
Abstract
Background
Inguinal and scrotal hernias are of great concern to pig producers, and lead to poor animal welfare and severe economic loss. Selection against these conditions is highly preferable, but at this time no gene, Quantitative Trait Loci (QTL), or mode of inheritance has been identified in pigs or in any other species. Therefore, a complete genome scan was performed in order to identify genomic regions affecting inguinal and scrotal hernias in pigs. Records from seedstock breeding farms were collected. No clinical examinations were executed on the pigs and there was therefore no distinction between inguinal and scrotal hernias. The genome scan utilised affected sib pairs (ASP), and the data was analysed using both an ASP test based on Non-parametric Linkage (NPL) analysis, and a Transmission Disequilibrium Test (TDT).
Results
Significant QTLs (p < 0.01) were detected on 8 out of 19 porcine chromosomes. The most promising QTLs, however, were detected in SSC1, SSC2, SSC5, SSC6, SSC15, SSC17 and SSCX; all of these regions showed either statistical significance with both statistical methods, or convincing significance with one of the methods. Haplotypes from these suggestive QTL regions were constructed and analysed with TDT. Of these, six different haplotypes were found to be differently transmitted (p < 0.01) to healthy and affected pigs. The most interesting result was one haplotype on SSC5 that was found to be transmitted to hernia pigs with four times higher frequency than to healthy pigs (p < 0.00005).
Conclusion
For the first time in any species, a genome scan has revealed suggestive QTLs for inguinal and scrotal hernias. While this study permitted the detection of chromosomal regions only, it is interesting to note that several promising candidate genes, including INSL3, MIS, and CGRP, are located within the highly significant QTL regions. Further studies are required in order to narrow down the suggestive QTL regions, investigate the candidate genes, and to confirm the suggestive QTLs in other populations. The haplotype associated with inguinal and scrotal hernias may help in achieving selection against the disorder.
Similar content being viewed by others

Background
The occurrence of hernias is a significant problem facing pig producers, and leads to poor animal welfare and severe economic loss. The most commonly occurring hernias are hernia inguinalis and hernia scrotalis which occur within the pig population at frequencies from 1.7% to 6.7% [1]. By definition, an inguinal hernia describes a situation where hernial contents are present in the inguinal canal, whereas scrotal hernia refers to a situation where hernial contents are present in the scrotum. In these conditions, most frequently the distal jejunum and ileum pass through the vaginal ring and enter the inguinal canal. Small colon and omentum can also herniate but this occurs less frequently. Inguinal and scrotal hernias are further subdivided, in human medicine, as indirect or direct. An indirect hernia refers to the passage of intestinal loops through the vaginal ring into the vaginal tunic. While a direct hernia refers to the passage of intestinal loops through a fascial defect near the vaginal ring, resulting in a situation where intestinal loops pass through the inguinal canal but are not covered by the vaginal tunic. The inguinal canal is a short passage running through the inferior part of the interior abdominal wall. Under normal conditions its function is to allow the testes, which develop in the lumbar region of the abdomen, to migrate to the scrotum. The testes descend from the anterior abdominal wall through the processus vaginalis via a propulsive force generated by muscles derived from the gubernaculums, a ligament linking the testes and scrotum. Development of inguinal and scrotal hernias usually results from failed obliteration of the processus vaginalis after descent of the testis [2], or from failed involution at the internal inguinal ring [3]. Complication may also occur, in the form of intestinal obstruction or strangulation [4]. Furthermore, in many cases the undescended testes are associated with a patent processus vaginalis because the processus vaginalis does not obliterate unless the testis reach the scrotum [5]. This particular form of inguinal and scrotal hernias is correlated with the occurrence of cryptorchism [1], and is a concern for the pig breeding industry as well.
Several studies have shown that genetic factors are involved in the development of inguinal and scrotal hernias [6, 7], however the mode of inheritance has not yet been clarified. Estimated h2 for these types of hernia range from 0.20 [8] to 0.86 [9]. Several studies have investigated genes involved in the control of testicular descent, obliteration of processus vaginalis and in the closing of the inguinal ring; by association, these genes may also be involved in the occurrence of inguinal and scrotal hernias. Specifically, Insulin-like receptor 3 (INSL3), Müllerian inhibiting substance (MIS), and relaxin are all involved in gubernacular growth [10], while the calcitonin gene-related peptide (CGRP) released from the genitofemoral nerve, may be responsible for failed fusion and disappearance of processus vaginalis [11, 12].
