
Abstract
Background
Phytoplankton diversity can be difficult to ascertain from morphological analyses, because of the existence of cryptic species and pico- and concealed phytoplankton. In-depth sequencing and metabarcoding can reveal microbial diversity, and identify novel diversity. However, there has been little comparison of metabarcoding and morphological datasets derived from the same samples, and metabarcoding studies covering total eukaryotic phytoplankton diversity are rare. In this study, the variable V7 region of the 18S rDNA gene was employed to explore eukaryotic phytoplankton diversity in 11 Chinese freshwater environments, and further compared with the dataset obtained through morphological identification.
Results
Annotation by the evolutionary placement algorithm (EPA) rather than alignment with the SILVA database improved the taxonomic resolution, with 346 of 524 phytoplankton operational taxonomic units (OTUs) being assigned to the genus or species level. The number of unassigned OTUs was greatly reduced from 259 to 178 OTUs by using the EPA in place of the SILVA database. Metabarcoding detected 3.5 times more OTUs than the number of morphospecies revealed by morphological identification; furthermore, the number of species and the Shannon–Wiener index inferred from the two methods were correlated. A total of 34 genera were identified via both methods, while 31 and 123 genera were detected solely in the morphological or metabarcoding dataset, respectively.
Conclusion
The dbRDA plot showed distinct separation of the phytoplankton communities between lakes and reservoirs according to the metabarcoding dataset. The same pattern was obtained on the basis of 10 environmental variables in the PCO ordination plot, while the separation of the populations based on morphological data was poor. However, 30 morphospecies contributed 70% of the community difference between lakes and reservoirs in the morphological dataset, while 11 morphospecies were not found by metabarcoding. Considering the limitations of each of the two methods, their combination could substantially improve phytoplankton community assessment.

Similar content being viewed by others


Background
Phytoplankton have traditionally been described and characterized based on morphological characteristics. However, their correct identification is often difficult or impossible, especially for cryptic species complexes, species with distinct sexual dimorphism, or species with multiple life stages [1, 2]. Photosynthetic pico-phytoplankton with a diameter below 3 μm diameter are usually ignored, although they are very important contributors to primary production due to exhibiting high cell-specific carbon fixation rates [3,4,5]. Trained experts often come to inconsistent conclusions concerning detailed species lists, which could be explained by differences in their experience and understanding of changing taxonomic system, and the different criteria used for species delimitation. Consequently, phytoplankton community composition based ecological assessment, and the suggested relationships between different taxa based on morphological characteristics will be misleading.
Molecular approaches can detect cryptic species, and have been treated as one of the most important approaches for the taxonomic revision of phytoplankton [6]. Many new and revised species, genera and taxonomic ranks higher than genera have been proposed every year [7]. Molecular techniques link a unique DNA sequence to a phytoplankton taxon based on sequence similarity to reference databases regardless of the dimensions, life stage, pleomorphism, or taxonomy of the phytoplankton [8]. Next-generation sequencing (NGS) technologies produce a large sequence dataset consisting of molecular markers, making them a promising tool for understanding microbial diversity in ecosystems. NGS allows automated sample handling and involves standard laboratory protocols, which increases the potential for comparisons between different research studies and facilitates large-scale in-depth monitoring programs and investigations [8].
NGS has been extensively used to investigate phytoplankton diversity [9], but has been limited largely to specific phytoplankton groups, including diatoms [8, 10] and dinoflagellates [11]. NGS reveals a greater number of identified diatom taxa than morphological analysis, and their presence can be subsequently verified by light microscopy [8]. Eiler et al. [12] investigated total phytoplankton diversity based on the 16S ribosomal DNA (rDNA) gene and compared it with the results of morphological identification. Unsatisfactorily, the detailed taxonomic lists presented low correspondence. A relatively low ratio of the number of phytoplankton reads and OTUs was observed, and the in-depth sequencing results were far from saturated.
There have been few studies targeting the full range of phytoplankton diversity via NGS methods, and comparisons of the morphologically and NGS-determined community composition have rarely been performed. In this study, high-throughput amplicon sequencing based on the 18S rDNA gene was performed to investigate total eukaryotic phytoplankton diversity, and the results were compared with datasets based on morphological identification. Furthermore, the variation of the phytoplankton composition explained by environmental variables was evaluated to discuss the compatibility and potential application of the metabarcoding approach.
Materials and methods
Sampling and microscopic observation
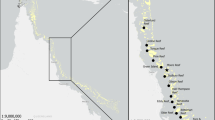
A total of 62 samples from 6 lakes and 5 reservoirs were surveyed during spring (Fig. 1, Additional file 1: Table S3). Water was collected 5 cm below the surface with a 5-L water sampler. The water (1.5 L) was preserved with 1.5% Lugol’s iodine solution [13], and after 48 h of sedimentation, a final 50 mL volume was concentrated for microscopic cell identification and counting. At least 50 random fields of view per sample were scanned using a 400× microscope (Eclipse 50i, Nikon, Japan). Morphological identification followed the descriptions of Hu and Wei [14]. The classification system proposed by Ruggiero et al. [15] was adopted (Additional file 1: Table S1). Aliquots of 100–200 mL of water, depending on algal density, were filtered through 0.2-μm nucleopore filters (Whatman, United Kingdom) for NGS analysis. The filters were stored at − 20 °C until being processed for NGS sequencing. An aliquot of 500 mL water was used for the assessment of environmental variables.

