, we also employed SVD to extract small number of features in our work.
By using SVD, the commuting matrix \(X \in \mathbb {R}^{m \times n}\) can be factorized into U, Σ and V such that
where \(U \in \mathbb {R}^{m \times m}\), \(\Sigma \in \mathbb {R}^{m \times n}\) and \(V \in \mathbb {R}^{n \times n}\). The diagonal entries of Σ are equal to the singular values of X (Other elements in Σ other than diagonal entries are 0). The columns of U and V are, respectively, left- and right- singular vectors for the corresponding singular values.
As is known to all, the magnitude of the singular values represents the importance of the corresponding vectors; and in Σ, the singular values are ordered in descending order. Moreover, in most cases, the sum of the first 10% or even 1% of the singular values is over 99% of the total sum of all singular values. Specifically in this drug-disease associations prediction problem, in the biomedical meaning, the most useful information about drug and disease features will be included in the first 10% even less singular values. In the process of dimensionality reduction, the useful data will not be lost, but the redundant information will be discarded. That is to say, we can use the top r singular values to approximate the matrix X:
where r≪min(m,n).
Row i in U can be used as latent features of drug di, and row j in V can be used as latent features of disease sj. As a result, the dimension of the latent feature vector of each drug-disease pair can be reduced to 2∗r. In this work, we will introduce a parameter latent_feature_percent far less than 1 (say 1%, 2%,...) to control the value of r such that r=latent_feature_percent×min(m,n).
Selection of likely negative samples from unlabeled drug-disease pairs
To build a prediction model by using supervised learning, we need both positive and negative samples. The known drug-disease treatment relations are positive samples. Being lack of validated negative samples, most methods simply select some of unlabeled samples as negative ones by random. However, the unlabeled samples are not necessarily negative, some of them may be positive samples that still remain uncovered by experiments [30]. Different with other methods, we try to find more reliable negative samples from the unlabeled ones in this work.
If a drug shares some proteins with a disease, then the drug may have potential to treat the disease. Intuitively, if a drug and a disease have no common related proteins, we can think the disease is not the indication of the drug, and thus the drug-disease pair is more likely a negative sample. By this means, we can select out more reliable negative samples from the unlabeled pairs based on the drug-protein and disease-protein interactions information. The procedure is listed in Algorithm 1.

Construction and ensemble of classifiers
The five meta paths we have selected to integrate heterogeneous data reflect different aspects of the drug-disease treatment relationship, such as two drugs with common proteins having similar indications, two drugs sharing one common indication also sharing another indication, and so on. Thus we can build five base classifiers for the prediction of drug-disease treatment relations from different sides. In our work, the base classifiers are built based on the Random Forest algorithm which was implemented by using the RandomForestClassifier function in the scikit-learn package [31], we set the number of trees as 256.
Since ensemble learning can often help to improve the performances [32, 33], after the five base classifiers are constructed, we can obtain an ensemble classifier. For an input of drug-disease pair, each base classifier outputs two probabilities indicating that the pair being negative and positive respectively. Since we want to know whether the pair has treatment relation, we only take the positive probability as considered in the ensemble model.
For a drug-disease pair x with unknown label, suppose the predicted score (probability) of each base classifier be hi(x),i=1,2,...5, we used average strategy to get the final score of the ensemble model:
If H(x) is greater than a predetermined threshold, then the sample x is predicted as the positive. Because F1-measure is a comprehensive metric, in this work, we let the program automatically determine the threshold value when F1-measure reaches the maximum value, which is the same strategy as the other researchers used.
Experiments and results
We perform 5-fold cross validation to evaluate our method. Since the filtered negative samples are more than the positive ones, we randomly select a subset from them that with size equal to the positives, and use the balanced data to train the models. We first select the appropriate number of features according to the relationship of the model performance and the feature number. Then we did three kinds of evaluation experiments: (1) We investigate whether our negative samples filtering strategy can help to improve the prediction performance; (2) We compare EMP-SVD with other state-of-the-art methods by using the same data; (3) We check the practicality of our method by doing case studies.
Evaluation metrics
Just as most other work, we performed 5-fold cross validation in the experiments. To evaluate performance of a method, there are some common metrics: Precison (PRE), Recall (REC), Accuracy (ACC), Matthews Correlation Coefficient (MCC) and F1-measure (F1). They can be calculated according to the following equations:
where TP, FP, TN and FN denote the number of true positive samples, false positive samples, true negative samples and false negative samples, respectively.