The aim of this study was to identify genomic regions affecting the frequency of inguinal and scrotal hernias in pigs. Records from seedstock breeding farms were used in this study. No clinical examinations were executed on the pigs and therefore, there was no distinction between inguinal and scrotal hernias. Examination of these regions may reveal causative genes responsible for the trait, which would be valuable information for use in Marker-Assisted Selection (MAS) schemes directed at decreasing hernia prevalence. Previously, parametric linkage analysis applied to large pedigrees containing many affected individuals has helped in the identification of genes with high penetrance [13]. However, for those diseases lacking a clear Mendelian inheritance pattern, or caused by several genes of low to moderate penetrance, nonparametric analysis can be a more robust and successful alternative. Crucially, linkage analysis requires correct assumptions to be made regarding the inheritance model and allele frequencies of the susceptibility alleles, and incorrect model specification will result in a loss of power and a bias in the estimation of recombination fraction [14]. Nonparametric methods, by comparison, require no such assumptions to be made, and we consider this to be better suited approach for our analysis of small sets of affected relatives. The Transmission/Disequilibrium Test (TDT) [15] was used to investigate whether the identified hernia-linked markers were associated with the trait on a population level. In the present article, we describe the results of the first published genome-wide scan of inguinal and scrotal hernias in pigs, as well as one of the first genome scans in livestock populations to use the affected sib-pair design.
Results
Linkage analysis
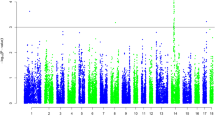
The linkage analysis was performed using an affected sib pair (ASP) test. Figure 1 and 2 shows the profiles of the multipoint NPL scores and the respective information content (Info) for each chromosome. The average information content was 0.52, which is a similar value to those found in previous studies in swine [16] and humans [17]. Two chromosomal regions had linkage score exceeding the nominal significance level of p < 0.001. The most significant regions were found on SSC2, with a multipoint NPL score of 3.16 at marker SW834, and on SSC15 which showed an NPL score of 3.47 at marker SW919. Four markers on SSC2 reached a significance level of p < 0.01 (NPL score larger than 2.33); these four markers spanned a chromosomal region of 40 cM. In addition, the linkage score exceeded the nominal significance level of p < 0.01 for six chromosomes, with highest maximum multipoint NPL scores of 2.35 at marker S0301 on SSC4, 2.45 at marker SW963 on SSC5, 2.92 at marker S0075 on SSC13, 2.47 at marker SW1891 on SSC17, 2.80 at marker SW1808 on SSC18, and 2.74 at marker SW259 on SSCX. Finally, the nominal significance level of p < 0.05 was reached on four chromosomal regions with highest maximum multipoint NPL scores of 2.06 at marker S0331 on SSC1, 1.68 at marker S0003 on SSC6, 2.20 at marker TNFB on SSC7, and 2.06 at marker SW2180 on SSC12. Results with significance level above 5% are presented in Table 1.

Linkage results of the total genome scan for inguinal and scrotal hernias, showing sus scrofa chromosomes (SSC) 1 to 12. The bold line shows multipoint NPL score (scale on left) while the dotted line shows the information content (scale on right) for each chromosome. The threshold values P = 0.05 and P = 0.01 are shown as horizontal lines at NPL = 1.64 and NPL = 2.33, respectively. Marker positions and names are shown on the X axis.

Linkage results of the total genome scan for inguinal and scrotal hernias, showing sus scrofa chromosomes (SSC) 13 to X. The bold line shows multipoint NPL score (scale on left) while the dotted line shows the information content (scale on right) for each chromosome. The threshold values P = 0.05 and P = 0.01 are shown as horizontal lines at NPL = 1.64 and NPL = 2.33, respectively. Marker positions and names are shown on the X axis.
TDT analysis
TDT analysis was applied to all loci in the data set. The significant loci are presented in Table 1. At the TDT-significant loci, TDT was also performed on the allele level, to identify individual hernia-associated alleles (Table 2). Markers displaying significance at a nominal value of p < 0.05 were found on SSC1, SSC5, SSC6, SSC7, SSC15, and SSC17. A chromosome-wide significance of p < 0.01 was found for the two markers; SW963 on SSC5 and SW1355 on SSC6. All TDT-significant markers were also significant in the ASP test, with the exception of SW1355 on chromosome 6, which was also the most significant marker found in the TDT.
The loci showing significance for both ASP and TDT, and the regions showing high significance (p < 0.005) with only ASP or TDT, were considered to be the most interesting for further analyses. Haplotypes from these regions were constructed and TDT analysis was used to identify those haplotypes most strongly associated with hernia. Six different haplotypes were found to be differently transmitted (p < 0.01). One haplotype on SSC5 was transmitted four times more frequently to pigs with hernias than to healthy pigs (p < 0.00005). Other haplotypes, showing an increased frequency of transmission to affected pigs (p < 0.01) were found on SSC6, SSC15 and SSC17. Table 3 shows the markers included in the haplotypes, the number of affected/non-affected animals with the transmitted/non-transmitted haplotype, and the p-values. Distributions belonging only to the significant haplotypes are shown.
