
Abstract
Mutations in TP53 lead to a defective G1 checkpoint and the dependence on checkpoint kinase 1 (Chk1) for G2 or S phase arrest in response to DNA damage. In preclinical studies, Chk1 inhibition resulted in enhanced cytotoxicity of several chemotherapeutic agents. The high frequency of TP53 mutations in triple negative breast cancer (TNBC: negative for estrogen receptor, progesterone receptor, and HER2) make Chk1 an attractive therapeutic target. UCN-01, a non-selective Chk1 inhibitor, combined with irinotecan demonstrated activity in advanced TNBC in our Phase I study. The goal of this trial was to further evaluate this treatment in women with TNBC. Patients with metastatic TNBC previously treated with anthracyclines and taxanes received irinotecan (100–125 mg/m2 IV days 1, 8, 15, 22) and UCN-01 (70 mg/m2 IV day 2, 35 mg/m2 day 23 and subsequent doses) every 42-day cycle. Peripheral blood mononuclear cells (PBMC) and tumor specimens were collected. Twenty five patients were enrolled. The overall response (complete response (CR) + partial response (PR)) rate was 4 %. The clinical benefit rate (CR + PR + stable disease ≥6 months) was 12 %. Since UCN-01 inhibits PDK1, phosphorylated ribosomal protein S6 (pS6) in PBMC was assessed. Although reduced 24 h post UCN-01, pS6 levels rose to baseline by day 8, indicating loss of UCN-01 bioavailability. Immunostains of γH2AX and pChk1S296 on serial tumor biopsies from four patients demonstrated an induction of DNA damage and Chk1 activation following irinotecan. However, Chk1 inhibition by UCN-01 was not observed in all tumors. Most tumors were basal-like (69 %), and carried mutations in TP53 (53 %). Median overall survival in patients with TP53 mutant tumors was poor compared to wild type (5.5 vs. 20.3 months, p = 0.004). This regimen had limited activity in TNBC. Inconsistent Chk1 inhibition was likely due to the pharmacokinetics of UCN-01. TP53 mutations were associated with a poor prognosis in metastatic TNBC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Approximately 15–20 % of patients with breast cancers are diagnosed with TNBC, which lacks the expression of estrogen receptor (ER) and progesterone receptor (PgR) and gene amplification of HER2 [1]. The median survival of those with metastatic TNBC is poor, ranging 9–13.3 months in retrospective studies [2, 3]. There is a significant unmet clinical need to identify new therapeutic targets and treatment options for this patient population [4].
Cell cycle checkpoints are mechanisms that arrest cell cycle progression in the presence of DNA damage to prevent the accumulation of mutations in normal cells but are exploited by cancer cells as a means of developing resistance to the cytotoxic effects of DNA damaging agents [5]. The majority of TNBCs carry mutations in TP53 [6–8], a gene that encodes the tumor suppressor protein p53 which is required for G1 checkpoint regulation in the presence of genotoxic stress [9]. However, cells with deficient p53 rely on checkpoint kinase 1 (Chk1) to activate S- and G2-checkpoints for cell cycle arrest [10–12]. When Chk1 is inhibited, p53-deficient cancer cells undergo “mitotic catastrophe” and apoptosis in response to DNA damage [5, 10–12]. Therefore, Chk1 inhibition in combination with DNA damaging agents is a potential synthetic lethal strategy to kill p53-deficient cells [13–16]. In addition, Chk1 transcript levels are found to be significantly elevated in TNBC compared to other breast cancer subtypes [17, 18], making it a feasible target in TNBC where TP53 mutations also occur at a high frequency.
UCN-01 (7-hydroxystaurosporine) was the first Chk1 inhibitor introduced in the clinic, although it also inhibits several other serine-threonine protein kinases, including certain protein kinase C isoenzymes; Cdk2, 4, and 6; and PDK1 [13, 19, 20]. Preclinical studies have shown that UCN-01 abrogates the S- and G2-checkpoints and enhances the cytotoxic effects of a variety of chemotherapeutic agents including SN-38, the active metabolite of camptothecin and irinotecan (a semisynthetic analog of camptothecin) in cancer cell lines including those of breast cancer [21–26]. Using human-in-mouse models of TNBC, we recently demonstrated that UCN-01 potentiated the cytotoxic effects of irinotecan in TP53 deficient human TNBC cells [27]. In our initial phase I study of UCN-01 and irinotecan in patients with refractory advanced solid tumor malignancies [28], two partial responses were observed in women with TNBC. Both tumors were deficient for p53. Therefore, we hypothesized that UCN-01 and irinotecan would be an effective regimen in metastatic TNBC. In this trial, we determined the response rate, toxicity, and pharmacodynamic effects of irinotecan in combination with UCN-01 and correlated these findings with TP53 mutation status, and breast cancer intrinsic subtype, in women with TNBC [29].
