
Abstract
We reviewed the current methodology and practices of the DNA metabarcoding approach using a universal PCR primer pair MiFish, which co-amplifies a short fragment of fish DNA (approx. 170 bp from the mitochondrial 12S rRNA gene) across a wide variety of taxa. This method has mostly been applied to biodiversity monitoring using environmental DNA (eDNA) shed from fish and, coupled with next-generation sequencing technologies, has enabled massively parallel sequencing of several hundred eDNA samples simultaneously. Since the publication of its technical outline in 2015, this method has been widely used in various aquatic environments in and around the six continents, and MiFish primers have demonstrably outperformed other competing primers. Here, we outline the technical progress in this method over the last 5 years and highlight some case studies on marine, freshwater, and estuarine fish communities. Additionally, we discuss various applications of MiFish metabarcoding to non-fish organisms, single-species detection systems, quantitative biodiversity monitoring, and bulk DNA samples other than eDNA. By recognizing the MiFish eDNA metabarcoding strengths and limitations, we argue that this method is useful for ecosystem conservation strategies and the sustainable use of fishery resources in “ecosystem-based fishery management” through continuous biodiversity monitoring at multiple sites.
Similar content being viewed by others
Introduction
In the past three decades biodiversity monitoring has been widely recognized as essential for ecosystem conservation and sustainable use of biological resources (Common and Norton 1994; Lepetz et al. 2009; Goodwin et al. 2018). Indeed, fisheries derive enormous benefits directly or indirectly from aquatic ecosystems, and those benefits are collectively referred to as ecosystem services (Costanza et al. 1997; Rapport et al. 1998; Barbier et al. 2011; Grizzetti et al. 2016). To maximize the benefits of these ecosystem services, fisheries management had previously focused on maximizing the catch of a single target species, often ignoring habitat, predators, and prey of the target species and other ecosystem components and interactions (Pikitch et al. 2014; Darling et al. 2017; Hansen et al. 2018). However, worldwide fisheries management is currently undergoing a paradigm shift from such a single-species approach to an ecosystem approach (Koslow 2009; Fogarty 2014; Brinson and Wallmo 2015; Long et al. 2017). In response to this trend, a subcommittee of the Science Council of Japan recently published a report advocating the ecosystem approach to fisheries management (https://www.scj.go.jp/ja/info/kohyo/pdf/kohyo-23-t248-2.pdf).
This approach is called “ecosystem-based fishery management,” and its overall objective is to sustain healthy ecosystems and the fisheries they support (Pikitch et al. 2014; Hill et al. 2020; DiBattista et al. 2020). The evaluation of ecosystem health inevitably requires continuous monitoring of biotic and abiotic components in ecosystems because we can only detect, for example, environmental degradation or biodiversity loss when temporal changes in ecosystems occur (Rapport et al. 1998; Halpern et al. 2015; Hoffmann et al. 2016). Such ecosystem changes have been recognized principally through physical attributes (e.g., water temperature and pH), as these attributes are easily measurable with great accuracy, and an international observation network has been constructed for continuous monitoring (Riser et al. 2016). Well-known examples include global warming and ocean acidification (Hoegh-Guldberg et al. 2007; Pandolfi et al. 2011; McCulloch et al. 2012; Six et al. 2013), both of which are causing ecosystem changes (Pörtner 2008; Robison 2009) and, in turn, have a serious impact on human life including fisheries (Sumaila et al. 2011; Quesne and Pinnegar 2012; Cheung et al. 2016; Stiasny et al. 2016).
In contrast, the enormous biodiversity in aquatic ecosystems, which include a myriad of unknown species, cannot automatically be monitored like physical attributes, making continuous biodiversity monitoring difficult (Fujikura et al. 2010; DiBattista et al. 2017; Alexander et al. 2019). Regarding fish, over 32,000 species are known from aquatic environments worldwide, and an average of around 400 new species were described annually between 2005 and 2014 (Nelson et al. 2016). Moreover, biodiversity monitoring of macroorganisms such as fishery resources is laborious, costly, and time consuming, often relying on direct capture of specimens through various kinds of netting, trapping, or fishing (Thomsen et al. 2012; Evans et al. 2017; Oka et al. 2020). These capture-based sampling methods are both invasive and destructive, and subsequent morphological identification of specimens based on taxonomic expertise is also required (Thomsen and Willerslev 2015; Andruszkiewicz et al. 2017b; Sigsgaard et al. 2019), which is increasingly scarce and declining in availability(Hopkins and Freckleton 2002). Additionally, sampling feasibility heavily depends on weather and water conditions. Therefore, it is practically impossible to continuously monitor biodiversity at multiple sites using the traditional approaches.