Discussion
Our genomewide scan for QTLs affecting incidence of hernia revealed 9 genomic regions on 8 chromosomes using ASP test (p < 0.01), and 6 genomic regions on 6 chromosomes using TDT (p < 0.01). Two of these regions, on SSC5 and SSC17, were significant (p < 0.01) when analysed by both statistical methods (Table 1). Five regions located on SSC1, SSC2, SSC6, SSC15 and SSCX were found to be highly significant (p < 0.005) from either the ASP test or the TDT. We consider these seven regions to be those most likely involved in genetic susceptibility to hernia. Two different statistical methods were used (ASP and TDT) in order to take advantage of their complementary qualities in detecting genomic regions affecting defect/disease traits. The ASP method tests for markers linked to genes affecting deformity status, while the TDT method tests for markers that are both linked to these genes and associated with a particular trait on a population level. Essentially, the TDT is a fine-mapping method requiring linkage disequilibrium (LD) between causative polymorphisms and the nearest markers. In this study however, the TDT has been used on families with more than one (affected or unaffected) offspring per parental pair; under such conditions the TDT is, to some extent, a test for linkage, although the power of the test still increases greatly with the amount of LD [18]. Therefore, for our application, the ASP and TDT are seen as two different tests for genes linked to the trait, possessing complementary strengths and weaknesses. In particular, the ASP is expected to be the more powerful test when the causative polymorphism is linked to, but not in LD with, the nearest marker, whereas the TDT would have greater power if some amount of LD were present. Other factors, such as penetrance and allele frequencies in causative polymorphisms and markers, may also lead to differences in power between the two tests [19]. Previous studies have shown that the two methods may give different results. For example, the TDT detected a very strong association between diabetes mellitus and a marker in the 5' region of the insulin gene [20, 21], although this marker had earlier been shown to have no association in an ASP-based test for linkage [22, 23]. In our study several genomic regions are highly significant for the ASP test but not for TDT (Table 1). As the ASP test can detect linkage over long distances, these results may be explained by markers being linked to, but not in close LD with, the causative polymorphisms. The negative TDT results should therefore not be interpreted as a negation of the findings from the ASP test, but rather as indications that the markers are not located in sufficient proximity to the causative polymorphisms. In fact, with the marker densities used in this study, ASP-positive but TDT-negative markers are expected. However, one marker that was strongly positive for the TDT test (SW1355 on SSC6, p < 0.0002), was negative for the ASP test. This may be explained by the fact that in contrast to the ASP, the TDT method can detect association with high power when homozygotes for a susceptibility allele have as little as 2 to 4-fold greater disease risk than homozygotes for the normal or wild-type allele, while parameters such as penetrance and allele frequencies may also favour the TDT rather than the ASP test [19].
The causative mutations we are seeking do not occur in isolation, but with a number of different mutations within the region. Linkage disequilibrium (LD) deteriorates over successive generation because of recombination, but the mutation will remain in LD with closely linked markers. It is therefore, possible to define a risk modifying haplotype in a region of the genome surrounding the disease mutation. Once multiple single-point associations within a region have been identified, haplotype based tests for disease associations may be performed. In this investigation, haplotypes consisting of marker alleles in the putative QTL regions were investigated using TDT analysis. Several haplotypes occurred with a higher frequency in the affected pigs compared to the unaffected pigs, indicating that these haplotypes are associated with the occurrence of inguinal and/or scrotal hernias in pigs.
While most porcine genes are still not mapped, the determination of homologous synteny blocks between human and porcine chromosomes allows us to propose some candidate genes for inguinal and scrotal hernias using comparative genomics [24–27]. The comparative regions and possible candidate genes within these regions are shown in Table 4. As mentioned previously, inguinal and scrotal hernias may arise from a failure to achieve complete closure of the processus vaginalis, and a failure of involution at the internal inguinal ring [3]. In addition, the genitofemoral nerve, the caudal ligament gubernaculum, and controllers of testicular descent may control subsequent closure of the processus vaginalis. Finally, regulation of collagen metabolism has been shown to play an important role in the development of inguinal and scrotal hernias [28, 29]. Using this information as a guide it is possible to anticipate that certain candidate genes may be located in some of the putative QTL regions. Three interesting candidate genes (INSL3, CGRP, MIS) have been mapped to homologous regions in the putative QTL on SSC2 [30–32]. From studies using transgenic knockout mice [33] and humans [34], INSL3 is believed to be the primary testicular hormone inducing gubernacular development, and has been associated with occurrences of inguinal hernia in female mice [7]. No significant associations were, however, observed between two polymorphisms in the INSL3 gene and inguinal hernia in pigs [35]. The CGRP is active in the induction of processus vaginalis fusion, a process involving substantial tissue remodelling and the characteristic transformation of processus vaginalis epithelium [6, 12]. Finally, MIS has been shown to be involved in the swelling reaction of gubernaculum occurring during the first phase of testicular descent [12, 36]. Moreover, MIS together with testosterone and INSL3, controls sex differentiation-like development of internal and external reproductive organs and the acquisition of male secondary sex characteristics [37]. Additional candidate genes were found in putative QTL regions using the network browser option in PubGene [38]. Estrogen receptor 1 (ESR1) and collagen typeIXα (COL9A1) are both candidate genes located to human (HAS) 6q25 region, which is comparative to the putative QTL on SSC1. Coveney et al. [39] demonstrated that estrogen has profound effects on development of the internal genitalia in male marsupials, preventing inguinal closure and interfering with testicular descent, and ESR1 knockout mice lack gubernaculum development in late stages of maturity [40]. Pathological changes in collagen are also involved in development of a hernia [41]. It is therefore interesting to notice that in cartilage, type IX collagen (COL9A1) molecules are covalently crosslinked to type II collagen molecules, which represents approximately 85% of the collagen contained in hyaline cartilage 3 [42]. Interestingly, the gene (COL2A1) coding for collagen type II has been mapped to a region in HSA12 which is comparative to the putative QTL region on SSC5. Candidate genes were also seen in the QTL regions on SSC6 (INSL5) and SSC7 (CYP19A1). No obvious candidate genes were seen in the genomic regions of SSC7q24-26, SSC15 and SSC17 or in the comparative genomic regions in human. The current comparative maps, however, do not allow precise prediction of the likely map position.