Patient population and methods
Patients
Eligible patients were at least 18 years of age with histologically confirmed breast cancer, negative for ER and PgR by immunohistochemistry and HER2 amplification by FISH and demonstrated an eastern cooperative oncology group (ECOG) performance status of 0–2. Patients had recurrent, measurable disease which was previously treated with an anthracycline and a taxane either in the adjuvant or metastatic setting and may have received up to three prior chemotherapeutic regimens for metastatic disease. Standard laboratory criteria were met and an oxygen saturation at least 90 % on room air at rest and after walking for 6 minutes was required. Prior chemotherapy or radiotherapy must have been completed at least 4 weeks prior to treatment. Patients with known sensitivity to UCN-01 or irinotecan, insulin-dependent diabetes mellitus or uncontrolled intercurrent illness, known diagnosis of Gilbert’s disease, or chronic unconjugated hyperbilirubinemia were excluded. The Cancer Therapy Evaluation Program (P5582), Division of Cancer Treatment and Diagnosis, National Cancer Institute (CTEP, NCI) and the institutional review boards at both Washington University School of Medicine and the University of Virginia Health System approved this protocol. All patients provided written informed consent prior to study entry.
Treatment plan and study design
Patients received irinotecan (Camptosar®, Pfizer Inc., New York, NY) 100 or 125 mg/m2 depending on the UGT1A1 genotype (patients with 7/7 allele were treated with 100 mg/m2, other patients 125 mg/m2), as a 90 min intravenous infusion on days 1, 8, 15, and 22 and UCN-01 (Kyowa Hakko Kogyo Co., Shizuoka, Japan and supplied by CTEP, NCI) as a 3 h continuous intravenous infusion on days 2 and 23 every 42 days. UCN-01 was administered at 70 mg/m2 day 2 and 35 mg/m2 day 23 and all subsequent doses. Dose modifications for irinotecan and UCN-01 included dose delays and reductions as previously described in the report on the phase I study of this combination [28]. Toxicities were graded according to common terminology criteria for adverse events (CTCAE) version 4.0. Antitumor response was evaluated by physical examination and/or imaging at baseline and following every cycle. Responses were evaluated by RECIST 1.0 [30].
Pharmacodynamic studies
Western blot analyses of peripheral blood mononuclear cells (PBMC) were collected at baseline, 24 h post irinotecan but prior to UCN-01 (day 2), 24 h post UCN-01 (day 3), and on day 8 prior to the second irinotecan administration during cycle 1 for western blot analyses of phosphorylated ribosomal protein S6 (pS6), S6, pAKTS473, AKT, pGSK3β, and GSK3β. The procedures of sample processing, western blot, and quantification of pS6 to S6 ratio were described in detail previously [28]. The primary antibodies for pS6S240/244, S6, pAKTS473, pGSK3β, and GSK3β were from Cell Signaling Technology, Inc., Danver, MA; and the primary antibodies for AKT and actin were from Santa Cruz Biotechnology, Delaware, CA, and Sigma Chemical Co., St. Louis, MO, respectively.
Immunofluorescent (IF) staining of tumors
Serial tumor biopsies of chest wall recurrences from five consented patients (patients 1, 3, 4, 6, and 11) were obtained at baseline, 24 h post irinotecan but prior to UCN-01 (day 2), 24 h post UCN-01 treatment (day 3) for immunofluorescent staining (IF) of pS6, cleaved caspase 3, phistone H3, pChk1S296, and γH2AX. Tumor biopsies were fixed in formalin, processed for hematoxylin and eosin staining, and reviewed by a pathologist to confirm cellularity prior to IF staining studies. Sample processing, antibodies and IF staining were performed as previously described [27]. The primary antibodies were pS6S240/244 (Cell Signaling Technology, Inc.), pChk1S296 (Epitomics, Inc., Burlingame, CA), cleaved caspase 3 (Cell Signaling Technology), phistone H3S28 (1:1,000, Sigma-Aldrich Co.), and γH2AXS139 (Cell Signaling Technology, Inc.). Tumor cells staining positive for phistone H3 and/or γH2AX or cleaved caspase 3 were counted in five randomly selected fields per tumor at 400X magnification. Between 225 and 1700 tumor cells were counted.
DNA extraction and TP53 sequencing of tumors
Tumor genomic DNA was isolated from 15 patients with metastatic breast cancer and analyzed for TP53 mutation by direct nucleotide sequencing of polymerase chain products of exons 4–9 as described [28].