Since Precision(PRE) and Recall(REC) have some conflicts, in general, a classifier gets a higher PRE will have a lower REC, and vise versa. To get a comprehensive performance, Area Under Precison-Recall Curve(AUPR) and Area Under Receiver Operating Characteristic Curve(AUC) are often used. AUPR takes both PRE and REC into account, AUC takes both the true positive rate(TPR, the same as REC) and the false positive rate (FPR) into account, so they are comprehensive metrics. At the same time, with the help of the curves we can intuitively find which classifier is better. Therefore, in this work, we adopted AUPR and AUC as the main metrics.
Determination of appropriate number of features
Parameters are often used in existing computational methods, which limits the generalization of a model. So, it will be better to use fewer parameters or to get an analytical solution.
In this work, we just need to determine the number of singular values (corresponding to the feature number that is controlled by the parameter latent_feature_percent) during the model construction, which is very different with most state-of-the-art methods. Just mentioned above r≪min(m,n), so we set latent_feature_percent as 1%, 2%, 3%,......, 20% respectively, and the performance curves of five base classifiers and the ensemble one with different latent_feature_percent are shown in Fig. 4. The results have shown that the performances of the ensemble classifier are better than other five base classifiers, illustrating that our ensemble rule is effective. Moreover, the performances of the six classifiers are robust across different parameter settings. Anyway, we set latent_feature_percent as 3% according to the curves in this work.

Influence of different latent_feature_percent on the a AUPR b AUC
We also find that the performances of classifiers based on meta-path-1 and meta-path-4 are the worst. Noticing that both meta-path-1 and meta-path-4 just take drug-disease interactions into consideration, while the other three meta paths contain more information on drug-protein or protein-disease interactions, we think integrating more interaction information into the meta path can help to improve the performance of the classifier.
Investigation of the filtering strategy of negative samples
Being lack of validated negative samples, most of the other methods randomly select unlabeled samples to be negative ones. However, the unlabeled samples are not necessarily negative, some of them may be positive samples still uncovered by experiments. So in this work we selected out more likely negative samples from unlabeled ones according to the common protein information (as described in Algorithm 1). As shown in Table 2, all the classifiers achieve better performances in most metrics when using our negative samples filtering strategy. We also noted that the improvement is little, which may due to the fact that the known drug-protein interactions and disease-protein interactions are too few (with density of 0.0034 and 0.0027, as shown in Table 1), resulting that very few proteins could be used in the filtering process. Anyway, our strategy for selecting more reliable negative samples is useful, feasible and interpretable. We believe that along with the increase of interactions data, we will get more reliable negative samples and thus achieve more great performance improvements.
Comparison with other methods
In this section, we compare EMP-SVD with state-of-the-art methods to demonstrate the superior performance of our method. PREDICT [11] and TL-HGBI method [12] are classical methods used to predict the drug-target and drug-disease interactions. MBiRW [13], LRSSL [17] and SCMFDD [20] are the methods proposed in these two years, and achieved high performance in the prediction of drug-disease interaction. So we choose these state-of-the-art methods to compare.
PREDICT calculates the score of a given drug-disease pair (dr,di) according to all the known drug-disease pairs \(\left (d_{r}^{\prime },d_{i}^{\prime }\right)\) associated with that given pair by equation \(Score(d_{r},d_{i})= {\underset {d_{r}^{\prime },d_{i}^{\prime } \neq d_{r},d_{i}}{\max }} \sqrt { S \left (d_{r},d_{r}^{\prime }\right) \times S \left (d_{i},d_{i}^{\prime }\right) }\), where \(S \left (d_{r},d_{r}^{\prime }\right)\) is drug-drug similarity and \(S\left (d_{i},d_{i}^{\prime }\right)\) is disease-disease similarity. TL-HGBI is a three layer heterogenous network model, which makes use of the similarities and interactions of drugs, diseases and targets by iterative update. MBiRW adjusts the similarities of drugs and diseases by correlation analysis and known drug-disease associations, then uses Bi-random walk algorithm to predict the potential drug-disease associations. LRSSL is a Laplacian regularized sparse subspace learning method used to predict the drug-disease associations which integrates drug chemical information, drug target domain information and target annotation information. SCMFDD is a similarity constrained matrix factorization method for the prediction of drug-disease associations by using known drug-disease interactions, drug features and disease semantic information.