Locations of 6 lakes and 5 reservoirs investigated in the study. Numbers in brackets represented number of sampling sites in each freshwater
TN (total nitrogen), TP (total phosphorus), ammonia and nitrate nitrogen were measured following Chinese standard methods [16]. Briefly, for TN determination, the sample was digested with alkaline potassium persulfate, and all available nitrogen was converted to nitrate, followed by scanning at wavelengths of 220 nm and 275 nm. Nitrate was directly tested at these wavelengths. For ammonia, the reaction of ammonia and ions with the sodium reagent produced a yellow–brown complex, which was then scanned at a wavelength of 420 nm. For TP, the sample was digested with potassium persulfate under neutral conditions, and all available phosphorus was converted to orthophosphate. Molybdophosphate was produced in the presence of ammonium molybdate and reduced by ascorbic acid to a blue complex, which was then scanned at a wavelength of 700 nm.
T (water temperature), DO (dissolved oxygen), conductivity and pH were obtained with a multiparameter meter (YSI 6820, Yellow Spring Instruments, USA). Transparency (Secchi depth, SD) was measured with a 20-cm diameter black and white Secchi disk.
DNA extraction and library construction
Genomic DNA extraction was performed according to the manual instructions of the PowerSoil®DNA Isolation Kit (MO BIO Laboratories, USA). The concentration and purity of the extracted genomic DNA was assessed with a NanoDrop spectrophotometer (ND-1000, NanoDrop Technologies, USA). The amplicon library was prepared through polymerase chain reaction (PCR) amplification of the V7 region of the 18S rDNA gene using the primers 960F (5′-GGCTTAATTTGACTCAACRCG-3′) [17] and NSR1438 (5′-GGGCATCACAGACCTGTTAT-3′) [18] combined with adapter sequences and barcode sequences. The primers were designed to balance primer usage frequencies and could reduce the frequency of cross-contamination [19]. Moreover, primers were chosen for a high proportion of the phytoplankton (over 40%) reads among total the eukaryotes obtained in Lake Bourget [20]. Two-step tailed PCR was performed in this study [21]. Each amplification reaction was performed in a 50-μL volume, which contained 25 μL 2 × Phusion® High-Fidelity PCR Master Mix with HF Buffer (New England Biolabs Inc., America) consisting of Phusion DNA Polymerase, deoxynucleotides and MgCl2, 10 μL High GC Enhancer, 1 μL of each primer at 10 μM, 11 μL ddH2O, and 2 μL template, with the following conditions: 95 °C for 3 min, then 15 cycles of 95 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min, and a final extension at 72 °C for 10 min. Negative controls showed no amplicons. The 260-bp-long PCR products were purified with the same volume of VAHTS™ DNA Clean Beads. A second round of PCR was then performed in a 40-μL reaction containing 20 μL of 2 × Phusion® High-Fidelity PCR Master Mix with HF Buffer, 8 μL ddH2O, 1 μL of each primer at 10 μM, and 10 μL of the PCR products from the first step. The thermal cycling conditions were as follows: initial denaturation at 98 °C for 30 s, followed by 10 cycles at 98 °C for 10 s, 55 °C for 30 s and 72 °C for 30 s, with a final extension at 72 °C for 5 min. The second round PCR products were purified with E.Z.N.A.® Gel Extraction Kit (OMEGA, America), and quantified using the Quant-iT™ dsDNA High Sensitivity Assay Kit with microplate reader. Finally, 150 ng PCR products were pooled for library construction.
Metabarcoding sequencing and analysis
Qualified amplicon libraries were sequenced on the Illumina HiSeq 2500 platform (2 × 250 paired ends) at Biomarker Technologies, China. The original paired-end reads were merged using FLASH v1.2.7 (http://ccb.jhu.edu/software/FLASH/) with a minimum overlap length of 10 bp and maximum mismatch ratio of 0.2 within overlapping regions. The raw reads were quality filtered to obtain high-quality clean reads using Trimmomatic v0.33 [22]. The window size was set as 50 bp with minimum average quality scores of 20. Chimera sequences were detected and removed using UCHIME v4.2 [23]. The final processed reads were clustered into OTUs based on 97% sequence similarity with USEARCH v10.0 [24]. OTUs comprising less than 0.005% of total reads were filtered from the dataset. The sequences with the greatest number of reads were selected as the representatives of each OTU and then compared against the SILVA database (Release 128, http://www.arb-silva.de, [25]) with a confidence threshold of 80% to identify OTUs belonging to phytoplankton with the RDP taxonomy assigner (v2.2) implemented in QIIME v1.8.0 [26]. OTUs annotated as fungi, protozoa, unassigned or other groups of eukaryotes were removed from further analysis.