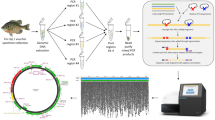
Environmental DNA (eDNA) is defined here as the extraorganismal genetic materials suspended in environmental samples, such as water and sediment. eDNA is shed from macroorganisms through feces, body mucus, blood, and sloughed tissue or scales and has emerged as an alternative data source for biodiversity monitoring (Bohmann et al. 2014; Kelly et al. 2014b; Cristescu 2014; Deiner et al. 2017). By filtering a certain amount of water, eDNA is concentrated and captured on the filter membrane, from which it is extracted and subjected to various molecular biology experiments for detection of organisms (Turner et al. 2015; Deiner et al. 2015; Miya et al. 2016). In particular, the eDNA metabarcoding approach enables simultaneous detection of multiple species using a high-throughput next-generation sequencing (NGS) platform (Taberlet et al. 2012; Thomsen et al. 2012; Miya et al. 2015). This approach co-amplifies a short fragment of eDNA from the target taxa (e.g., fishes) using a set of universal primers through PCR (Fig. 1a) and then appends various adapters and index sequences to both ends of the amplified fragments (amplicons) (Fig. 1b). Various combinations of different index sequences enable massively parallel sequencing using the NGS platform (MiSeq system, Illumina Inc., San Diego, CA, USA), with an output comprising tens of millions to billions of amplicons from multiple sampling sites. After data preprocessing and subsequent taxonomic assignment using a bioinformatics pipeline, a tentative taxonomic list is available for each sampling site (Miya et al. 2015; Sato et al. 2018).

(From Miya et al. 2015 with slight modifications)
Schematic representation of the paired-end library preparation using a two-step tailed PCR for MiSeq sequencing ((MiSeq system, Illumina Inc.). a First-round PCR (1st PCR) to amplify a region of interest using a pair of MiFish primers (blue) with overhang adapter sequences (orange), b second-round PCR to add dual indices (red) and sequencing adapters (green).
One of the most crucial steps to determine the success or failure of eDNA metabarcoding is the first-round PCR (1st PCR; Fig. 1a). The 1st PCR co-amplifies a short fragment of sparse eDNA from across target taxa using a set of universal PCR primers to an amount that can be sequenced by an NGS platform (Miya and Sado 2019a). For successful eDNA metabarcoding, the PCR primer pair and the amplified intervening eDNA fragments should meet the following requirements: (1) the designed universal PCR primers should unbiasedly amplify gene fragments across the target taxa without PCR dropouts, and (2) the amplified fragments should contain sufficient variations to allow unambiguous taxonomic assignment. In addition to these requirements, (3) the primers should be specific to the target taxa (e.g., fishes); otherwise, DNA from non-target taxa (prokaryotic and non‐target eukaryotic DNAs) will predominate in the amplified products. Such predominant amplification of non-specific taxa has been demonstrated to compromise eDNA metabarcoding by wasting sequencing effort (Collins et al. 2019). Among a number of universal primers for the fish eDNA metabarcoding, MiFish primers (Miya et al. 2015) best satisfy the above criteria and have been shown to outperform other competing primers in several ways (Bylemans et al. 2018a; Collins et al. 2019; Zhang et al. 2020). Actually, MiFish primers have been used for biodiversity monitoring of fishes in various aquatic environments in and around six continents worldwide (Figs. 2, 3).

Approximate geographic location of the study sites where MiFish metabarcoding has been conducted using environmental DNA (eDNA) and bulk DNA samples. Multiple studies conducted in the same area are indicated by a single symbol. For details on the references, see text. For details on the study sites in Japan, see Fig. 3

Approximate geographic location of the study sites where MiFish eDNA metabarcoding has been conducted in Japan. Multiple studies conducted in the same area (e.g., Lake Biwa and adjacent areas) are indicated by a single symbol
In addition to these individual case studies plotted on the two maps (Figs. 2, 3), two global initiatives are attempting eDNA metabarcoding using MiFish primers (hereafter called MiFish eDNA metabarcoding). The first initiative, DNAqua-Net, has been supported by the EU COST (Co-Operation in Science and Technology) Action program since 2016, and has set up a network of 39 countries across the EU and beyond to develop the next-generation of aquatic biomonitoring technology (Leese et al. 2016, 2018). One of the DNAqua-Net working groups (W3) is developing field and laboratory protocols for biomonitoring using DNA, and has discussed and tested MiFish eDNA metabarcoding in the December 2018 and December 2019 DNAqua-Net newsletters (https://dnaqua.net/newsletter/; Weigand et al. 2019). Weigand et al. (2019) implicitly stated, “the 12S marker has a high potential to become the gold standard for regular eDNA-based fish monitoring in the future.” To date, the UK and Swedish governmental agencies have commissioned environmental survey projects using eDNA metabarcoding of fishes, and two private environmental research firms have conducted the surveys using MiFish eDNA metabarcoding (Tang et al. 2018; Hellström and Blomstrand 2019; Näslund et al. 2019; Tang 2020). The second initiative involves several universities and research institutes in the California Current region in a joint initiative to consider using MiFish eDNA metabarcoding to supplement traditional capture-based sampling to improve the efficiency of biodiversity monitoring in that region (Kacev et al. 2018). As part of this effort, the MiFish eDNA metabarcoding library preparation protocol has been published online (Pitz et al. 2020), and the California Current specific reference database (FishCARD) has been assembled for more accurate taxonomic assignment in MiFish eDNA metabarcoding (Gold et al. 2020). Thus, these two global initiatives in different geographic regions are expected to use MiFish eDNA metabarcoding in their future studies.