To our knowledge, this is the first report of a whole genome scan for markers associated with inguinal and scrotal hernias, and one of the first genetic studies using ASP in livestock. The results reported in this study will encourage us to perform follow-up studies using finemapping approaches in an extended genetic material. Our goal is to discover the causal mutations underlying inguinal and/or scrotal hernias, and to look for shared ancestral haplotypes among pigs with these types of hernias. Ultimately we wish to understand the epistatic interactions between inguinal/scrotal hernia genes, and the interactions between genes and their environment, and their impact on a variety of relevant cofactors associated with this defect. We believe the data contained in the present study represents an important and useful basis to further search for candidate genes causing inguinal and scrotal hernias in pigs. The outcome from these inquiries may also be of relevance to human hernia conditions.
Conclusion
For the first time in any species, a genome scan has revealed suggestive QTLs for inguinal and scrotal hernias. Significant QTLs were detected in 8 out of 19 porcine chromosomes (p < 0.01). However, the most promising QTLs were found on SSC1, SSC2, SSC5, SSC6, SSC15, SSC17 and SSCX. These regions were found to be significant when using both statistical methods (ASP and TDT), or with a convincing high significance for either of the methods. Haplotypes from the suggestive QTL regions were constructed and used in TDT analysis. Six different haplotypes were found to be differently transmitted when contrasting hernia pigs with healthy pigs (p < 0.01). One haplotype on SSC5 was transmitted four times more frequently to hernia pigs than to healthy pigs (p < 0.00005). Only chromosomal regions could be detected in this study, but it is noteworthy that several promising candidate genes, including INSL3, MIS, and CGRP, are all located in a highly significant QTL region. Further studies must be performed to narrow down the suggestive QTL regions, investigate the candidate genes, and finally to confirm the suggestive QTLs in other populations.
Methods
Animals and phenotypic records
Due to a generally low incidence of hernia inguinalis or hernia scrotalis, it was necessary to utilise a collection system involving all the breeding farms in Norsvin (Hamar, Norway) in order to access a sufficient number of affected sib pairs (ASP). The power calculations and minimum number of affected sib pairs required for this study was evaluated based on a method suggested by Simianer and Stricker [43]. Blood samples were collected from a total of 100 full- and half-sib Norwegian Landrace piglets displaying inguinal and/or scrotal hernia, along with blood from 51 phenotypically unaffected full- and half-sibs. To supplement these samples, DNA from 27 fullsib Danish Landrace piglets affected with inguinal and/or scrotal hernias, and 16 unaffected siblings, was provided from Danish Slaughterhouses (Denmark). In total there were 194 samples, distributed on 52 litters containing 103 affected sib pairs, with each litter containing 2 to 5 affected piglets. Two affected siblings from one litters gives 1 ASP, three affected siblings from one litters gives 3 ASP, and so on. In addition to affected animals and 1 or 2 unaffected siblings, we obtained blood specimens from all available parents (31 sires and 46 dams). In several cases these were parent to more than one affected litter. Samples were obtained from 35 pig breeding farms altogether, and several sires were used on different farms.
When a farmer reported an affected animal, diagnostic procedures were performed by a breeding consultant from Norsvin. Without clinical examination, it is impossible to distinguish direct hernia from indirect hernia, or inguinal hernia from scrotal hernia. No clinical examinations were executed, so the diagnosis of hernia includes both inguinal and scrotal hernias, as well as both indirect and direct hernias. In human and horses, inguinal and scrotal hernias are almost exclusively of the indirect type [44], therefore we expect most of the hernias to be of the indirect type in pigs also.