The phytoplankton OTUs identified using SILVA were further delimited with the evolutionary placement algorithm (EPA) to improve the taxonomic resolution [27] because half of these OTUs were assigned to taxonomic ranks higher than the genus level. For this purpose, first, the reference phylogenetic tree was computed with published reference sequences longer than 1000 bp selected from the GenBank database for EPA assignments. Detailed information on the published reference sequences for each phylum/class is shown in Additional file 1: Table S2. The sequences for the reference tree were aligned using online MAFFT v7.429 with the autoselect algorithm depending on the size of reference sequences (https://mafft.cbrc.jp/alignment/server/), and trimmed using trimAI v2.0 [28], the resulting alignment was called reference alignment. The tree was computed with RAxML-HPC v8.0 in XSEDE at the CIPRES Science Gateway (http://www.phylo.org/portal2/). The reference tree was calculated using the GTRGAMMA model, and RaxML was allowed to halt bootstrapping automatically [29]. Second, every representative read obtained from the phytoplankton OTUs in this study was aligned to the reference alignment with MAFFT v7.429 online (https://mafft.cbrc.jp/alignment/server/add.html), and the alignment length was kept the same as the reference alignment. Then, EPA was performed to assign our phytoplankton OTUs onto the reference phylogenetic trees [27, 30]. The resulting tree containing the reference sequences and our phytoplankton OTUs was visualized with Archaeopteryx Treeviewer v0.970 [31]. The phytoplankton OTUs were manually delimited based on the branch where they were affiliated in the phylogenetic backbone tree and the classification likelihood weights supporting the branch. When the branch contained a mix of taxa from 2 or more genera, they were delimited at family or higher taxonomic ranks and were recognized as unassigned reads. The EPA analyses were performed individually for every phylum/class, and 9 reference phylogenetic trees were constructed for Dictyochophyceae, Bacillariophyceae, Dinophyceae, Chrysophyceae, Cryptophyceae, Eustigmatophyceae, Chlorophyta/charophyte, Euglenales, and Xanthophyceae, while the remaining 5 classes with only 11 phytoplankton OTUs were included in a single phylogenetic tree.
Finally, 524 phytoplankton OTUs were used to calculate alpha diversity metrics including rarefaction curves, OTU diversity, the Chao1 index, the Simpson index, and the Shannon–Wiener index with Mothur v1.30 [32].
Statistical analysis
The morphological and metabarcoding datasets of phytoplankton abundance were standardized prior to analysis, followed by fourth root or square root transformation, respectively. Environmental variables other than DO, T, and pH were log(x + 1) transformed. All transformed datasets met the requirements for homogeneity and normality of variance inferred from the histogram plots. Randomization/permutation procedure analysis of similarities (ANOSIM) was carried out to evaluate significant differences in the community composition between groups. The ANOSIM statistic R is calculated from the differences in the between-group and within-group mean rank similarities, and can reveal the degree of separation between groups. R values approaching 1 indicate complete separation, while R values close to 0 indicate no separation. The similarity percentage (SIMPER) routine was used to discriminate dominant species within or between groups. Principal coordinate analysis (PCO) was performed to investigate environmental variables correlated with lakes or reservoirs. Distance-based linear models (DISTLMs) provided quantitative measures and tests of phytoplankton community variation according to environmental variable, and were visualized in distance-based redundancy analysis (dbRDA) ordination plots. Marginal and sequential tests were performed to quantify the contribution of 10 environmental variables, both individually and together, to the variation in the phytoplankton community. The dbRDA was constrained to identify the linear combinations of the predictor variables that explained the greatest amount of variation in the data cloud. The environmental variables that presented correlations > 0.4 or < − 0.4 with the dbRDA axis are shown in the dbRDA plot. All of the abovementioned analyses were performed with PRIMER v7 [33].
Results
Environmental variables
The average pH was higher in all examined reservoirs (9.08–11.59) except for the Changtan Reservoir (7.15) than in the lakes (8.34–9). The average T and DO values showed no difference between the lakes and reservoirs within the ranges of 19.7 to 23.3 °C and 7.2 to 13.6 mg/L, respectively, with the exception of a relatively lower T of 9.9 °C in the Changtan Reservoir. The average conductivity exhibited obviously higher values in lakes (245–539 μS/cm) than in reservoirs (37–95 μS/cm). Reservoirs (1.0–2.6 m) showed higher average transparency than lakes (0.2–0.7 m) except for Lake Gucheng (1.7 m). In lakes, TP (3.36–13.45 μmol/L) values were much higher than those in reservoirs (0.52–3 μmol/L). TN presented little difference and exhibited overlapping values between lakes (40.71–191.43 μmol/L) and reservoirs (35.86–104.93 μmol/L). Much higher ammonia nitrogen values were observed in lakes (13.86–51.29 μmol/L) than in reservoirs (6–14.14 μmol/L), except for Foziling Reservoir (31.29 μmol/L). Apart from the Changtan Reservoir (35.5 μmol/L), the other reservoirs (1.86–2.5 μmol/L) displayed lower average nitrate nitrogen values than the lakes (2.29–63.64 μmol/L). The reservoirs presented much higher TN and TP ratios (7.4–51.2) than the lakes, which showed low values (2.6–8.2) (Additional file 1: Fig. S1).
The first two axes of the PCO explained 57.3% of the total variation in 10 environmental variables, showing that the differences were clearly distinguishable between 6 lakes and 5 reservoirs in the ordination plot. TN, ammonia nitrogen, TP, T and conductivity presented strong positive relationships with PCO axis1 that were indicative of the samples from lakes, while SD and TN and TP ratio had negative relationships with PCO axis1 which were indicative of samples in reservoirs. DO and pH exhibited strong positive relationships with PCO axis2, which separated the samples from the Foziling Reservoir and the Bailianya Reservoir from the samples from the other 3 reservoirs (Fig. 2).

Vector overlay on the PCO of the environmental variables datasets, showing environmental variables with vectors longer than 0.5
Morphological identification
A total of 150 taxa (132 at the species level) from 8 eukaryotic phytoplankton groups were identified from 62 samples, with Chlorophyta (69) and Bacillariophyceae (55) contributing 87% of the total phytoplankton diversity. In each sample, 1 to 38 morphospecies were detected (Fig. 3). Four species exhibited high frequencies at 11 freshwaters sites, including 1 from phylum Chlorophyta (Chlorella vulgaris), 2 from class Bacillariophyceae (Synedra acus, Cyclotella) and 2 from class Cryptophyceae (Cryptomons ovata, Cryptomons erosa), all of which were detected in over 40 samples. The total phytoplankton density ranged from 1.16 × 105 to 8.2 × 107 cells/L in each sample. Chlorophyta and Bacillariophyceae dominated phytoplankton abundance, together accounting for 43.2% to 100% (avg. 87.0%) of the observed abundance, except in Lake Yangcheng sample 01, where euglenoids contributed 79.0% of the total abundance (Fig. 3).