In Japan, several governmental projects have recently employed MiFish eDNA metabarcoding for biodiversity monitoring (Minamoto et al. 2020). One example of such a project is a pilot study called the National Census on River Environments, initiated by the Ministry of Land, Infrastructure, Transport and Tourism of Japan, which uses MiFish eDNA metabarcoding for regular river surveys (Kitagawa et al. 2020). A second example is the promotion of the Biodiversity Center of Japan, Ministry of the Environment to standardize MiFish eDNA metabarcoding for monitoring endangered freshwater fishes (https://www.biodic.go.jp/edna/edna_top.html). Further, the Fishery Agency of Japan, Ministry of Agriculture, Forestry, and Fisheries has recently initiated a new project employing eDNA technologies to monitor fishery resources throughout the Pacific Ocean, with the aim to expand the target species from 50 to 200 in the next 5 years (https://www.maff.go.jp/j/budget/attach/pdf/31kettei-99.pdf). In response to these government-led national trends, several private environmental research firms have also begun to utilize MiFish eDNA metabarcoding (e.g., https://ideacon.jp/technology/inet/vol53/vol53_alls.pdf).
In this review paper we primarily focus on MiFish eDNA metabarcoding and review the (1) performance comparisons with competing PCR primers; (2) taxonomic optimization of the primers; (3) desired laboratory settings and revised experimental protocols (Miya and Sado 2019a) that lead to successful MiFish eDNA metabarcoding without carryover contaminations; (4) new bioinformatics pipeline and reference database; (5) and empirical studies conducted in various aquatic environments, including marine, freshwater, and estuary ecosytems. Additionally, we discuss (6) the developments of new techniques using MiFish primers and applications of the MiFish metabarcoding methods to (7) other organisms and (8) bulk DNA samples.
This review is based on published information from 283 studies and documents citing Miya et al. (2015) as of 22 August 2020 (Google Scholar: https://scholar.google.com), supplemented by additional information from websites and several colleagues around the world.
Performance of MiFish primers
In this section, we briefly describe the background of MiFish primer developments (Miya et al. 2015) and introduce three recent studies on in silico and in vitro comparisons of the primer performance (Bylemans et al. 2018a; Collins et al. 2019; Zhang et al. 2020).
Miya et al. (2015) developed a set of universal PCR primers (MiFish-U/E) for metabarcoding eDNA from fishes. The primers were designed with reference to the whole mitochondrial genome (mitogenome) sequences from 880 fish species (Fig. 4), supplemented by partial mitogenome sequences from 160 elasmobranchs (sharks and rays). Visual inspection of the aligned sequences found two conserved regions across the 880 species in the 12S rRNA gene with an intervening hypervariable region with an average length of 172 (range 163–185) bp, leading the authors to design a new primer pair based on these two conserved regions.

Schematic representation of the aligned nucleotide sequences from a hypervariable region of the mitochondrial 12S rRNA gene (approx. 172 bp) located between the two conserved regions (priming sites of MiFish primers)
During primer design process on the conserved regions, Miya et al. (2015) considered a number of “technical tips” that enhance primer annealing to the template without requiring degenerate bases (Palumbi 1996). First, primers include some G/C at the 3ʹ ends to strengthen primer–template annealing at this position, with the caveat that a string of either Gs or Cs at the 3ʹ end be avoided. Second, taking into consideration the unconventional base pairing in the T/G bond, the designed primers use G rather than A when the template is variably C or T, and T rather than C when the template is A or G. The G/C content of the primers falls between 40 and 60%, with the primers having an almost identical melting temperature. These “technical tips” have proven useful for designing primers for mitochondrial genomics of fishes (Miya and Nishida 2015), and Miya et al. (2015) found that degenerate primers did not amplify the target eDNA when they were used with long adapter sequences in the tailed PCR.