RNA preparation, RT-PCR, and assignment of molecular subtyping of tumors
The details on RNA extraction, RT-PCR, and PAM50 Breast Cancer Intrinsic Classifier™ (PAM50, ARUP Laboratories, Salt Lake City, Utah) molecular subtyping assignment were according to previously published methods [31].
Statistical considerations
The primary endpoints for the study were the evaluation of efficacy including the overall response rate defined as complete response and partial response (CR + PR), clinical benefit rate [CR + PR + SD (stable disease)], time to treatment failure (TTF), overall survival (OS), and the evaluation of the side effect profile of this combination. The study was designed to accrue a minimum of 20 patients to have adequate precision to estimate the true overall response rate. Precision was defined by the width of a 95 % exact binomial confidence interval with an expected observed rate of at least 5/20 or a minimum lower bound of 8 %.
OS was measured from start of treatment date until date of death from any cause or last follow, whichever occurred first. TTF was measured from start of treatment date until date of treatment discontinuation due to adverse events; physician discretion; patient withdrawal; progression; death from any cause; or last follow up, whichever occurred first.
Adverse events were tabulated by severity and frequency of events. Point estimates and 95 % exact binomial confidence intervals were calculated for all response rates. The Kaplan–Meier product limit estimator was used to estimate time to event distributions which included OS and TTF. The logrank test was used to test for differences in time to event distributions with a two-sided test.
Graphical methods were used to display pharmacodynamic measures, and analyses were considered exploratory. Repeated measure models were used to estimate the pattern of changes over time for log transformed pS6 and S6 ratio measures.
Results
Patient characteristics
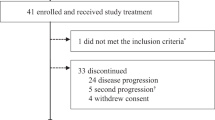
Between January 2007 and November 2010, 25 patients with metastatic TNBC were enrolled. Patient characteristics are summarized in Table 1. Patients had prior treatment with an anthracycline and a taxane and the median number of prior regimens was 3 (ranges 1–4).
Toxicity
Twenty of 25 patients completed at least one cycle of treatment (range 1–6). All 25 patients were evaluated for treatment-related adverse events (Table 2). The most common hematological toxicities were neutropenia and anemia. Only 1 patient developed febrile neutropenia. The most common non-hematological toxicities were gastrointestinal toxicities including nausea/vomiting and diarrhea. Two patients developed colitis while on study (1 due to Clostridium difficile). Laboratory abnormalities were observed and were most often associated with gastrointestinal toxicities. Two treatment-related elevations in AST/ALT were observed. One patient with baseline grade 1 AST/ALT and liver metastases developed a grade 2 fever, grade 3 hyperglycemia, and transient grade 3 ALT/AST elevation within 24 h following cycle 1, day 2 UCN-01 administration. The patient’s serum levels of ALT/AST decreased to baseline level (grade 1) within 2 days and she completed the cycle without further transaminase elevations. The other patient developed a grade 3 ALT following cycle 1 week 1 and grade 4 ALT/grade 3 AST during cycle 2 week 4. As per protocol, this patient was removed from study due to grade 4 ALT. This patient’s transaminase elevations resolved to grade 1 2 weeks after the last treatment. Several patients had toxicities related to UCN-01. Two patients experienced hypoxia (grade 2 and grade 3, respectively), and five patients experienced grade 1 or 2 hypotension during the UCN-01 infusion that responded to intravenous fluid. As per protocol, the patient with grade 3 hypoxia, associated with grade 2 hypotension, was removed from study. Hyperglycemia occurred in over half of the patients, with grade 3 or 4 in five patients.
Efficacy
Among the 25 patients enrolled, only 22 were evaluated for response as one withdrew consent and two went off study during cycle 1 due to toxicities, one due to elevated transaminases and the other as a result of hypoxia during the UCN-01 infusion (Table 3). No complete responses were observed. One PR lasting 24 weeks occurred in a patient (patient 15) with liver and lung metastasis. This patient was previously treated with neoadjuvant FEC-T (5-Fluorouracil, epirubicin, cyclophosphamide followed by docetaxel) at initial diagnosis and, at disease progression, with capecitabine followed by paclitaxel and bevacizumab. Tumor from this patient was found to be basal-like, and carry a TP53 mutation (Table 4). The overall response rate (CR + PR) was 1 in 25 [4, 95 % CI (0, 20 %)]. Two patients had prolonged disease stabilization that lasted for 37 and 28 weeks, respectively. The clinical benefit rate (CR + PR + SD > 6 months) was 3 in 25 [12, 95 % CI (3, 31 %)]. The median TTF and OS were 1.7 months, 95 % CI (1.2, 2.6 months) and 11.3 months, 95 % CI (3.8, 19.0 months), respectively, as shown in Fig. 1a, b. The study did not meet the pre-defined efficacy criteria for further evaluation.