The species diversity (a) and abundance (b) of 8 eukaryotic phytoplankton groups identified in morphological method. Number on the X axis indicates the number of samples in each freshwater
Metabarcoding to determine phytoplankton diversity and novel OTUs
A total of 40,526 to 74,292 reads (avg. 71,421) with average lengths ranging from 223 to 238 base pairs were obtained from each sample (Additional file 1: Table S3). Finally, 524 eukaryotic phytoplankton OTUs from 15 phytoplankton groups were determined with the RDP classifier against the SILVA database and EPA, with the number of phytoplankton reads ranging from 4892 (6.6%) to 59,550 (79.6%) (avg. 34,962 [48.7%]). The number of phytoplankton reads surpassed 12,000 in all but two samples, from Lake Taihu and the Mozitan Reservoir. The rarefaction curves of the majority of samples approached the plateau curve (Additional file 1: Fig. S2), and good coverage (≥ 0.992) of OTU richness was observed for each sample (Additional file 1: Table S3), indicating that the number of phytoplankton reads approached saturation in most samples.
Using the RDP classifier against the SILVA database, 265 OTUs were identified at the genus and species levels, and a large number of OTUs (up to 259 (49.4%) OTUs with 875,271 (40.4%) reads) were annotated to taxonomic ranks higher than the generic level and were identified as unassigned OTUs. EPA identified 346 OTUs at the genus and species levels, and 178 (34%) unassigned OTUs with 686,352 (31.7%) reads were also identified (Table 1). The EPA improved the resolution of the assignment of OTUs.
Each sample contained 56 to 278 phytoplankton OTUs (avg. 178), and the most diversity was observed in Lake Changdang, Lake Gucheng, Lake Baima, and Lake Yangcheng, all of which presented OTU numbers surpassing 182 (Fig. 4). Chlorophyta was the most diverse, with 222 OTUs (42.3%), followed by Chrysophyceae (87, 16.6%), Bacillariophyceae (55, 10.5%), Cryptophyceae (48, 9.1%), Dinophyceae (44, 8.4%), Dictyochophyceae (24, 4.6%), and Eustigmatophyceae (17, 3.2%). The other 9 phytoplankton groups presented low diversity, including Euglenales, Xanthophyceae, Haptophyta, Raphidophyceae, Bolidophyceae, Charophyta, Schizocladiophyceae, Leucocryptea, and Picophagophyceae, all of which showed fewer than 10 OTUs and contributed 5.3% of the total phytoplankton diversity. Chlorophyta (avg. 44.7%), Bacillariophyceae (avg. 18.8%), and Cryptophyceae (avg. 18.4%) dominated or codominated the abundance values in all samples with the exception of those from the Changtan Reservoir, where Chlorophyta (avg. 41.6%) and Dinophyceae (avg. 41.7%) exhibited the highest abundance.

The species diversity (a) and proportion of abundance (b) of 15 eukaryotic phytoplankton groups determined by metabarcoding approach. Number on the X axis indicated the number of samples in each freshwater
Comparison of the phytoplankton community identified with the two methods
A significantly (p < 0.001) higher number of phytoplankton OTUs (56–278) was found compared to the number of morphospecies (1–38) (Additional file 1: Fig. S3), and these measures were positively correlated (R2 = 0.13, P = 0.003) (Fig. 5a). The community composition determined by metabarcoding exhibited higher Shannon–Wiener index values (ave. 2.66) than that obtained through morphological identification (ave. 1.78), and the Shannon–Wiener index values determined by these two methods were positively correlated (R2 = 0.349, P < 0.001) (Fig. 5b, c).

Phytoplankton diversity compared between metabarcoding and morphological methods. Linear regression analysis of the number of species (a); Shannon–Wiener index (b) and linear regression analysis (c). The calculation of Shannon–Wiener index was based on cell numbers in morphological dataset and number of reads in metabarcoding dataset
The phytoplankton communities characterized by morphological and metabarcoding methods were compared at the generic level due to morphological and molecular complexity. The comparison was performed for 8 phytoplankton groups identified according to the morphological data. These 8 groups were Chlorophyta, Chrysophyceae, Cryptophyceae, Bacillariophyceae, Dinophyceae, Euglenales, Xanthophyceae, and Charophyta. The two methods detected 34 genera in common, while 31 and 123 genera were specific to the morphological and metabarcoding datasets, respectively (Fig. 6). The genera identified by morphological methods in Chrysophyceae, Cryptophyceae, and Dinophyceae were all found in the metabarcoding dataset, while 31 genera from the other 5 phytoplankton groups were not fully detected by metabarcoding (Fig. 6). The detailed list and information on the genera obtained via both methods and those only identified in the morphological or metabarcoding datasets are presented in Additional file 1: Tables S4, S5 and S6, respectively. The morphological and metabarcoding datasets contained 27 and 54 genera with more than 2 morphospecies or OTUs, respectively (Additional file 1: Tables S4–S6).