Genotyping
Genomic DNA was extracted from a total of 282 pigs (parents and offspring). 130 microsatellite markers from the autosomal chromosomes and 7 microsatellites from the X/Y chromosome were selected from the USMARC Genome Database (2000), based on position, ease of scoring and number of alleles [45]. The markers were amplified using PCR; the reaction volume was 10 μl, containing 50 ng swine genomic DNA template, 10 × PCR buffer, 15 mM MgCl2, 2 mM dNTP, 0.01 μm of each primer and 5U Gold Taq polymerase (Applied Biosystems, Foster City, CA, USA). The PCR program included an initial denaturation step of 10 sec at 95°C, followed by 40 cycles of 30 sec at 94°C, and 30 sec at 55°C-62°C, with a final extension step of 5 min at 73°C. Fragment lengths were determined either by 1) electrophoresing PCR product through a 6% denaturating polyacrylamide gel in an ABI-377 DNA sequencer (ABI, Perkin Elmer, Foster City, CA, USA), where each forward primer was 5'-labeled with one of three fluorophores (TET, HEX or 6-FAM), or 2) with capillary electrophoresis on an ABI 3730 DNA sequencer (ABI, Perkin Elmer, Foster City, CA, USA), each forward primer 5'-labeled with one of four fluorophores (NED, PET, VIC or 6-FAM). Genescan and Genotyper version 3.7 software or GeneMapper 3.0 (ABI, Perkin Elmer, Foster City, CA, USA) was used for genotype scoring. A computer program was written in house to control for Mendelian inconsistencies in the marker data.
Statistical analysis
Marker map
The family material used in the study was not suitable for constructing genetic maps. Therefore the order of the 137 microsatellites chosen for the genome scan, and the genetic distances between them, were taken from the USMARC Genome Database (2000). The markers were evenly distributed across the 18 autosomal chromosomes and the X-chromosome, including one marker in the pseudoautosomal region. The total length of the map spanned by the marker set was 2335 cM. Average marker heterozygosity was 0.57 and the mean intermarker spacing was 17 cM. The locations of markers are shown in Figure 1 and 2.
Allele frequencies
A non-parametric linkage analysis using the software ALLEGRO 1.0 [46], requires allele frequencies for markers if one parent is unknown and the transmission of alleles are not unambiguously traceable from the marker genotypes of the other parent and offspring. In our animal material some of the dams and a few sires were missing. Marker allele frequencies in the population were estimated from our data set using a computer program made for this purpose.
Linkage analysis
Two different test statistics were applied for linkage analysis using the software package ALLEGRO 1.0 [46]. The first statistic was numbers of shared alleles (NSA), i.e. the number of times that two affected full-sibs displayed identical alleles at the two flanking markers. Under the null hypothesis of no correlation between marker genotypes and defect status, two affected full-sibs would be expected to share half their alleles. Under the alternative hypothesis of a linked gene affecting defect prevalence, the proportion of alleles being identical by descent (and accordingly, identical by state) would be expected to be higher. The second test statistic considers how many of the shared alleles of an affected sib-pair are identical by the descent (IBD). Multipoint linkage analysis of the genome scan data was performed with the non-parametric affected relative method of ALLEGRO 1.0 [46], which also provides multipoint NPL scores and the corresponding p-values [47].
Transmission/Disequilibrium test
Transmission/Disequilibrium Tests [21] were carried out according to Lazzeroni and Lange [18]. At every locus, the TDT statistic was calculated as
where and are the number of times allele j has been passed on from an heterozygous parent to an affected or unaffected offspring, respectively, and are the number of times allele j has not been passed on from an heterozygous parent (carrying allele j) to an affected or unaffected offspring, respectively, and l is the number of alleles at the locus. The TDT test thus took both affected and non-affected siblings into account. A permutation test was carried out according to Lazzeroni and Lange [18], to correct for multiple loci and multiple alleles per locus. For each iteration of the permutation procedure, a permuted data set was made by sampling, for each parent-offspring pair, a haplotype inherited by the offspring from that parent. The two possible, and equally likely, haplotypes were i) the haplotype inherited in the real data, and ii) the haplotype not inherited in the real data. In cases where inheritance from parents to offspring could not be determined in the real data set, because offspring and both parents were heterozygous and identical, linkage phase in the offspring was randomly assigned. TDT statistics were then calculated as above, using the permuted data set. Following the last of m permutations, TDT statistics t i for all loci i (from the real data set and all permutations) were converted to p-values using the empirical estimate;
where a ki = 1 if T ki ≥ t i and a ki = 0 otherwise (meaning that p i (t i ) is the rate at which the permuted test statistic was larger than the true test statistic). Finally, adjusted p-values (accounting for multiple loci) for the real data were calculated as:
where b ki = 1 if p i (t i ) ≥ min m , with min m being the smallest p-value obtained across loci for permutation m. A Visual Basic program was written to perform this analysis, since, to our knowledge, the permutation option and the option of using both affected and unaffected offspring is not available in any publicly available programs.
In addition to the locus-level TDT described above, TDT was also performed on individual alleles, at single loci or haplotypes. The TDT statistic (T) was calculated using equation 1, and nominal significance levels were used, T being distributed approximately as χ2 with one degree of freedom. For haplotype TDT, only those haplotypes falling in the significant marker regions from the TDT or ASP tests were considered. Haplotypes were retrieved from ALLEGRO output.
Information content
The information content (Info) supplied by ALLEGRO is displayed in Figure 1 and 2. The information content ranges from 0 to 1 (with 1 reflecting complete knowledge of inheritance) and provides a measure of to what extend the information present in the pedigrees could be extracted from the available genotypes, compared to a fully informative situation. It therefore allows an overview of which chromosomes were sufficiently covered by markers and in which chromosomal regions additional markers might increase the power of the QTL study considerably.