Patient outcome. Kaplan–Meier survival curves on time to treatment failure (TTF) (a) and OS (b) for all patients and Kaplan–Meier survival curves on TTF (c) and OS (d) by tumor TP53 mutation status
Pharmacodynamic studies
PBMCs were collected to evaluate the bioavailability of UCN-01 over time. Since UCN-01 inhibits PDK1, treatment with UCN-01 was expected to reduce levels of pS6, pAKT, and pGSK3β [32, 33]. The levels of the phosphorylated S6, AKT, and GSK3β were decreased 24 h after UCN-01 on day 3, but returned to baseline on day 8 in most patients (n = 24, data not shown). Representative western blots pre- and post-therapy are shown for two patients (Fig. 2a). The ratio of pS6 to S6 was quantified at baseline (n = 24), day 1 (n = 24), day 3 (n = 22), and day 8 (n = 20) (Fig. 2b). Compared to baseline, the ratio of pS6 to S6 was decreased significantly (p < 0.001) 24 h after UCN-01 on day 3, but not significantly different on day 8, indicating that UCN-01 activity was not present by day 8.

Pharmacodynamic effects of UCN-01 on PBMC (a, b) and tumor tissue (c–h). PBMC were collected at baseline (BL), 24 h post irinotecan but prior to UCN-01 on day 2 (D2), 24 h post UCN-01 on day 3 (D3), and on day 8 (D8) prior to the second irinotecan treatment during cycle 1. PBMC were lysed and analyzed by western blotting with antibodies specific for total and phosphorylated forms of S6 ribosomal protein (S6), AKT, and GSK3ß with actin as a loading control. Representative western blots from two patients (4 and 11) are shown (a). The ratio of pS6 to total S6 protein level was plotted at each time point (b). Optional serial tumor biopsies were collected before study drug therapy at baseline (BL), 24 h post irinotecan but prior to UCN-01 on day 2 (D2), and 24 h post UCN-01 on day 3 (D3) from 4 patients (1, 4, 6, and 11). Representative immunofluorescent (IF) staining images on tumors from patient 11 are shown in (c). The quantitation of the IF analysis on the γH2AX (d), pChk1S296 (e), phistone H3 (f), cleaved caspase 3 (g), and pS6 (h), for each patient is shown in the lower panels. CC3 cleaved caspase 3
To evaluate the molecular effects of UCN-01, serial tumor biopsies were collected at three time points including baseline, day 2 (24 h post irinotecan), and day 3 (24 h post UCN-01) in five patients (1, 3, 4, 6, and 11) with chest wall recurrences. Tumors from patients 1 and 11 were mutant for TP53, while those from patients 3, 4, and 6 were wild type for TP53 (Table 4). Tumor biopsies from patient 3 were not submitted for further analysis because there were no tumor cells in the baseline sample. Immunostaining of γH2AX (marker for double strand DNA breaks), pChk1S296 (marker of Chk1 activation), phistone H3 (marker for mitosis), cleaved caspase 3 (CC3) (marker for apoptosis), and pS6 were performed. As expected from treatment with irinotecan, there was an increase in double strand DNA breaks (↑ γH2AX) (Fig. 2c, d) and activation of Chk1 (↑ pChk1S296) (Fig. 2c, e) in all tumors on day 2 biopsy (representative staining from patient 11’s tumor). phistone H3 staining was low at baseline (1–2 %); therefore, the anticipated further reduction of phistone H3 due to cell cycle arrest from irinotecan was difficult to demonstrate (Fig. 2c, f). Despite the anticipated pharmacodynamic effects of irinotecan, there was little induction of apoptosis on day 2 (Fig. 2c, g). Following UCN-01 therapy, pS6 was reduced in all tumors (Fig. 2c, h). However, loss of Chk1S296 was only observed in tumors from patients 6 (TP53 WT (wild type)) and 11 (TP53 deletion) (Fig. 2c, e). Tumor from patient 11, which had a deletion in TP53, demonstrated the most dramatic increase in apoptosis with close to 10 % of tumor cells stained positive for cleaved caspase 3 on day 3 (Fig. 2g). The same tumor demonstrated a slight increase in phistone H3 (Fig 2f) and an increase in γH2AX (Fig. 2d) following combination therapy, suggesting cell cycle abrogation and enhanced DNA damage from the addition of UCN-01. The patient had a best response of stable disease and a TTF of 2.6 months. Interestingly, tumor from patient 6, which was WT (wild type) for TP53, showed the most dramatic increase in phistone H3 (Fig. 2f) following UCN-01, suggesting cell cycle abrogation, but this patient had a best response of PD and a TTF of 1.6 months on study, indicating ineffective therapy.