Genera co-detected or solely found with metabarcoding and morphological determination
As inferred via ANOSIM, the phytoplankton communities in the lakes and reservoirs were clearly separated from each other in the metabarcoding dataset (global R = 0.421, P = 0.001), while their separation according to the morphological data was much poorer (global R = 0.185, P = 0.001). A total of 8 and 6 species, from 3 phytoplankton groups in both cases, contributed 70% of the abundance in lakes and reservoirs, respectively, in the morphological dataset (Fig. 7; Additional file 1: Table S7), while 58 and 42 OTUs from 8 and 9 phytoplankton groups contributed 70% of the abundance in lakes and reservoirs according to the metabarcoding dataset (Fig. 7; Additional file 1: Table S8). A total of 30 morphospecies and 117 OTUs contributed 70% of the difference between phytoplankton that allowed discrimination between lakes and reservoirs in the respective morphological (Fig. 7; Additional file 1: Table S9) and metabarcoding datasets (Fig. 7; Additional file 1: Table S10).

Taxa and species contributing 70% accumulation of phytoplankton abundance in lakes and reservoirs (a); species contributing 70% community difference of phytoplankton abundance between lakes and reservoirs (b). Note: numbers on the bars represented the number of morphospecies or OTUs in the morphological or metabarcoding datasets
Phytoplankton community explained by environmental variables
The sequential tests of the 10 environmental variables explained a total of 36.4% and 44.1% of the variation in the phytoplankton composition according to the morphological and metabarcoding methods, respectively (Additional file 1: Table S11). The first two dbRDA axes captured 50.8% of the variability in the fitted model, and 18.5% of the total phytoplankton variation in the morphological dataset, showing incomplete separation of the samples between lakes and reservoirs except for the samples from the Changtan Reservoir, which exhibited much greater similarity of the phytoplankton composition to the samples from lakes. Conductivity presented a strong negative relationship with dbRDA axis1, which was indicative of the samples from lakes, whereas pH exhibited a strong positive relationship with dbRDA axis1, which was indicative of the samples from reservoirs. The community in lakes exhibited much dissimilarity, TN, and nitrate presented strong positive relationships with dbRDA axis2 and separated the samples from Lake Tai from those of the other 5 lakes (Fig. 8).

Distance-based redundancy analysis (dbRDA) ordination plot for the fitted model of phytoplankton community in morphological and metabarcoding datasets versus environmental variables of correlations greater than 0.4 were shown on the plot
The first two dbRDA axes captured 60.1% of the variability in the fitted model and 26.5% of the total phytoplankton variation in the metabarcoding dataset, showing clear separation of the samples from lakes and reservoirs. The conductivity exhibited a strong positive relationship with dbRDA axis1 which was indicative of samples in lakes, while SD had strong negative relationship with dbRDA axis1, which was indicative of the samples in reservoirs. The community in reservoirs exhibited much dissimilarity, pH, and TN and TP ratio exhibited strong positive relationships with dbRDA axis2 and separated the samples from the Changtan Reservoirs from those of the other 4 reservoirs. The communities in the lakes exhibited less dissimilarity and T presented a strong positive relationship with dbRDA axis2, which separated the samples from Lake Gucheng from those of the other 5 lakes (Fig. 8).
Discussion
NGS facilitates the investigation of environmental microbial diversity by producing a large dataset of molecular marker sequences for target microorganisms. Metabarcoding has been employed to investigate eukaryotic phytoplankton diversity [8,9,10,11]; however, research involving metabarcoding efforts to reveal phytoplankton communities and their correspondence with morphological identification results has rarely been reported. In this study, total eukaryotic phytoplankton diversity was explored via the metabarcoding of 18S rDNA gene amplicons, and the results were further compared with those of morphological identification.
Sequencing efforts and applicability of the metabarcoding method
Half of the reads were assigned to eukaryotic phytoplankton in our samples. The number of phytoplankton reads varied widely among the samples, ranging from 4892 (6.6%) to 59,550 (80.0%) (Additional file 1: Table S3). Although the sequencing efforts were poor in 2 samples, the percentages of phytoplankton reads retrieved from 60 samples were high (avg. 34,962, 48.7%), and the majority of samples approached the plateau curve (Additional file 1: Fig. S2). Moreover, good coverage (≥ 0.992) of OTU richness was observed for each sample (Additional file 1: Table S3), indicating that the number of phytoplankton reads was approaching saturation in most samples and that the performance of the 18S rDNA primers selected for revealing phytoplankton diversity in the environment was good.
Comparatively, the obtained ratio and the sequencing effort achieved for phytoplankton far surpassed those in Ref. [12], with a 60-fold increase in phytoplankton coverage being observed for our samples (avg. 34,962). In Ref. [12], in which the authors attempted to identify total phytoplankton diversity, including that of cyanobacteria, using 16S rDNA plastid gene amplicons, 120 out of 259 samples produced fewer than 100 phytoplankton reads, and the average number of phytoplankton reads analyzed in the remaining 139 samples was 596. We suggest that the chloroplast 16S rDNA gene used in Ref. [12] might not be an appropriate choice for detecting eukaryotic phytoplankton diversity. First, there is a bias toward bacteria when common primers targeting this gene are used in an attempt to cover a wide spectrum of taxa, thus reducing the sequencing efforts aimed at the phytoplankton diversity. Second, eukaryotic phytoplankton acquire their chloroplasts via endosymbiosis; endosymbiont origins are diverse; and endosymbionts are permanently or temporarily retained in host cells. Diatom, cryptophyte, and haptophyte algae have been reported to serve as endosymbiont chloroplasts in diverse dinoflagellate species [34]. Therefore, the chloroplast 16S rDNA gene might not truly reflect host phytoplankton diversity.