The hypervariable region between the two conservative regions contains sufficient information to identify fishes to taxonomic family, genus, and species, with the exceptionn of some closely related congeners. Miya et al. (2015) performed in silico comparisons of taxonomic discriminatory power between the hypervariable regions amplified by MiFish primers (hereafter also referred to as MiFish sequences) and one of the ecoPrimer pairs (Riaz et al. 2011). The latter primer pair (12SV5) was used in a previous metabarcoding study of fishes (Kelly et al. 2014a), which attempted to estimate an artificial fish fauna using eDNA in the large tank at the Monterey Bay Aquarium. Miya et al. (2015) found that MiFish primers (average length 172 bp) have more variation than the 12SV5 primer pair (average length 106 bp) and outperformed the latter in more unambiguous taxonomic assignment.
To provide an in silico workflow for the evaluation of metabarcoding primers at the ecoregion scale, Bylemans et al. (2018a) used the custom reference database and available software programs to evaluate the performance of eight existing and seven newly developed metabarcoding primers assessing freshwater fish biodiversity of the Murray-Darling Basin, Australia. Of the 15 primer pairs, five passed the initial screening based on consistent high scores in taxonomic discriminatory power, specificity, and taxonomic coverage for each primer pair. Among these, three primer pairs on the mitochondrial 12S rRNA gene (MiFish-U/Teleo/AcMDB07) were used for further eDNA metabarcoding analyses of an artificial DNA community and two eDNA samples from natural rivers. The results were somewhat ambiguous, and each of the three primer pairs was found to have advantages and disadvantages. Consequently, these authors (2018a) were uncertain about which primer could be used to most effectively reproduce fish compositions contained in the three samples. However, in a subsequent empirical study monitoring riverine fish communities from the same water basin, MiFish primers were chosen for eDNA metabarcoding analyses based on the results from an independent in silico primer evaluation of the local fish diversity (Bylemans et al. 2018b), suggesting that overall performance of MiFish primers was better than that of the other two primer pairs.
In a case study of marine and freshwater fishes from the British Isles, Collins et al. (2019) evaluated the in silico performance of 12 PCR primer pairs from four mitochondrial genes, i.e., cytochrome oxidase I (COI), cytochrome b (cyt b), and 12S and 16S rRNAs, regarding reference library coverage, taxonomic discriminatory power, and primer universality. Based on the in silico comparisons, they selected four primer pairs with higher performance, i.e., three COI primer pairs (Leray-XT, SeaDNA-mid, SeaDNA-short) and one 12S primer pair (MiFish-U), which were then used for metabarcoding analyses using eDNA samples from five estuarine and coastal sites around the English Channel and North Sea. Collins et al. (2019) examined the metabarcoding results regarding reproducibility and congruence using independent datasets derived from traditional capture-based methods. MiSeq sequencing and subsequent bioinformatic analyses showed that despite having the least total raw reads, the MiFish-U primers produced the greatest number and proportion of usable fish reads (76% of processed reads) compared to those of the three COI primers (0.1–4.4% of processed reads) (Table 1). Species richness estimates at all sampling sites were consistently the highest with MiFish-U (Fig. 5) despite the deficiencies in the reference library, at only 61% species coverage. Collins et al. (2019) concluded that eDNA metabarcoding for restricted taxonomic groups (e.g., fishes) using degenerate COI primers results in excessive volumes of “wasted” sequencing effort through the co-amplification of prokaryotic and non-target eukaryotic DNAs. Furthermore, higher specificity to fishes in MiFish-U primers was recently demonstrated in an empirical study on Lake Tanganyika’s fish communities (96.9 vs. 32.5–49.4% in the other three primer pairs used), where cichlids exhibit explosive radiation among the 400 fish species recorded in the lake (Doble et al. 2019).