In summary, data on pChk1S296 indicated an inconsistent effect of UCN-01 on Chk1, with one (patient 11) of two TP53 mutant tumors demonstrating the anticipated cell cycle pharmacodynamic effect (loss of pS296) of UCN-01. However, given that active Chk1 (p296-phosphorylated form) is targeted for ubiquitin-mediated proteolysis, there may have been less total Chk1 in this tumor sample [34–36].
Predictive biomarker analysis
To investigate the molecular characteristics of the TNBC and their potential predictive value in prognosis and therapeutic response, tumor specimens for TP53 mutation and molecular subtype by PAM50 were analyzed (Table 4). Among the 25 patients enrolled, one withdrew consent and a specimen was not requested, three were without available archival tumor specimens, and five had archival tumor specimens with limited cellularity, and one each had poor quality DNA and RNA, respectively. Among the 15 patients with sufficient specimens for PAM50 analysis, there were ten basal-like, one basal/HER2-enriched, two HER2-enriched, and two luminal B. Among the 15 patients with sufficient tumors for analysis, eight (53 %) carried mutations in TP53. In this limited dataset, there was no significant difference in TTF between the two populations (Fig. 1c), but a significantly worse OS was observed in patients with mutations in TP53. The median OS was 5.5 (95 % CI: 2, 11) months and 20.3 (95 % CI: 3, 24) months in patients with and without TP53 mutation (p = 0.004), respectively (Fig. 1d).
Discussion
Irinotecan administered weekly as a single agent has been shown to induce an overall response rate of 5–23 % in unselected patients with metastatic breast cancers resistant to anthracycline and/or taxane [37, 38]. In this study, women with resistant TNBC had previously received both an anthracycline and a taxane and had a median of 3 chemotherapy regimens. The overall response rate to irinotecan in combination with UCN-01 was 4 % and TTF was 1.7 months in our study. Given the low response rate observed, UCN-01, when administered on a day 2 and 23 schedule in combination with weekly irinotecan (days 1, 8, 15, 22), did not appear to potentiate the cytotoxic effects of irinotecan.
The pharmacodynamics studies on serially collected PBMC and tumor biopsies revealed limited bioavailability of UCN-01 to tissue, likely as a result of the high affinity binding of this agent to the α1-acid glycoprotein (AAG) [28, 39–41]. Despite the higher dose of UCN-01 on day 2 compared to subsequent doses, the initial suppression of pS6 in PBMC on day 3 was no longer detected by day 8, a similar finding from our initial phase I study of this combination [28]. In addition, the inhibitory effect of UCN-01 on tumor tissue Chk1, which was activated by irinotecan in all tumors on day 2, was inconsistent although pS6 was inhibited on day 3. These data support the notion that UCN-01 was not able to achieve the desired Chk1 inhibition, likely due to the limited tissue bioavailability with the current dosing and scheduling. Interestingly, patient 11’s tumor, which had a deletion in TP53 and a reduction in pChk1 following UCN-01, had a dramatic increase in apoptosis following UCN-01, indicating inhibition of cell cycle checkpoint and potentiation of the cytotoxic effects of irinotecan. As anticipated, enhanced apoptosis was not observed in the tumor from patient 6, which was wild type for TP53, although there was also a reduction in pChk1 following UCN-01 in this tumor. Further studies of Chk1 inhibitors with improved pharmacokinetic properties and selectivity for Chk1 are needed [13].
Analysis of γH2AX, pChk1, and cleaved caspase 3 on serial biopsies obtained from four patients at baseline and on day 2 after irinotecan indicated that although irinotecan induced anticipated DNA damage and activation of checkpoint pathways, the effect on apoptosis was minimal. The failure to induce cell death following DNA damage indicates mechanisms in place in these tumors to counteract the apoptotic effect of irinotecan, and these resistance mechanisms remain to be elucidated. These data highlight the need for a better understanding of treatment resistance to chemotherapy for TNBC.
As reported in the literature [42, 43], we observed a significant heterogeneity in TNBC in this study. Most of the tumors were basal-like by PAM50 breast cancer molecular subtyping; other subtypes included HER2-enriched and luminal B. The frequency of each subtype is consistent with previous reports [43–45]. Despite its small sample size, this study demonstrates that TP53 mutation is a robust predictor of worse clinical outcome in patients with metastatic TNBC. In a single institution small retrospective study of 135 patients with early stage breast cancer, including 32 patients with TNBC treated with adjuvant anthracycline-based chemotherapy, p53 status assessed by IHC was strongly predictive of relapse free survival and OS only in the TNBC cases [46]. Similar findings were obtained in another retrospective study that used IHC analysis [47]. Our study provides important confirmatory evidence that TP53 mutation is a predictor of worse outcome in the anthracycline and taxane resistant metastatic TNBC patient population. The significant difference in outcome indicates a need to stratify patients based on TP53 status in the design of clinical trials in patients with metastatic TNBC. Though we were not able to obtain specimens on all patients, the biomarker analyses of PBMC, archival tumor, and biopsy specimens that were obtained have provided important insights into the biology of TNBC when treated with UCN-01 in combination with irinotecan.