We obtained the source code of PREDICT, TL-HGBI and SCMFDD from the authors, the code of MBiRW, LRSSL are publicly available, and the parameters were set according to their papers. The parameter latent_feature_percent in EMP-SVD was set 3%. To be fair, the five parts data were kept the same division in all methods when conducting 5-fold cross validation.
As shown in Table 3, compared with other five state-of-the-art methods which make use of several kinds of similarities as well as the interaction data, the proposed classifier EMP-SVD only uses the known interaction data but achieves better performances in most metrics, especially the comprehensive metrics (AUPR and AUC). To make it more intuitively, we plotted the Precison-Recall Curve and ROC curve, which are shown in Fig. 5a and b, respectively. The AUPR and AUC of the proposed EMP-SVD are 0.956 and 0.951, respectively, better than the compared methods. Hence, it shows the simplicity and effectiveness of our method.

a Precision-Recall Curve b ROC Curve of EMP-SVD and compared methods
Case studies
Here, we test the practicality of EMP-SVD for predicting unknown associations. Except for training set composing of the known 1827 drug-disease associations and randomly selected 1827 negative samples by using our strategy, we used the trained EMP-SVD model to predict the associations for other unknown drug-disease pairs, and validate the results by literature investigation.
The new predicted top 20 drug-disease associations are shown in Table 4. We checked them carefully by literature validation and found that 13 of the top 20 predicted associations have been reported in the literatures. And these predicted associations were not originally in our data set, but we could find it out by our method, thus showing the practicality of our proposed EMP-SVD.
It should be noted that Triamcinolone (DrugBank ID: DB00620) and Betamethasone (DrugBank ID: DB00443), as glucocorticoid, are commonly used in the treatment of various skin diseases such as “Eczema” [34–36], and we find that their predicted associations include the disease “Growth Retardation, Small And Puffy Hands And Feet, And Eczema” (OMIM ID:233810). During the process of literature validation, we also find a case of growth retardation and Cushing’s syndrome due to excessive application of betamethasone-17-valerate ointment [37]. In a responsible attitude, we think that whether they can be used to treat the disease “Growth Retardation, Small And Puffy Hands And Feet, And Eczema”, or the usage and dosage should be further carefully studied by the chemists and doctors, especially should be with caution when used on children and pregnant women.
In more details, we checked the predicted potential indications of drug “Amitriptyline” (DrugBank ID: DB00321). Amitriptyline is a tricyclic antidepressant which is often used to treat symptoms of depression with the brand name: Vanatrip, Elavil, Endep. As shown in Table 5, we can find literature evidences to support 8 diseases in the top 10 predictions for Amitriptyline.
Breast cancer is a relatively common malignant tumor for female, which seriously endangers women’s health and life safety. To discover the potential drugs is of great value. So we also checked the drug list that have been predicted to treat the disease “Breast Cancer” (OMIM ID: 114480). In the top 10 drugs, as shown in Table 6, we found that 8 have been reported to be used in the clinical treatment.
Therefore, the case studies have further shown the practicality of the proposed method EMP-SVD.
Conclusions and discussions
To uncover the potential drug-disease associations is an important step in drug development, but it is time-consuming and costly to uncover them by wet experiments. Along with the accumulation of drug and disease related multi-omics data, as well as the development of machine learning techniques, more and more computational methods have been proposed to predict the potential drug-disease associations. To help the prediction, many methods integrate multiple source of data, including drugs, diseases, targets, side effects, and so on. They achieved good performances and could provide a helpful reference to the drug development. Most of them need the similarities of drug and disease related data. However, the similarity data can not be easily obtained, and people often need to customize a program to crawl data and to compute the similarities to satisfy their own need. Even worse, some features needed to calculate the similarity are unknown or unavailable. These methods will not work facing the incomplete data. Besides, being lack of validated negative samples in the prediction of drug-disease associations, most of the machine learning based methods assume the unlabeled samples to be negative ones in the training of the model. Such strategy may input errors because there may be positive samples uncovered in the unlabeled samples. What’s more, most of the existing methods use many parameters in the data integration and the model construction. The parameters are difficult to tune, which limits the generalization ability of the method.