Box plots of fish species richness as estimated by four primer pairs at five sampling locations (four estuaries and one coastal water in UK; Collins et al. 2019). The horizontal bar, boxes, and whiskers represent the median value, 25–75th percentiles, and values < 1.5-fold the interquartile range (IQR), respectively, while dots represent the outlying data points and crosses represent the cumulative number of species. COI Cytochrome oxidase I. The original figure was kindly provided by Rupert A. Collins and Stefano Mariani (Collins et al. 2019)
Zhang et al. (2020) recently compiled a list of 22 primer sets for fish eDNA metabarcoding based on a literature survey, which performed more comprehensive in silico and in vitro comparisons of the primer performance. Those primer sets targeted the mitochondrial 12S rRNA, 16S rRNA, cyt b, and COI genes (7, 6, 7, and 2 primers, respectively), exhibiting considerable differences in the amplified taxonomic ranges and proportions, richness, species discriminatory power, and community compositions (Zhang et al. 2020). For example, the number of fish species detected from eDNA from freshwaters in Beijing, China ranged from 0 to 66, with the 12S primers consistently detecting greater fish diversity than the other primers. Specifically, the top six primer sets that recovered the greatest number of fish taxa were all 12S primers, of which four primers, including MiFish, showed outstanding detection of fish diversity. Zhang et al. (2020) also found that the results from in silico PCR and in vitro tests did not always agree, leading these authors to argue that primer choice for biodiversity monitoring should not be based solely on in silico evaluation. We favor their argument, as in silico evaluation of PCR primers only considers primer/template mismatches and does not accommodate the technical tips described above (Ficetola et al. 2010; Elbrecht and Leese 2017; Taberlet et al. 2018), which enhance primer annealing to the template.
These three case studies used pure freshwater, estuarine, and coastal fish communities, each of which exhibit remarkably different taxonomic compositions across the planet, the equator, and different continents. The consistently higher performance of MiFish primers in these three studies suggests the versatility of this primer pair. However, these studies were conducted in temperate latitudes with a moderate species diversity around 20–60 species (Bylemans et al. 2018a; Collins et al. 2019; Zhang et al. 2020). To validate the versatility of MiFish primers at broader taxonomic and geographic scales, primer performance should be evaluated in different aquatic ecosystems with much higher taxonomic diversity than that in these studies, at lower latitudes, such as tropical rivers (Cilleros et al. 2019; Sales et al. 2019) and coral reefs (Oka et al. 2020), where more than hundreds of species are expected to occur (Jerde et al. 2019).
Taxonomic optimization of the primers
Although the overall performance of MiFish primers is demonstrably higher than that of the competing primers (Bylemans et al. 2018a; Collins et al. 2019; Zhang et al. 2020), there are still several challenges to overcome. The first challenge is “PCR dropouts,” i.e., amplification failures of fish DNAs contained in the eDNA extracts, which inevitably result in false negatives in eDNA metabarcoding. The second challenge is a lack of variations in the MiFish sequences (hypervariable region intervening with MiFish primers) among closely related congeners, which inevitably results in underestimation of species richness. In the following text we briefly discuss these two challenges with some examples from our own research group.
Regarding PCR dropouts, Miya et al. (2015) noted that the original MiFish primers did not amplify eDNAs from a number of elasmobranchs (sharks and rays) reared in an aquarium tank. They suspected that absence of the elasmobranch sequences in the results from MiFish eDNA metabarcoding originated from PCR bias owing to primer–template mismatches. Miya et al. (2015) designed a new primer pair (MiFish-E) to accommodate sequence variations in the priming sites of elasmobranchs (E) and then went on to successfully detect all of the 17 elasmobranch species reared in the tank with the new primers. However, a subsequent study revealed that, in contrast to MiFish-U, the detection probabilities of elasmobranch species remarkably decreased with increasing annealing temperature in MiFish-E (Fig. 6) (Doi et al. 2019), and the primers were further modified by adding one extra base at the 5´ ends of both the forward (-F) and reverse (-R) primers to increase their melting temperatures, thereby mitigating the temperature dependency of the detection rate, i.e., MiFish-E2 (Miya and Sado 2019a). The effectiveness of such modifications for the existing primers should be tested in a future study.

Relationships between detection probabilities and annealing temperatures during the 1st PCR separately shown for bony and cartilaginous fish (Doi et al. 2019). Detection probabilities were estimated by applying the multispecies occupancy modeling framework. Note that the detection probabilities of cartilaginous fish remarkably decrease with increasing annealing temperature ≥ 62 °C, while those of bony fish are stable ≥ 58 °C. Filled circles and error bars indicate medians and 95% credible intervals of the posterior distribution, respectively
An additional example related to PCR dropouts includes a newly optimized primers for ten species of sea sculpins from the four genera Pseudoblennius, Furcina, Ocynectes, and Vellitor (Scorpaeniformes: Cottidae), which are common and tiny reef-associated fish species along the Pacific coast of central Japan (Miya and Sado 2019a). While examining eDNA metabarcoding results from the coastal waters along the Boso Peninsula, NGS reads from several common species were either scarce or completely lacking in a list of detected species. Subsequent Sanger sequencing for the MiFish priming sites clearly demonstrated that there were one to six primer/template mismatches for MiFish-U (Fig. 7a), which were comparable to those observed in MiFish-E. Newly optimized primers (MiFish-U2) accommodate the sequence variations among these ten species of sea sculpins, and a series of later experiments provided reasonable results that more accurately reflect the regional fish community compositions (M. Miya, unpublished data). In our laboratory, multiplex 1st PCR has been performed to amplify eDNA from the local marine fish fauna with a primer mix of MiFish-U-F/R/MiFish-E–F/R/MiFish-U2-F/R = 2:1:1 (Miya and Sado 2019a).