Conclusions
In this trial, we examined the therapeutic potential of UCN-01, the first identified Chk1 inhibitor, in combination with irinotecan, in patients with metastatic TNBC. Although the clinical activity was unimpressive most likely due to the pharmacokinetic property of UCN-01, we developed effective pharmacodynamic markers to evaluate target inhibition for this class of agents. Our data indicated that effective Chk1 inhibition could enhance chemotherapy-induced apoptosis in TP53 mutant tumors. Furthermore, we demonstrated that TNBC is molecularly heterogeneous and TP53 mutant TNBC was associated with a particularly poor survival, indicating a need to stratify patient populations in clinical trials. These results provide a foundation for future trials of Chk1 inhibitors in TNBC.
Abbreviations
- TNBC:
-
Triple negative breast cancer
- PBMC:
-
Peripheral blood mononuclear cells
- CR:
-
Complete response
- PR:
-
Partial response
- OS:
-
Overall survival
- ER:
-
Estrogen receptor
- PgR:
-
Progesterone receptor
- Chk1:
-
Checkpoint kinase 1
- ECOG:
-
Eastern cooperative oncology group
- CTEP:
-
Cancer therapy evaluation program
- NCI:
-
National cancer institute
- ANC:
-
Absolute neutrophil count
- AST:
-
Aspartate aminotransferase
- ALT:
-
Alanine aminotransferase
- CTCAE:
-
Common terminology criteria for adverse events
- pS6:
-
Phosphorylated ribosomal protein S6
- IF:
-
Immunofluorescent
- IHC:
-
Immunohistochemistry
- HRP:
-
Horseradish peroxidase
- SD:
-
Stable disease
- TTF:
-
Time to treatment failure
- FEC-T:
-
5-Fluorouracil, epirubicin, cyclophosphamide followed by docetaxel
- CB:
-
Clinical benefit
- WT:
-
Wild type
- CC3:
-
Cleaved caspase 3
References
Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med 363(20):1938–1948
Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA (2007) Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 13(15 Pt 1):4429–4434
Kassam F, Enright K, Dent R, Dranitsaris G, Myers J, Flynn C, Fralick M, Kumar R, Clemons M (2009) Survival outcomes for patients with metastatic triple-negative breast cancer: implications for clinical practice and trial design. Clin Breast Cancer 9(1):29–33
Gelmon K, Dent R, Mackey JR, Laing K, McLeod D, Verma S (2012) Targeting triple-negative breast cancer: optimising therapeutic outcomes. Ann Oncol 23(9):2223–2234 April 19
Eastman A (2004) Cell cycle checkpoints and their impact on anticancer therapeutic strategies. J Cell Biochem 91(2):223–231
Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S et al (2006) Race, breast cancer subtypes, and survival in the Carolina breast cancer study. JAMA 295(21):2492–2502
Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490(7418):61–70
Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G et al (2012) The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486(7403):395–399
Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW (1991) Participation of p53 protein in the cellular response to DNA damage. Cancer Res 51(23 Pt 1):6304–6311
Xiao Z, Chen Z, Gunasekera AH, Sowin TJ, Rosenberg SH, Fesik S, Zhang H (2003) Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem 278(24):21767–21773
Zhao H, Watkins JL, Piwnica-Worms H (2002) Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci USA 99(23):14795–14800
Sorensen CS, Syljuasen RG, Falck J, Schroeder T, Ronnstrand L, Khanna KK, Zhou BB, Bartek J, Lukas J (2003) Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 3(3):247–258
Ma CX, Janetka JW, Piwnica-Worms H (2011) Death by releasing the breaks: Chk1 inhibitors as cancer therapeutics. Trends Mol Med 17(2):88–96
Dai Y, Grant S (2010) New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res 16(2):376–383
Tse AN, Carvajal R, Schwartz GK (2007) Targeting checkpoint kinase 1 in cancer therapeutics. Clin Cancer Res 13(7):1955–1960
Chen T, Stephens PA, Middleton FK, Curtin NJ (2012) Targeting the S and G2 checkpoint to treat cancer. Drug Discov Today 17(5–6):194–202
Verlinden L, Vanden Bempt I, Eelen G, Drijkoningen M, Verlinden I, Marchal K, De Wolf-Peeters C, Christiaens MR, Michiels L, Bouillon R et al (2007) The E2F-regulated gene Chk1 is highly expressed in triple-negative estrogen receptor/progesterone receptor/HER-2 breast carcinomas. Cancer Res 67(14):6574–6581