In this work, we proposed a method named EMP-SVD to predict drug-disease interactions based on ensemble meta paths and singular value decomposition. Five meta paths from source node (drug) to end node (disease) were selected to integrate the interaction information of drugs, proteins and diseases. Then the commuting matrices of these meta paths were calculated out, each element indicates the number of path instances between the corresponding drug and disease pair. By using singular value decomposition on the commuting matrices, we can extract small number of latent features of drugs and diseases. In order to get reliable negative samples, we selected those unlabeled samples as negative under the assumption that if a drug and a disease have no common proteins, then there is smaller probability for them to be treatment relationship. Based on each meta path we first built a base classifier, and then combined them to get an ensemble classifier. The experiments results have shown that our proposed EMP-SVD method outperformed several state-of-the-art methods. Better than other methods, EMP-SVD has few parameters and very easy to set. Further more, case studies have shown the predicted new associations could be useful for further biomedical research, which demonstrate the practicality of our method.
Although there are meta path based methods in social network and some other networks, to the best of our knowledge, it is the first work in the prediction of drug-disease associations by using ensemble meta paths and singular value decomposition. Different with many existing methods, we do not need the similarity data which are not easily obtained or sometimes unavailable or unknown. Instead, we just use the interaction data which can be easily accessed in many databases to build the prediction model. The other advantage of method is that there is only one parameter that can easily set. Though we use ensemble strategy to improve the performance, each of the five base classifiers can independently act as the model as well to predict the drug-disease interactions. Since there are many computational methods to predict the target proteins for a new drug such as docking methods. For a new drug which has no known interactions with any diseases, we still can predict its interacted diseases by building classifier using meta-path-2 by making use of drug-protein and protein-disease interactions.
Though the results of our methods are promising, there are still some limitations. Firstly, we only use the information of drugs, proteins and diseases, there are many other information could also be integrated in the further work, such as the information of side effects, pathways, tissues, and so on. Secondly, we only make use of common proteins to select out the negative samples, some other information such as gene expression data can also be used for this purpose. Or we can directly build the model by positive and unlabeled samples based learning method. We will address these issues in the future study.
Abbreviations
- ACC:
-
Accuracy
- AUC:
-
Area under ROC curve
- AUPR:
-
Area under Precison-Recall curve
- FN:
-
False negative
- FP:
-
False positive
- FPR:
-
False positive rate
- MCC:
-
Matthews correlation coefficient
- OMIM:
-
Online Mendelian inheritance in man
- PRE:
-
Precision
- REC:
-
Recall
- ROC:
-
Receiver operating characteristic
- SMILES:
-
Simplified molecular input line entry specification
- SVD:
-
Singular value decomposition
- SVM:
-
Support vector machine
- TN:
-
True negative
- TP:
-
True positive
- TPR:
-
True positive rate
- UMLS:
-
Unified medical language system
References
Adams CP, Brantner VV. Estimating the cost of new drug development: is it really 802 million?Health Aff. 2006; 25(2):420–8.
Wu G, Liu J, Wang C. Semi-supervised graph cut algorithm for drug repositioning by integrating drug, disease and genomic associations. In: 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). Institute of Electrical and Electronics Engineers Inc.: 2016. p. 223–228.
Wishart DS, Feunang YD, Guo AC, et al.Drugbank 5.0: a major update to the drugbank database for 2018. Nucleic Acids Res. 2017; 46(D1):1074–82.
Bodenreider O. The unified medical language system (umls): integrating biomedical terminology. Nucleic Acids Res. 2004; 32(suppl_1):267–70.
Hurle M, Yang L, Xie Q, et al.Computational drug repositioning: from data to therapeutics. Clin Pharmacol Ther. 2013; 93(4):335–41.
Varothai S, Bergfeld WF. Androgenetic alopecia: an evidence-based treatment update. Am J Clin Dermatol. 2014; 15(3):217–30.
Novac N. Challenges and opportunities of drug repositioning. Trends Pharmacol Sci. 2013; 34(5):267–72.
Sirota M, Dudley JT, Kim J, et al.Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med. 2011; 3(96):96ra77.
Jahchan NS, Dudley JT, Mazur PK, et al.A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov. 2013; 3(12):1364–77.