Aligned l-strand nucleotide sequences of priming sites of MiFish-U-F/R with ten species of sea sculpins (a), four osmerid fishes (b), and three species of lampreys (c). Note that the MiFish-U-R sequence is the reverse/complement of the actual primer sequence from 5ʹ–3ʹ ends. Dots represent nucleotide sequences identical to those of the corresponding first lines. F Forward, R reverse
These two examples strongly suggest that it is essential to perform preliminary experiments using MiFish primers based on a few water samples from the target study area before starting the actual survey. By comparing the preliminary results with direct observations through capture-based sampling, underwater visual censuses, and/or historical records through literature survey, apparent false-negative species (or groups of species) can be found among common species that are lacking in the preliminary results. In freshwater fishes of Japan, some species of lampreys (Lethenteron and Lampetra) and some osmerid fishes (Plecoglossus altivelis and Hypomesus spp.) are underrepresented in the MiFish eDNA metabarcoding data in comparisons with their known distributions and abundances, possibly owing to primer/template mismatches (Fig. 7b, c), and modified primers to accommodate their unique variations effectively increase their detections (M. Miya, unpublished data). More recently, a similar example was reported for cryptic seahorse taxa (family Syngnathidae) from Perth, Australia, in which false negatives were apparent with the existing primer sets, including MiFish, although the newly developed primers could detect these sea horse taxa (Nester et al. 2020). This finding is, however, in contrast with the consistent detections of two species of seahorse in the UK using MiFish primers (Tang et al. 2018).
Regarding the lack of variations in MiFish sequences, Miya et al. (2015) found that nucleotide differences in the MiFish sequences from tunas (seven species of Thunnus, members of Scombridae) were so small that the bioinformatics pipeline was unable to assign assembled reads to the correct species (Fig. 8). After visually inspecting all mitogenome sequences from the seven tuna species, they found a region with sufficient interspecific variations among constituent species and subsequently designed genus-specific primers (MiFish-tuna-F/R) to amplify a portion of the ND5 gene (180 bp). Although the new primers were designed based on ND5 instead of 12S, the two primer pairs (MiFish-U and MiFish-tuna) were successfully used simultaneously in multiplex PCR, resulting in the correct species assignment (Miya et al. 2015). For multiplex PCR, it is important to match the amplicon sizes from the two primer pairs (172 vs. 180 bp in this example) for successful library preparation and MiSeq sequencing.

(From Miya et al. 2015, with slight modifications)
Neighbor-joining trees of the seven tuna species based on the amplified regions using MiFish-U (12S rRNA gene) and MiFish-tuna (ND5 gene) primer pairs. The two trees were constructed using MEGA7 (Kumar et al. 2016). The two tuna species contained in the aquarium tank (yellowfin and Pacific bluefin) are highlighted in bold. Distances were calculated using the Kimura’s two-parameter model of base substitution with gaps being deleted. Numbers beside the internal branches are bootstrap probabilities based on 300 pseudo-replicates, and branch lengths are proportional to substitutions per site.
Similarly, the freshwater eel genus Anguilla also showed a lack of interspecific variations, and Takeuchi et al. (2019) designed a new primer pair (MiEel) on the mitochondrial ATP6 gene with reference to the aligned mitogenome sequences from 16 constituent species. The new primer pair amplifies a variable region with sufficient interspecific variations (5–22 nucleotide differences and 1–3 nucleotide differences between three subspecies pairs), and preliminary experiments using natural waters from two rivers successfully detected Japanese freshwater eel Anguilla japonica using multiplex PCR with MiFish-U (Takeuchi et al. 2019). Takeuchi et al. (2019) argue that the MiEel metabarcoding would be particularly effective for documenting species distributions and for surveying spawning grounds where co-occurrence of multiple Anguilla species might be expected. Indeed, eight Anguilla species co-occur within and near the Indonesian archipelago and six species are known to spawn in the western South Pacific (Takeuchi et al. 2019).
A similar example can be found in some salmonid species, and a new primer pair was developed on the mitochondrial ND2 gene (Morita et al. 2019). Considering those species groups exhibiting explosive radiation with extremely little interspecific differences in the coding regions of mitogenomes, such as freshwater family Cichlidae (Kocher 2004), rockfishes of the genus Sebastes (Johns and Avise 1998), and pufferfishes of the genus Takifugu (Yamanoue et al. 2009), alternative genetic markers should be explored for their correct taxonomic assignment to species through eDNA metabarcoding analysis. Recently, a new primer pair has been developed for Lake Tanganyika’s highly diverse cichlids using a hypervariable region of the mitochondrial control region (Doble et al. 2019).