EPA improves taxon resolution
The reference reads were assigned a place in the reference backbone phylogenetic tree and could be delimited corresponding to the branch on which they were found. On the branch, we could check and revise the taxonomic names of the published reference sequences to guarantee that accurate assignments were obtained for a large number of misidentified algal cultures, and their sequences were deposited in a public database. Novel phytoplankton diversity estimates may vary with the approach and the gene database selected for the taxonomic assignment of OTUs. Novel phytoplankton diversity was largely reduced, and up to 66% of OTUs were assigned to the genus and species levels using the EPA instead of the SILVA database. Although the SILVA database contains highly quality controlled 18S rDNA gene sequences [35], it may not include enough representatives at low taxonomic ranks [36]. The curated SILVA database is composed of numerous environmental samples, and these sequences are predicted rather than representing the authoritative taxonomy [37]. Many of these sequences are annotated as unnamed or uncultured species [38], and the assignment of metabarcoding data via the SILVA database might result in many OTUs that are unresolved at the genus/species level in many studies. A highly quality controlled database incorporating sequences from diverse phytoplankton groups is needed to investigate phytoplankton diversity with metabarcoding data.
Metabarcoding detects diverse phytoplankton
Due to its high sequencing depth, metabarcoding can detect rare species [39], cryptic species [6], pico-phytoplankton [40], and uncultured eukaryotic phytoplankton [4]. Metabarcoding detected 3.5 times more phytoplankton OTUs than morphospecies in our 62 samples considering that each OTU represented one species at 97% sequence similarity. Metabarcoding and morphological data detected 15 and 8 phytoplankton groups, respectively, and the rare Haptophyta, Raphidophyceae, Bolidophyceae, Leucocryptea, and Picophagophyceae were not detected in the latter (Figs. 3 and 4), leading to an underestimation of phytoplankton diversity in the environment according to morphological identification.
These two methods differed greatly in the number of phytoplankton groups detected and the estimated abundance of phytoplankton groups. This difference could be ascribed to the fact that in-depth sequencing based metabarcoding retrieved higher number of sequences compared with counting according to morphological methods, and that rare species and pico-phytoplankton are easily ignored [41]. Second, molecular methods may reveal concealed phytoplankton diversity when morphologically identical taxa exhibit distinct genetic variation [42, 43]. However, the phytoplankton diversity identified under these two methods is highly correlated, including the number of species identified and the Shannon–Wiener index (Fig. 5).
A total of 34 genera were detected by both methods, and 31 and 123 genera were detected solely in the morphological and metabarcoding datasets, respectively (Fig. 6; Additional file 1: Tables S4–S6). Genera from 5 phytoplankton groups identified by morphological evaluation were detected almost in the metabarcoding dataset, while within Chlorophyta and Bacillariophyta, 19 and 8 genera, respectively, were not found in the metabarcoding dataset (Fig. 6). The identification of genera by morphological methods that are not found via metabarcoding techniques could arise from primer bias [44] or morphological misidentification due to limited diacritical characteristics [7]. The metabarcoding dataset was assigned by the EPA but not the Protist Ribosomal Reference database (PR2) because many more OTUs (66.0% vs 22.1%) were identified at the genus and species levels. Moreover, the genera identified by morphology that were missing from the metabarcoding dataset with our selected GenBank database were not resolved by analysis at the PR2 database (data not shown).
Metabarcoding community pattern consistent with environmental variables
As inferred from the dbRDA ordination plot, the phytoplankton communities identified in the samples from 6 lakes and 5 reservoirs were clearly separated from each other in the metabarcoding dataset, while the separation in the morphological dataset was indistinct for the community composition in the Changtan Reservoir, which showed much greater similarity with samples from lakes (Fig. 8). This was further corroborated by ANOSIM, which showed that the phytoplankton communities of lakes and reservoirs were much more distinctly separated from each other according to the metabarcoding dataset (global R = 0.421, P = 0.001) than that in morphological dataset (global R = 0.185, P = 0.001).
There were many dissimilarities in the phytoplankton communities among the samples from the 5 reservoirs according to the metabarcoding dataset, and the same pattern could be seen in the PCO ordination plot of the 10 environmental variables, suggesting that the phytoplankton community is reflected in environmental changes. In the morphological dataset, many dissimilarities were observed in the samples from different lakes, according to which Lake Taihu was separated from the other 5 lakes. The results following the elucidation of hidden diversity (including rare species, pico-phytoplankton, and concealed phytoplankton) revealed by metabarcoding approached the size of the actual phytoplankton community, and the obtained community composition could reflect changes in environmental variables.
The first dbRDA axis separated the samples from lakes and reservoirs from each other according to both methods, although the separation was much weaker for the morphological dataset. Conductivity and SD were strongly correlated with lakes and reservoirs, respectively, according to the metabarcoding dataset, while for the morphological dataset, conductivity and pH were strongly correlated with lakes and reservoirs, respectively, along the first dbRDA axis (Fig. 8). Additionally, in the PCO plot inferred from environmental variables, conductivity and SD strongly were correlated with lakes and reservoirs along the first PCO axis, while pH was correlated with the second PCO axis, which separated the Foziling Reservoir and the Bailianya Reservoir from the other 3 reservoirs. These results showed a consistent pattern in which the same separation pattern of lakes and reservoirs was inferred from the environmental variables and the phytoplankton communities revealed by the metabarcoding dataset, as well as the community variation explained by the environmental variables.