Speers C, Tsimelzon A, Sexton K, Herrick AM, Gutierrez C, Culhane A, Quackenbush J, Hilsenbeck S, Chang J, Brown P (2009) Identification of novel kinase targets for the treatment of estrogen receptor-negative breast cancer. Clin Cancer Res 15(20):6327–6340
Busby EC, Leistritz DF, Abraham RT, Karnitz LM, Sarkaria JN (2000) The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res 60(8):2108–2112
Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O’Connor PM, Piwnica-Worms H (2000) The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem 275(8):5600–5605
Jones CB, Clements MK, Wasi S, Daoud SS (2000) Enhancement of camptothecin-induced cytotoxicity with UCN-01 in breast cancer cells: abrogation of S/G(2) arrest. Cancer Chemother Pharmacol 45(3):252–258
Monks A, Harris ED, Vaigro-Wolff A, Hose CD, Connelly JW, Sausville EA (2000) UCN-01 enhances the in vitro toxicity of clinical agents in human tumor cell lines. Invest New Drugs 18(2):95–107
Sugiyama K, Shimizu M, Akiyama T, Tamaoki T, Yamaguchi K, Takahashi R, Eastman A, Akinaga S (2000) UCN-01 selectively enhances mitomycin C cytotoxicity in p53 defective cells which is mediated through S and/or G(2) checkpoint abrogation. Int J Cancer 85(5):703–709
Jones CB, Clements MK, Redkar A, Daoud SS (2000) UCN-01 and camptothecin induce DNA double-strand breaks in p53 mutant tumor cells, but not in normal or p53 negative epithelial cells. Int J Oncol 17(5):1043–1051
Shao RG, Cao CX, Shimizu T, O’Connor PM, Kohn KW, Pommier Y (1997) Abrogation of an S-phase checkpoint and potentiation of camptothecin cytotoxicity by 7-hydroxystaurosporine (UCN-01) in human cancer cell lines, possibly influenced by p53 function. Cancer Res 57(18):4029–4035
Xie X, Sasai K, Shibuya K, Tachiiri S, Nihei K, Ohnishi T, Hiraoka M (2000) p53 status plays no role in radiosensitizing effects of SN-38, a camptothecin derivative. Cancer Chemother Pharmacol 45(5):362–368
Ma CX, Cai S, Li S, Ryan CE, Guo Z, Schaiff WT, Lin L, Hoog J, Goiffon RJ, Prat A et al (2012) Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J Clin Invest 122(4):1541–1552
Fracasso PM, Williams KJ, Chen RC, Picus J, Ma CX, Ellis MJ, Tan BR, Pluard TJ, Adkins DR, Naughton MJ et al (2011) A phase I study of UCN-01 in combination with irinotecan in patients with resistant solid tumor malignancies. Cancer Chemother Pharmacol 67(6):1225–1237
Marty B, Maire V, Gravier E, Rigaill G, Vincent-Salomon A, Kappler M, Lebigot I, Djelti F, Tourdes A, Gestraud P et al (2008) Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res 10(6):R101
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European organization for research and treatment of cancer, national cancer institute of the United States, national cancer institute of Canada. J Natl Cancer Inst 92(3):205–216
Parker JS, Mullins M, Cheang MCU, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z et al (2009) Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 27(8):1160–1167
Templeton DJ (2001) Protein kinases: getting NEKed for S6K activation. Curr Biol 11(15):R596–R599
Mora A, Komander D, van Aalten DM, Alessi DR (2004) PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol 15(2):161–170
Leung-Pineda V, Huh J, Piwnica-Worms H (2009) DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res 69:2630–2637
Zhang YW, Brognard J, Coughlin C, You Z, Dolled-Filhart M, Aslanian A, Manning G, Abraham RT, Hunter T (2009) The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol Cell 35(4):442–453
Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, Abraham RT (2005) Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell 19(5):607–618
Perez EA, Hillman DW, Mailliard JA, Ingle JN, Ryan JM, Fitch TR, Rowland KM, Kardinal CG, Krook JE, Kugler JW et al (2004) Randomized phase II study of two irinotecan schedules for patients with metastatic breast cancer refractory to an anthracycline, a taxane, or both. J Clin Oncol 22(14):2849–2855
Shigeoka Y, Itoh K, Igarashi T, Ishizawa K, Saeki T, Fujii H, Minami H, Imoto S, Sasaki Y (2001) Clinical effect of irinotecan in advanced and metastatic breast cancer patients previously treated with doxorubicin- and docetaxel-containing regimens. Jpn J Clin Oncol 31(8):370–374