Desired laboratory settings and revised experimental protocols
The original experimental protocols for MiFish eDNA metabarcoding were developed using seawater samples from four aquarium tanks in the Churaumi Aquarium, Okinawa, Japan. The contents of these tanks represent the remarkable taxonomic diversity of marine fishes in a variety of tanks that resemble the environmental settings in the subtropical western North Pacific (Miya et al. 2015). Thus, species composition and species richness in the four tanks were similar to those of the surrounding waters, while the population density of the reared fish species was significantly higher than that of the surrounding waters. Owing to the high density of fish species, the concentrations of eDNA extracted from those tank waters were consistently high, making it easy to perform a series of molecular experiments from library preparation (PCR amplification of MiFish sequences and addition of three adapter sequences at both ends of the fragments; Fig. 1) to massively parallel sequencing using an NGS platform (Illumina MiSeq). Subsequent empirical studies using natural waters from various aquatic environments, including coastal and oceanic waters, deep-sea, rivers, or lakes, across a wide range of latitudes and altitudes revealed that experimental protocols should be optimized for natural waters, where the fish population density is relatively sparse and eDNA concentration is unknown before the experiments. Taking this result into account, the experimental protocols have been revised in our laboratory; details of the latest information can be found in a newly published manual entitled “Environmental DNA Sampling and Experiment Manual” (The eDNA Society 2019). In addition, we came to recognize that eDNA metabarcoding is extremely susceptible to exogenous DNA, which seriously contaminates every experimental step from DNA extraction to library preparation. In the following text we summarize the optimal laboratory settings and revised experimental protocols for MiFish eDNA metabarcoding.
One of the pivotal issues for successful eDNA metabarcoding is to set up an experimental environment that is less susceptible to exogenous DNA. There are various possible sources of such exogenous DNA, but all of them have serious, adverse effects on eDNA metabarcoding owing to the detection of false positives (Miya et al. 2015). To avoid the possible effects of exogenous DNA, the laboratory performing eDNA metabarcoding should be divided into at least three rooms solely dedicated to (1) eDNA extraction, (2) PCR preparation (pre-PCR), and (3) PCR amplification and checking (PCR and post-PCR) (Fig. 9). To avoid carryover contamination, each of these rooms requires its own refrigerators, freezers, centrifuges, lab coats, pipettes, a Milli-Q water dispenser, etc. (Sefers and Schmitz 2018). In addition to such a spatial separation of each experimental step in dedicated rooms, the “one-way rule” should be strictly followed to avoid carryover contamination by bringing amplicons (amplified DNA fragments) from room 3 (PCR and post-PCR) to the other rooms (The eDNA Society 2019). Therefore, after performing the PCR amplification and checking, the same personnel should not conduct an additional eDNA experiments on the same day. Similarly, the use of DNA-free laboratory equipment is essential. Prior to the molecular experiments, all equipment should be new and unused or completely decontaminated and DNA-free. For more details about general precautions for the contamination-free eDNA experiments, see the eDNA Society manual (2019).

An example of three physically separated laboratory rooms solely dedicated to eDNA extraction (a), PCR preparation (pre-PCR) (b), and PCR and PCR checking (post-PCR) (c) in the Natural History Museum and Institute, Chiba
While optimizing the eDNA metabarcoding protocol to accommodate natural seawaters with varying concentrations of fish eDNA, Miya et al. (2016) noted that MiFish primers occasionally failed to amplify the target region for subsequent library preparation. One of the most likely reasons for this unsuccessful PCR amplification was the lack of adequate amounts of template DNA contained in a small volume of water filtered (e.g., 1 L). Although eDNA concentration from a specific taxonomic group is unknown before amplification, filtration of large water volumes (> 1 L) would be a simple and effective way to collect more eDNA from the aquatic environments with scarce fish abundance and biomass, such as open-ocean and deep-sea ecosystems (Miya et al. 2016). Relative to the disk fiber filters conventionally used in a number of fish eDNA studies (Rees et al. 2014), filter cartridges (Sterivex©; Sigma-Aldrich, St. Louis, MO, USA) have the advantage of accommodating larger water volumes before becoming clogged (Walsh et al. 2009). In addition, they are individually packaged and sterile, and several steps of the experimental workflow can be performed in the filter housing, thus reducing the probability of contaminations from the laboratory (Miya et al. 2016). This latter feature is critical for eDNA metabarcoding, in which the risk of contaminations remains among the greatest experimental challenges (Pedersen et al. 2014; Thomsen and Willerslev 2015). Considering this, Miya et al. (2016) developed a protocol for the filtration of water samples with the Sterivex cartridge and for eDNA extraction without having to cut open the housing to remove the filter. Using the newly developed protocol with the Sterivex cartridge and previously used protocols with the GF/F glass microfiber filters (Whatman plc, Maidstone, UK), Miya et al. (2016) performed MiFish eDNA metabarcoding analysis of a huge aquarium tank (7500 m3) with known species composition and found that the number of detected species using the Sterivex cartridges was significantly higher than that using the GF/F filters (Fig. 10). This finding is consistent with those of subsequent studies comparing the effects of filtration methods on eDNA capture efficiency (Spens et al. 2016; Li et al. 2018; Capo et al. 2020). More recently, Oka et al. (2020) developed a parallel filtering system using an aspirator, which allows simultaneous filtration of 14 water samples contained in 1-L plastic bags (Fig. 11) and is less prone to filter clogging.