The combination of the two methods revealed the actual phytoplankton community
The metabarcoding technique is a promising tool for revealing phytoplankton communities, which can reflect and be clearly separated by the changes in environmental variables in different freshwater environments as shown in Ref. [12] and by our study (Figs. 2 and 8). However, 31 genera from Chlorophyta, Bacillariophyceae, Euglenida, Dinophyceae, and Xanthophyceae that were identified in the morphological dataset were not detected under the metabarcoding method in our study (Fig. 6, Additional file 1: Table S5). The 1 to 77 reference sequences were obtained for 25 genera that were solely detected in the morphological dataset (Additional file 1: Table S5). This showed the gaps existed for only 6 genera in the reference database, while reference sequences were present in 25 genera. Thus, we demonstrated that bias could be introduced in the metabarcoding processes by DNA extraction and PCR procedures [45, 46] or primer bias [44], which might result in the absence of these genera in our metabarcoding dataset. Moreover, 30 species contributed 70% of the differences between the communities of lakes and reservoirs according to the morphological dataset, while 11 morphospecies were not identified by the metabarcoding method (Fig. 7, Additional file 1: Table S9), showing that this method was flawed in revealing the actual phytoplankton community. The morphological and metabarcoding approaches were complementary, and combining these two methods could substantially improve phytoplankton community assessment.
Conclusion and perspective
The metabarcoding technique was shown to be more valuable for assessing pico-phytoplankton and novel phytoplankton diversity than morphological identification due to the in-depth sequencing that is achieved. It is a promising tool for revealing the phytoplankton community, which can reflect and be clearly separated by the differences in environmental variables between lakes and reservoirs, but their separation in the morphological dataset was poor. Moreover, the number of OTUs (species) and the Shannon–Wiener index were much higher for the metabarcoding dataset than the morphological dataset and were strongly correlated under each method. However, biases could be introduced during the metabarcoding processes by the DNA extraction and PCR procedures or primer bias, as 31 genera identified in the morphological dataset were not detected by the metabarcoding method. Drawbacks exist for both of these methods. In the future, morphological and metabarcoding methods should be combined to reveal the phytoplankton community in the environment.
Availability of data and materials
The original metabarcoding datasets from 62 samples have been deposited at GenBank and are publicly available under the project name “PRJNA506128” with the accession number SRP169841.
Abbreviations
- EPA:
-
Evolutionary placement algorithm
- NGS:
-
Next-generation sequencing
- TN:
-
Total nitrogen
- TP:
-
Total phosphorus
- T:
-
Water temperature
- DO:
-
Dissolved oxygen
- SD:
-
Secchi depth
- ANOSIM:
-
Randomization/permutation procedure for the analysis of similarities
- SIMPER:
-
The similarity percentage
- PCO:
-
The principal coordinate analysis
- DISTLM:
-
The distance-based linear models
- dbRDA:
-
The distance-based redundancy analysis
References
Hebert PD, Penton EH, Burns JM, Janzen DH, Hallwachs W (2004) Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc Natl Acad Sci USA 101(41):14812–14817
Mahon AR, Barnes MA, Senapati S, Feder JL, Darling JA, Chang H, Lodge DM (2011) Molecular detection of invasive species in heterogeneous mixtures using a microfluidic carbon nanotube platform. PLoS ONE 6:e17280
Li WKW (1994) Primary productivity of prochlorophytes, cyanobacteria, and eucaryotic ultraphytoplankton: measurements from flow cytometric sorting. Limnol Oceanogr 39:169–175
Cuvelier ML, Allen AE, Monier A, McCrow JP, Messié M, Tringe SG et al (2010) Targeted metagenomics and ecology of globally important uncultured eukaryotic phytoplankton. Proc Natl Acad Sci USA 107(33):14679–14684
Jardillier L, Zubkov MV, Pearman J, Scanlan DJ (2010) Significant CO2 fixation by small prymnesiophytes in the subtropical and tropical northeast Atlantic Ocean. ISME J 4:1180–1192
Kermarrec L, Bouchez A, Rimet F, Humbert JF (2013) First evidence of the existence of semi-cryptic species and of a phylogeographic structure in the Gomphonema parvulum (Kützing) Kützing complex (Bacillariophyta). Protist 164(5):686–705
Hoffmann L, Komárek J, Kaštovský J (2005) System of cyanoprokaryotes (cyanobacteria)-state in 2004. Arch Hydrobiol Suppl Algol Stud 117:95–115
Zimmermann J, Glöckner G, Jahn R, Enke N, Gemeinholzer B (2015) Metabarcoding vs morphological identification to assess diatom diversity in environmental studies. Mol Ecol Resour 15(3):526–554
De Vargas C, Audic S, Henry N, Decelle J, Mahé F, Logares R et al (2015) Eukaryotic plankton diversity in the sunlit ocean. Science 348(6237):1261605
Mortágua A, Vasselon V, Oliveira R, Elias C, Chardon C, Bouchez A et al (2019) Applicability of DNA metabarcoding approach in the bioassessment of Portuguese rivers using diatoms. Ecol Indic 106:105470
Lin S, Zhang H, Hou Y, Zhuang Y, Miranda L (2009) High-level diversity of dinoflagellates in the natural environment revealed by assessment of mitochondrial cox1 and cob genes for dinoflagellate DNA barcoding. Appl Environ Microb 75(5):1279–1290
Eiler A, Drakare S, Bertilsson S, Pernthaler J, Peura S, Rofner C, Simek K, Yang Y, Znachor P, Lindström ES (2013) Unveiling distribution patterns of freshwater phytoplankton by a next generation sequencing based approach. PLoS ONE 8(1):e53516