Abbreviations
- ASP:
-
affected sib pair
- LD:
-
linkage disequilibrium
- NPL:
-
non-parametric linkage score
- TDT:
-
transmission/disequilibrium test
- NSA:
-
numbers of shared alleles
- info:
-
information content
- INSL3:
-
Insulin like receptor 3
- MIS:
-
Müllerian inhibiting substance
- CGRP:
-
Calcitonin gene-related peptide
- ESR1:
-
estrogen receptor 1
- COL9A1:
-
collagen typeIXα
- COL2A1:
-
collagen type II
- INSL5:
-
Insulin-like hormone 5
- CYP19A1:
-
cytochrome P450 family19A1
- MAS:
-
master assisted selection.
References
Thaller G, Dempfle L, Hoeschele I: Maximum likelihood analysis of rare binary traits under different modes of inheritance. Genetics. 1996, 143: 1819-29.
Clarnette TD, Lam SKL, Hutson JM: Ventriculo-peritoneal shunts in children reveal the natural history of closure of the processus vaginalis. J Pediatr Surg. 1998, 33: 413-416. 10.1016/S0022-3468(98)90080-X.
Clarnette TD, Hutson JM: Is the ascending testis actually 'stationary'? Normal elongation of the spermatic cord is prevented by a fibrous remnant of the processus vaginalis. Pediatr Surg Int. 1997, 12: 155-157.
Perrott CA: Inguinal Hernias: Room for a Better Understanding. Diagnostics. 2003, 48-50.
Tanyel FC: Obliteration of processus vaginalis: aberrations in the regulatory mechanism result in an inguinal hernia, hydrocele or undescended testis. Turk J Pediatr. 2004, 18-27. Suppl 46
Cook BJ, Hasthorpe S, Hutson JM: Fusion of Childhood Inguinal Hernia Induced by HGF and CGRP Via an Epithelial Transition. J Pediatr Surg. 2000, 35: 77-81.
Koskimies P, Suvanto M, Nokkala E, Huhtaniemi IT, McLuskey A, Themmen APN, Poutanen M: Female mice carrying a ubiquitin promoter-Insl3 transgene have descended ovaries and inguinal hernias but normal fertility. Molecul Cell Endocrin. 2003, 206: 159-166. 10.1016/S0303-7207(02)00425-2.
Althoff H, Hofstadter F, Oehmichen W: "Heart myxoma syndrome" a hereditary disease? (in German). Pathologe. 1988, 9: 204-10.
Mikami H, Fredeen HT: A genetic study of cryptorchidism and scrotal hernia in pigs. Can J Genet Cytol. 1979, 21: 9-19.
Kubota Y, Temelcos C, Bathgate RAD, Smith KJ, Scott D, Zhao C, Hutson JM: The role of insulin 3, testosterone, Müllerian inhibiting substance and relaxin in rat gubernacular growth. Mol Human Reprod. 2002, 8: 900-905. 10.1093/molehr/8.10.900.
Clarnette TD, Rowe DR, Hasthorpe S, Hutson JM: Incomplete disappearance of the processus vaginalis as a cause of ascending testis. J Urol. 1997, 157: 1889-91. 10.1016/S0022-5347(01)64894-8.
Hutson JM, Albano FR, Paxton G, Sugita Y, Connor R, Clarnette TD, Gray AZ, Watts LM, Farmer PJ, Hasthorpe S: In vitro fusion of human inguinal hernia with associated epithelial transformation. Cells tissues organs. 2000, 166: 249-258. 10.1159/000016738.
Xu J, Dimitrov L, Chang BL, Adams TS, Turner AR, Meyers DA, Eeles RA, Easton DF, Foulkes WD, Simard J, Giles GG, Hopper JL, Mahle L, Moller P, Bishop T, Evans C, Edwards S, Meitz J, Bullock S, Hope Q, Hsieh CL, Halpern J, Balise RN, Oakley-Girvan I, Whittemore AS, Ewing CM, Gielzak M, Isaacs SD, Walsh PC, Wiley KE, Isaacs WB, Thibodeau SN, McDonnell SK, Cunningham JM, Zarfas KE, Hebbring S, Schaid DJ, Friedrichsen DM, Deutsch K, Kolb S, Badzioch M, Jarvik GP, Janer M, Hood L, Ostrander EA, Stanford JL, Lange EM, Beebe-Dimmer JL, Mohai CE, Cooney KA, Ikonen T, Baffoe-Bonnie A, Fredriksson H, Matikainen MP, Tammela TLj, Bailey-Wilson J, Schleutker J, Maier C, Herkommer K, Hoegel JJ, Vogel W, Paiss T, Wiklund F, Emanuelsson M, Stenman E, Jonsson BA, Gronberg H, Camp NJ, Farnham J, Cannon-Albright LA, Seminara D: A combined genomewide linkage scan of 1,233 families for prostate cancer-susceptibility genes conducted by the international consortium for prostate cancer genetics. Am J Hum Genet. 2005, 77: 219-29. 10.1086/432377.