Jimeno A, Rudek MA, Purcell T, Laheru DA, Messersmith WA, Dancey J, Carducci MA, Baker SD, Hidalgo M, Donehower RC (2008) Phase I and pharmacokinetic study of UCN-01 in combination with irinotecan in patients with solid tumors. Cancer Chemother Pharmacol 61(3):423–433
Perez RP, Lewis LD, Beelen AP, Olszanski AJ, Johnston N, Rhodes CH, Beaulieu B, Ernstoff MS, Eastman A (2006) Modulation of cell cycle progression in human tumors: a pharmacokinetic and tumor molecular pharmacodynamic study of cisplatin plus the Chk1 inhibitor UCN-01 (NSC 638850). Clin Cancer Res 12(23):7079–7085
Sausville EA, Arbuck SG, Messmann R, Headlee D, Bauer KS, Lush RM, Murgo A, Figg WD, Lahusen T, Jaken S et al (2001) Phase I trial of 72-hour continuous infusion UCN-01 in patients with refractory neoplasms. J Clin Oncol 19(8):2319–2333
Irshad S, Ellis P, Tutt A (2011) Molecular heterogeneity of triple-negative breast cancer and its clinical implications. Curr Opin Oncol 23(6):566–577
Metzger-Filho O, Tutt A, de Azambuja E, Saini KS, Viale G, Loi S, Bradbury I, Bliss JM, Azim HA Jr, Ellis P et al (2012) Dissecting the heterogeneity of triple-negative breast cancer. J Clin Oncol 30(15):1879–1887
Carey LA, Rugo HS, Marcom PK, Mayer EL, Esteva FJ, Ma CX, Liu MC, Storniolo AM, Rimawi MF, Forero-Torres A et al (2012) TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol 30(21):2615–2623 June 4
Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM (2010) Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res 12(5):R68
Chae BJ, Bae JS, Lee A, Park WC, Seo YJ, Song BJ, Kim JS, Jung SS (2009) p53 as a specific prognostic factor in triple-negative breast cancer. Jpn J Clin Oncol 39(4):217–224
Biganzoli E, Coradini D, Ambrogi F, Garibaldi JM, Lisboa P, Soria D, Green AR, Pedriali M, Piantelli M, Querzoli P et al (2011) p53 status identifies two subgroups of triple-negative breast cancers with distinct biological features. Jpn J Clin Oncol 41(2):172–179
Acknowledgments
We wish to thank the patients and their families for participation in this study. We also thank the nurses, clinical research and regulatory coordinators at Washington University Siteman Cancer Center and the Emily Couric Clinical Cancer Center at the University of Virginia Health System for their care of the patients on this study. Dr. Mark A. Watson, Director, Tissue Procurement Core; Alvin J. Siteman Cancer Center at Washington University School of Medicine; and Barnes-Jewish Hospital and his staff members, especially Ms. Vicky Holtschlag, for tissue acquisition and processing. We would like to thank Ms. Jennifer Stratman and Ms. Shelly Audrain from the Laboratory and Genomic Medicine, Department of Pathology at Washington University School of Medicine for tumor TP53 sequencing. We would like to thank Dr. Katherine Deschryver for reviewing the pathology slides. We would like to thank members of the SPECS team, Sherri Davies, Elaine Mardis, Jacqueline Snider, and Tammi Vickery, for assistance with the PAM50 Breast Cancer Intrinsic Classifer™ molecular subtype analysis. Grant Support: NIH R21 CA128369 (P. M. F.), Susan G. Komen Foundation KG08155 (C.X.M. and H.P.-W.), and NIH U01 CA114722 (M.J.E).
Conflict of interest
Dr. Matthew J. Ellis and Dr. Philip S. Bernard disclose Patent and licensing regarding PAM50, Bioclassifier LLC. All other co-authors disclose no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Helen Piwnica-Worms and Paula M. Fracasso contributed equally to this study.
Preliminary results of this study were presented at the American Society of Clinical Oncology Meeting 2012 and published in abstract form in J Clin Oncol 30, 2012 (suppl; abstr 3047).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Ma, C.X., Ellis, M.J.C., Petroni, G.R. et al. A phase II study of UCN-01 in combination with irinotecan in patients with metastatic triple negative breast cancer. Breast Cancer Res Treat 137, 483–492 (2013). https://doi.org/10.1007/s10549-012-2378-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-012-2378-9