Peyvandipour A, Saberian N, Shafi A, et al.A novel computational approach for drug repurposing using systems biology. Bioinformatics. 2018; 34(16):2817–2825.
Gottlieb A, Stein GY, Ruppin E, et al.Predict: a method for inferring novel drug indications with application to personalized medicine. Mol Syst Biol. 2011; 7(1):496.
Wang W, Yang S, Zhang X, et al.Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics. 2014; 30(20):2923–30.
Luo H, Wang J, Li M, et al.Drug repositioning based on comprehensive similarity measures and bi-random walk algorithm. Bioinformatics. 2016; 32(17):2664–2671.
Martínez V, Navarro C, Cano C, et al.Drugnet: Network-based drug–disease prioritization by integrating heterogeneous data. Artif Intell Med. 2015; 63(1):41–9.
Wu G, Liu J, Wang C. Predicting drug-disease interactions by semi-supervised graph cut algorithm and three-layer data integration. BMC Med Genet. 2017; 10(5):79.
Moghadam H, Rahgozar M, Gharaghani S. Scoring multiple features to predict drug disease associations using information fusion and aggregation. SAR QSAR Environ Res. 2016; 27(8):609–28.
Liang X, Zhang P, Yan L, et al.Lrssl: predict and interpret drug-disease associations based on data integration using sparse subspace learning. Bioinformatics (Oxford, England). 2017; 33(8):1187–1196.
Zhang W, Yue X, Liu F, et al.A unified frame of predicting side effects of drugs by using linear neighborhood similarity. BMC Syst Biol. 2017; 11(6):101.
Zhang W, Yue X, Huang F, et al.Predicting drug-disease associations and their therapeutic function based on the drug-disease association bipartite network. Methods. 2018; 145:51–59.
Zhang W, Yue X, Lin W, et al.Predicting drug-disease associations by using similarity constrained matrix factorization. BMC Bioinforma. 2018; 19(1):233.
Zhang W, Yue X, Chen Y, et al.Predicting drug-disease associations based on the known association bipartite network. In: 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). Institute of Electrical and Electronics Engineers Inc.: 2017. p. 503–9.
Knox C, Law V, Jewison T, et al.Drugbank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2011; 39(suppl 1):1035–41.
Law V, Knox C, Djoumbou Y, et al.Drugbank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2013; 42(D1):1091–7.
Hamosh A, Scott AF, Amberger JS, et al.Online mendelian inheritance in man (omim), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005; 33(suppl 1):514–7.
Weininger D. Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules. J Chem Inf Comput Sci. 1988; 28(1):31–6.
O’Boyle NM, Banck M, James CA, et al.Open babel: An open chemical toolbox. J Cheminformatics. 2011; 3(1):33.
Smith TF, Waterman MS, Burks C. The statistical distribution of nucleic acid similarities. Nucleic Acids Res. 1985; 13(2):645–56.
Van Driel MA, Bruggeman J, Vriend G, et al.A text-mining analysis of the human phenome. Eur J Hum Genet. 2006; 14(5):535–42.
Sun Y, Han J, Yan X, et al.Pathsim: Meta path-based top-k similarity search in heterogeneous information networks. Proc VLDB Endowment. 2011; 4(11):992–1003.
Wu G, Liu J, Min W. Prediction of drug-disease treatment relations based on positive and unlabeled samples. J Intell Fuzzy Syst. 2018; 35(2):1363–73.
Pedregosa F, Varoquaux G, Gramfort A, et al.Scikit-learn: Machine learning in python. J Mach Learn Res. 2011; 12(Oct):2825–30.
Guan D, Yuan W, Lee YK, et al.A review of ensemble learning based feature selection. Iete Tech Rev. 2014; 31(3):190–8.
Zhang W, Shi J, Tang G, Wu W, Yue X, Li D. Predicting small rnas in bacteria via sequence learning ensemble method. In: 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). Institute of Electrical and Electronics Engineers Inc.: 2017. p. 643–7.
Keczkes K, Frain-Bell W, Honeyman A, Sprunt G. The effect on adrenal function of treatment of eczema and psoriasis with triamcinolone acetonide. Br J Dermatol. 1967; 79(8–9):475–86.