Clerget-Darpoux F, Bonaiti-Pellie C, Hochez J: Effects of misspecifying genetic parameters in lod score analysis. Biometrics. 1986, 42: 393-9.
Spielman RS, Ewans WJ: The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet. 1996, 59: 983-9.
Wiedemann S, Fries R, Thaller G: Geneome-wide scan for Anal Atresia in swine identifies linkage and association with a chromosome region on SSC 1. Genetics. 2005, 171: 1207-17. 10.1534/genetics.104.032805.
Laitinen T, Daly MJ, Rioux JD, Kauppi P, Laprise C, Petays T, Green T, Cargill M, Haahtela T, Lander ES, Laitinen LA, Hudson TJ, Kere J: A susceptibility locus for asthma-related traits on chromosome 7 revealed by genome-wide scan in a founder population. Nat Genet. 2001, 28: 87-91. 10.1038/88319.
Lazzeroni LC, Lange K: A conditional inference framework for the Transmission/Disequilibrium Test. Human Heredity. 1998, 48: 67-81. 10.1159/000022784.
McGinnis RE: Hidden linkage: a comparison of the affected sib pair (ASP) test and transmission/disequilibrium test (TDT). Ann Hum Genet. 1998, 62: 159-79. 10.1046/j.1469-1809.1998.6220159.x.
McGinnis RE, Spielman RS, Ewens WJ: Linkage between the insulin gene (IG) region and susceptibility to insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet. 1991, A476-Suppl 49
Spielman RS, McGinnis RE, Ewens WJ: Transmission test for linkage disequilibrium: The insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet. 1993, 52: 506-516.
Spielman RS, Baur MP, Clerget-Darpoux F: Genetic analysis of IDDM: summary of GAW5 IDDM results. Genet Epidemiol. 1989, 6: 43-58. 10.1002/gepi.1370060111.
Cox NJ, Spielman RS: The insulin gene and susceptibility to IDDM. Genet Epidemiol. 1989, 6: 65-69. 10.1002/gepi.1370060113.
Goureau A, Yerle M, Schmitz A, Riquet J, Milan D, Pinton P, Frelat G, Gellin J: Human and porcine correspondence of chromosome segments using bidirectional chromosome painting. Genomics. 1996, 36: 252-62. 10.1006/geno.1996.0460.
Goureau A, Vignoles M, Pinton P, Gellin J, Yerle M: Improvement of comparative map between porcine chromosomes 1 and 7 and human chromosomes 6, 14, and 15 by using human YACs. Mamm Genome. 2000, 11: 796-9. 10.1007/s003350010158.
Yerle M, Lahbib-Mansais Y, Goureau A, Tosser-Klopp G, Pinton P, Sun HS, Tuggle CK, Gellin J: Contribution to the comparative map between humans and pigs. Anim Biotechnol. 1999, 10: 93-7.
Sun HS, Tuggle CK, Goureau A, Fitzsimmons CJ, Pinton P, Chardon P, Yerle M: Precise mapping of breakpoints in conserved synteny between human chromosome 1 and pig chromosomes 4, 6 and 9. Anim Genet. 2002, 33: 91-6. 10.1046/j.1365-2052.2002.00819.x.
Rosch R, Klinge U, Si Z, Junge K, Klosterhalfen B, Schumpelick V: A role for the collagen I/III and MMP-1/13 genes in primary inguinal hernia?. BMC Medical Genetics. 2002, 3: 2-10.1186/1471-2350-3-2.
Rodrigues Junior AJ, Rodrigues CJ, da Cunha AC, Yoo J: Quantitative analysis of collagen and elastic fibers in the transversalis fascia in direct and indirect inguinal hernia. Rev Hosp Clin Fac Med Sao Paulo. 2002, 57: 265-70.
Rettenberger C, Burkhardt E, Adham IM, Engel W, Fries R, Klett C, Hameister H: Assignment of the Leydig insulin-like hormone to porcine chromosome 2q12-q13 by somatic cell hybrid analysis and fluorescence in situ hybridization. Mamm Genome. 1994, 5: 307-9. 10.1007/BF00389546.
Lahbib-Mansais Y, Barbosa A, Yerle M, Parma P, Milan D, Pailhoux E, Gellin J, Cotinot C: Mapping in pig of genes involved in sexual differentiation: AMH, WT1, FTZF1, SOX2, SOX9, AHC, and placental and embryonic CYP19. Cytogenet Cell Genet. 1997, 76: 109-14.
Knorr C, Kollers S, Fries R, Brenig B: Assignment of the CALC-A/alpha-CGRP gene (CALCA) to porcine chromosome SSC2p13-->p11 by fluorescence in situ hybridization and by analysis of somatic cell and radiation hybrid panels. Cytogenet Genome Res. 2002, 97: 140-10.1159/000064050.