Zhou GJ, Zhao XM, Bi YH, Hu ZY (2011) Effects of silver carp (Hypophthalmichthys molitrix) on spring phytoplankton community structure of Three-Gorges Reservoir (China): results from an enclosure experiment. J Limnol 70(1):26–32
Hu HJ, Wei YX (2006) The freshwater algae of China: systematics, taxonomy and ecology. Science Press, China (in Chinese)
Ruggiero MA, Gordon DP, Orrell TM, Bailly N, Bourgoin T, Brusca RC, Cavalier-Smith T, Guiry MD, Kirk PM (2015) A higher level classification of all living organisms. PLoS ONE 10(4):e0119248
China Environmental Protection Administration (2006) Water and wastewater monitoring and analysis methods, 4th edn. China Environmental Science Press, Beijing
Gast RJ, Dennett MR, Caron DA (2004) Characterization of Protistan assemblages in the Ross Sea, Antarctica, by denaturing gradient gel electrophoresis. Appl Environ Microb 70:2028–2037
Van de Peer Y, De RP, Wuyts J, Winkelmans T, De Wachter R (2000) The European small subunit ribosomal RNA database. Nucleic Acids Res 28:175–176
Esling P, Lejzerowicz F, Pawlowski J (2015) Accurate multiplexing and filtering for high-throughput amplicon sequencing. Nucleic Acids Res 43:2513–2524
Capo E, Debroas D, Arnaud F, Perga ME, Chardon C, Domaizon I (2017) Tracking a century of changes in microbial eukaryotic diversity in lakes driven by nutrient enrichment and climate warming. Environ Microbiol 19(7):2873–2892
Miya M, Sato Y, Fukunaga T, Sado T, Poulsen JY, Sato K, Minamoto T, Yamamoto S, Yamanaka H, Araki H, Kondoh M, Iwasaki W (2015) MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: detection of more than 230 subtropical marine species. Roy Soc Open Sci 2(7):150088
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10(10):996–998
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO (2012) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microb 73:5261–5267
Berger SA, Krompass D, Stamatakis A (2011) Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum likelihood. Syst Biol 60(3):291–302
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25(15):1972–1973
Stamatakis A (2015) Using RAxML to infer phylogenies. Curr Protoc Bioinform 51(1):6–14
Berger SA, Stamatakis A (2011) Aligning short reads to reference alignments and trees. Bioinformatics 27(15):2068–2075
Han MV, Zmasek CM (2009) phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinform 10(1):356
Schloss PD, Westcot SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb 75:7537–7541
Clarke KR, Gorley RN (2015) PRIMERv7: user manual/tutorial. PRIMER-E, Plymouth
Kim E, Archibald JM (2010) Plastid evolution: gene transfer and the maintenance of ‘stolen’ organelles. BMC Biol 8:73
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196
Federhen S (2011) The NCBI taxonomy database. Nucleic Acids Res 40:136–143
Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, Schweer T, Peplies J, Ludwig W, Glöckner FO (2014) The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res 42(D1):D643–D648
Yarzaet P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer KH, Whitman WB, Euzéby J, Amann R, Rosselló-Móra R (2014) Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12(9):35
Zhan A, Hulák M, Sylvester F, Huang X, Adebayo AA, Abbott CL, Adamowicz SJ, Heath DD, Cristescu ME, MacIsaac HJ (2013) High sensitivity of 454 pyrosequencing for detection of rare species in aquatic communities. Methods Ecol Evol 4(6):558–565
Moon-van der Staay SY, De Wachter R, Vaulot D (2001) Oceanic 18S rDNA sequences from picoplankton reveal unsuspected eukaryotic diversity. Nature 409(6820):607
Stoeck T, Behnke A, Christen R, Amaral-Zettler L, Rodriguez-Mora MJ, Chistoserdov A, Orsi W, Edgcomb VP (2009) Massively parallel tag sequencing reveals the complexity of anaerobic marine protistan communities. BMC Biol 7:72
Trobajo R, Mann DG, Clavero E, Evans KM, Vanormelingen P, McGregor RC (2010) The use of partial cox1, rbcL and LSU rDNA sequences for phylogenetics and species identification within the Nitzschia palea species complex (Bacillariophyceae). Eur J Phycol 45:413–425
Abarca N, Enke N, Zimmermann J, Jahn R (2014) Does the cosmopolitan diatom Gomphonema parvulum (Kützing) Kützing have a biogeography? PLoS ONE 9:e86885
Tedersoo L, Anslan S, Bahram M, Põlme S, Riit T, Liiv I, Kõljalg U, Kisand V, Nilsson H, Hildebrand F, Bork P, Abarenkov K (2015) Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 10(1):1–43
Martin-Laurent F, Philippot L, Hallet S, Chaussod R, Germon JC, Soulas G, Crtroux G (2001) DNA extraction from soils: old bias for new microbial diversity analysis methods. Appl Environ Microb 67:2354–2359
Acinas SG, Sarma-Rupavtarm R, Klepac-Ceraj V, Polz MF (2005) PCR- induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl Environ Microb 71:8966–8969
Acknowledgements
This work was supported by the National Key Research and Development program of China [No. 2017YFA0605003] and the National Natural Science Foundation of China [Nos. 51922010, 41521003] and the National Science Foundation for Young Scientists of China (No. 31700404).
Author information
Authors and Affiliations
Contributions
XL analyzed the data and wrote the paper. SH and BX revised the paper. HZ, CM and ZH collected the samples and analyzed the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Additional figures and tables.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huo, S., Li, X., Xi, B. et al. Combining morphological and metabarcoding approaches reveals the freshwater eukaryotic phytoplankton community. Environ Sci Eur 32, 37 (2020). https://doi.org/10.1186/s12302-020-00321-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12302-020-00321-w