Schmied C, Piletta P-A, Saurat J-H. Treatment of eczema with a mixture of triamcinolone acetonide and retinoic acid a double-blind study. Dermatology. 1993; 187(4):263–7.
Granlund H, Erkko P, Eriksson E, Reitamo S. Comparison of cyclosporine and topical betamethasone-17, 21-dipropionate in the treatment of severe chronic hand eczema. Acta Derm Venereol. 1996; 76(5):371–6.
Vermeer B, Heremans G. A case of growth retardation and cushings syndrome due to excessive application of betamethasone-17-valerate ointment. Dermatology. 1974; 149(5):299–304.
Sillanpää M, Pihlaja T. Oxcarbazepine (gp 47 680) in the treatment of intractable seizures. Acta Paediatr Hung. 1988; 29(3-4):359–64.
Virdi SK, Kanwar AS. Generalized morphea, lichen sclerosis et atrophicus associated with oral submucosal fibrosis in an adult male. Indian J Dermatol Venereol Leprology. 2009; 75(1):56.
Willemze R, Peters W, Van Hennik M, et al.Intermediate and high-dose ara-c and m-amsa (or daunorubicin) as remission and consolidation treatment for patients with relapsed acute leukaemia and lymphoblastic non-hodgkin lymphoma. Scand J Haematol. 1985; 34(1):83–7.
Richardson DS, Kelsey SM, Johnson SA, et al.Early evaluation of liposomal daunorubicin (daunoxome Ⓡ, nexstar) in the treatment of relapsed and refractory lymphoma. Investig New Drugs. 1997; 15(3):247–53.
Gustafson MP, Lin Y, New KC, et al.Systemic immune suppression in glioblastoma: the interplay between cd14+ hla-drlo/neg monocytes, tumor factors, and dexamethasone. Neuro-Oncol. 2010; 12(7):631–44.
Hultén K, Jaup B, Stenquist B, Engstrand L. Combination treatment with ranitidine is highly eff icient against helicobacter pylori despite negative impact of macrolide resistance. Helicobacter. 1997; 2(4):188–93.
Greenbaum-Lefkoe B, Rosenstock JG, Belasco JB, et al.Syndrome of inappropriate antidiuretic hormone secretion. a complication of high-dose intravenous melphalan. Cancer. 1985; 55(1):44–6.
Winick NJ, McKenna RW, Shuster JJ, et al.Secondary acute myeloid leukemia in children with acute lymphoblastic leukemia treated with etoposide. J Clin Oncol. 1993; 11(2):209–17.
Marks DI, Forman SJ, Blume KG, et al.A comparison of cyclophosphamide and total body irradiation with etoposide and total body irradiation as conditioning regimens for patients undergoing sibling allografting for acute lymphoblastic leukemia in first or second complete remission. Biol Blood Marrow Transplant. 2006; 12(4):438–53.
Christiaens G, Nieuwenhuis H, von Dem Borne AK, et al.Idiopathic thrombocytopenic purpura in pregnancy: a randomized trial on the effect of antenatal low dose corticosteroids on neonatal platelet count. BJOG: Int J Obstet Gynaecol. 1990; 97(10):893–8.
Ratain M, Rowley J. Therapy-related acute myeloid leukemia secondary to inhibitors of topoisomerase ii: from the bedside to the target genes. Ann Oncol. 1992; 3(2):107–11.
Rivera GK, Pui C-H, Abromowitch M, et al.Improved outcome in childhood acute lymphoblastic leukaemia with reinforced early treatment and rotational combination chemotherapy. Lancet. 1991; 337(8733):61–6.
Kamp O, Sieswerda GT, Visser CA. Comparison of effects on systolic and diastolic left ventricular function of nebivolol versus atenolol in patients with uncomplicated essential hypertension. Am J Cardiol. 2003; 92(3):344–8.
Williams B, MacDonald TM, Morant S, et al.Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (pathway-2): a randomised, double-blind, crossover trial. Lancet. 2015; 386(10008):2059–68.
Allen RP, Bharmal M, Calloway M. Prevalence and disease burden of primary restless legs syndrome: results of a general population survey in the united states. Mov Disord. 2011; 26(1):114–20.
Barrickman LL, Perry PJ, Allen A, et al.Bupropion versus methylphenidate in the treatment of attention-deficit hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 1995; 34(5):649–57.