Nef S, Parada LF: Cryptorchidism in mice mutant for Insl3. Nat Genet. 1999, 22: 295-9. 10.1038/10364.
Zimmermann S, Steding G, Emmen JM, Brinkmann AO, Nayernia K, Holstein AF, Engel W, Adham IM: Targeted disruption of the Insl3 gene causes bilateral cryptorchidism. Mol Endocrinol. 1999, 13: 681-91. 10.1210/me.13.5.681.
Knorr C, Täubert H, Peters U, Brenig B: Characterization of two SNPs (Single-Nucleotide Polymorphisms) in the Porcine INSL3 Gene and their exclusion as a common genetic basis of Hernia Inguinalis in Pigs. Biochem Gen. 2004, 42: 11-19. 10.1023/B:BIGI.0000012140.41292.a9.
Hutson JM, Davidson PM, Reece LA, Baker ML, Zhou B: Failure of gubernacular development in the persistent Müllerian duct syndrome allows herniation of the testes. Pediatr Surg Int. 1994, 9: 544-546.
Viger RS, Silversides DW, Tremblay JJ: New insights into the regulation of mammalian sex determination and male sex differentiation. Vitam Horm. 2005, 70: 387-413. 10.1016/S0083-6729(05)70013-3.
Website titel. [http://www.pubgene.uio.no/]
Coveney D, Shaw G, Renfree MB: Effects of oestrogen treatment on testicular descent, inguinal closure and prostatic development in a male marsupial, Macropus eugenii. Reproduction. 2002, 124: 73-83. 10.1530/rep.0.1240073.
Barlett JE, Washburn T, Eddy EM, Korach KS, Temelcos C, Hutson JM: Early development of the gubernaculum and cremaster sac in estrogen receptor knockout mice. Urol Res. 2001, 29: 163-167. 10.1007/s002400100180.
Bendavid R: The unified theory of hernia formation. Hernia. 2004, 8: 171-6.
Eyre DR, Apon S, Wu J-J, Ericsson LH, Walsh KA: Collagen type IX: evidence for covalent linkages to type II collagen in cartilage. FEBS Lett. 1987, 220: 337-341. 10.1016/0014-5793(87)80842-6.
Simianer H, Stricker C: Mapping of Qualitative Trait Loci. Genomics and Biotechnology in Livestock Breeding. Edited by: Schellander K, Li N, Neeteson AM, Wimmers K. 2002, Aachen: Shaker Verlag, 47-58. ISBN 3-8322-0246-3
Graf JL, Caty MG, Martin DJ, Glick PL: Pediatric hernias. Semin Ultrasound CT MR. 2002, 23: 197-200. 10.1016/S0887-2171(02)90006-5.
Website title. [http://www.ncbi.nlm.nih.gov/mapview/]
Gudbjartsson DF, Jonasson K, Frigge M, Kong A: Allegro, a new computer program for multipoint linkage analysis. Nature. 2000, 25: 12-13.
Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES: Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996, 58: 1347-63.
Lopez-Corrales NL, Beattie CW, Rohrer GA: Cytogenetic assignment of 53 microsatellites from the USDA-MARC porcine genetic map. Cytogenet Cell Genet. 1999, 84: 140-4. 10.1159/000015241.
Robic A, Riquet J, Yerle M, Milan D, Lahbib-Mansais Y, Dubut-Fontana C, Gellin J: Porcine linkage and cytogenetic maps integrated by regional mapping of 100 microsatellites on somatic cell hybrid panel. Mamm Genome. 1996, 7: 438-45. 10.1007/s003359900129.
Yerle M, Lahbib-Mansais Y, Pinton A, Goureau A, Milan D, Gellin J: The cytogenetic map of the domestic pig. Mamm Genome. 1997, 8: 592-607. 10.1007/s003359900512.
Acknowledgements
We want to thank Emma Stubsjøen in Norsvin for a valuable contribution in organising the sample collection, and the technicians in Norsvin for taking blood samples. Thanks also to Arne Roseth and Brita Dalsgard for technical assistance in the lab. We are also very grateful to Heine Kristensen and Danish Slaughterhouses for providing us blood samples from hernia pigs in Denmark, to Olav Eik-Nes and Håvard Tajet for their contribution in initiating this project, and to Matthew Kent for valuable comments to the paper. NORSVIN (the Pig Breeders Association in Norway), the Research Council of Norway and the Swedish "Stiftelsen Lantbruksforskning" provided financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
EG coordinated the study, organised and carried out molecular genetic work, performed the statistical analysis of QTL mapping and drafted the paper. MM carried out molecular genetic work. HT was involved in the statistical analysis. HS was involved in planning the project, power calculations, supervising the statistical analysis and writing the paper. SL was involved in planning the project and writing the paper. TM performed the TDT statistical analysis and contributed in writing the paper.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Grindflek, E., Moe, M., Taubert, H. et al. Genome-wide linkage analysis of inguinal hernia in pigs using affected sib pairs. BMC Genet 7, 25 (2006). https://doi.org/10.1186/1471-2156-7-25
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-7-25