Sinha R, Raut S. Management of nocturnal enuresis-myths and facts. World J Nephrol. 2016; 5(4):328.
Jeyakumar A, Brickman TM, Haben M. Effectiveness of amitriptyline versus cough suppressants in the treatment of chronic cough resulting from postviral vagal neuropathy. Laryngoscope. 2006; 116(12):2108–12.
Di Lorenzo C, Ambrosini A, Coppola G, Pierelli F. Heat stress disorders and headache: a case of new daily persistent headache secondary to heat stroke. J Neurol Neurosurg Psychiatry. 2008; 79(5):610–1.
Gawin FH, Markoff RA. Panic anxiety after abrupt discontinuation of amitriptyline. Am J Psychiatry. 1981; 138(1):117–118.
Beckman TJ, Mynderse LA. Evaluation and medical management of benign prostatic hyperplasia. In: Mayo Clinic Proceedings, vol. 80. Elsevier Inc.: 2005. p. 1356–62.
Cassidy J, Merrick MV, Smyth JF, Leonard RC. Cardiotoxicity of mitozantrone assessed by stress and resting nuclear ventriculography. Eur J Cancer. 1988; 24(5):935–8.
Yap H-Y, Blumenschein GR, Tashima CK, et al.Combination chemotherapy with vincristine and methotrexate for advanced refractory breast cancer. Cancer. 1979; 44(1):32–4.
Holland JF, Scharlau C, Gailani S, et al.Vincristine treatment of advanced cancer: a cooperative study of 392 cases. Cancer Res. 1973; 33(6):1258–64.
Gnant M, Mlineritsch B, Schippinger W, et al.Endocrine therapy plus zoledronic acid in premenopausal breast cancer. N Engl J Med. 2009; 360(7):679–91.
Mego M, Sycova-Mila Z, Obertova J, et al.Intrathecal administration of trastuzumab with cytarabine and methotrexate in breast cancer patients with leptomeningeal carcinomatosis. Breast. 2011; 20(5):478–80.
Hines SL, Mincey BA, Sloan JA, et al.Risedronate, breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2009; 27(7):1047.
Fenner MH, Elstner E. Peroxisome proliferator-activated receptor- γ ligands for the treatment of breast cancer. Expert Opin Investig Drugs. 2005; 14(6):557–68.
Ma CX, Ellis MJ, Petroni GR, et al.A phase ii study of ucn-01 in combination with irinotecan in patients with metastatic triple negative breast cancer. Breast Cancer Res Treat. 2013; 137(2):483–92.
Thamake SI, Raut SL, Gryczynski Z, et al.Alendronate coated poly-lactic-co-glycolic acid (plga) nanoparticles for active targeting of metastatic breast cancer. Biomaterials. 2012; 33(29):7164–73.
Vaughan W, Reed E, Edwards B, Kessinger A. High-dose cyclophosphamide, thiotepa and hydroxyurea with autologous hematopoietic stem cell rescue: an effective consolidation chemotherapy regimen for early metastatic breast cancer. Bone Marrow Transplant. 1994; 13(5):619–24.
Acknowledgements
The authors thank the editors and the anonymous reviewers for their helpful comments and suggestions on the quality improvement of our present paper.
Funding
Publication of this article was sponsored by grant from the National Science Foundation of China [61272274, 60970063]; the program for New Century Excellent Talents in Universities [NCET-10-0644]; and the National Science Foundation of Jiangsu Province [BK20161249].
Availability of data and materials
The data supporting the results of this research paper are included within this article.
About this supplement
This article has been published as part of BMC Bioinformatics Volume 20 Supplement 3, 2019: Selected articles from the 17th Asia Pacific Bioinformatics Conference (APBC 2019): bioinformatics. The full contents of the supplement are available online at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-20-supplement-3.
Author information
Authors and Affiliations
Contributions
GW, JL and XY developed the methodology. GW and XY executed the experiments, JL provided guidance and supervision. JL and GW wrote this paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License(http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver(http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wu, G., Liu, J. & Yue, X. Prediction of drug-disease associations based on ensemble meta paths and singular value decomposition. BMC Bioinformatics 20 (Suppl 3), 134 (2019). https://doi.org/10.1186/s12859-019-2644-5
Published:
DOI: https://doi.org/10.1186/s12859-019-2644-5