(From Miya et al. 2016, with slight modifications)
Boxplot showing relationships between number of detected species and filtered water volumes in MiFish metabarcoding analysis of eDNA from the Kuroshio Tank, Okinawa Churaumi Aquarium (Miya et al. 2016). The horizontal bar, upper and lower edges of the box, and vertical bars represent the median inter-quartiles, and 1.5× quartiles, respectively, and the dots represent outliers. Sterivex Sterivex filter cartridges, GF/F GF/F glass microfiber filters

Hand-made instrument capable of filtering up to 14 Sterivex cartridges simultaneously (Oka et al. 2020). This filtration device is less likely to cause filter clogging and can filter 1 L of highly turbid water samples within 1 h. Photo was kindly provided by Shin-ichiro Oka
For MiSeq paired-end sequencing (sequencing from both ends of the amplicon), a two-step tailed PCR approach is now employed to construct dual-indexed libraries (Fig. 1). Ideally, the constructed libraries solely consist of an amplified hypervariable region from fish (MiFish sequence) intervened with primer and three adapter sequences at both ends (Fig. 1b). However, in unsuccessful MiSeq sequencing with virtually no usable fish reads, MiFish sequences may be missing between the two primer/adapter regions or be predominantly replaced by sequences from non-fish organisms. To avoid such failures in the library preparation, it is important to: (1) purify the 1st PCR products to eliminate redundant tailed primers and the primer dimers (hybridized tailed-primer molecules) using either a spin column or beads; (2) quantify the purified 1st PCR products using an automated electrophoresis system, such as BioAnalyzer or TapeStation (both Agilent Technologies, Inc., San Diego, CA, USA) (Fig. 12); (3) dilute the quantified 1st PCR products to a fixed concentration (e.g., 0.1 ng/µl) as templates for the second-round PCR (2nd PCR); and (4) size-select the 2nd PCR products at around 370 bp using gel electrophoresis to remove extra bands at around 440 bp (presumed to be a product derived from the 16S rRNA gene of a microorganism; see also Gold et al. 2020). Note that those extra bands are more prominent in waters that are susceptible to anthropogenic effects, such as estuaries and bay areas near the big cities, than in other areas. Strictly following these four steps ensures high-quality outputs from MiSeq paired-end sequencing for MiFish eDNA metabarcoding, comprising mostly fish with almost constant read numbers (Miya and Sado 2019a). An example can be found in Table 2 from Oka et al. (2020).

Screenshot of the automatically launched analysis file for 11 column-purified 1st PCR products from MiFish eDNA metabarcoding by the the Agilent 2200 TapeStation. The upper-left panel shows a gel electrophoresis image from the 11 samples, the upper-right panel is an electropherogram of lane 11, and the lower panel shows the results of the quantification, such as concentration (pg/µl) and peak molarity (pmol/l). Based on this quantification, PCR products were diluted to 0.1 ng/µl for subsequent use as template for the second-round PCR (2nd PCR) in our laboratory (Miya and Sado 2019a)
It is generally known that eDNA metabarcoding is prone to species detection errors that may occur at sequential steps in field sampling, laboratory experiments, and bioinformatics pipelines (Doi et al. 2019). Doi et al. (2019) examined the effects of the number of replicates in filtration and 1st PCR (repeated extractions and amplifications for the same samples) on detection probabilities of species across a range of PCR annealing temperatures using an aquarium tank water with known species composition. Their analysis showed consistently high detection rates at the filtration step and relatively low and variable detection rates in the 1st PCR step, which favored increasing the number of PCR replicates rather than that of filter replicates. Therefore, in practice, it would be a good choice to perform the 1st PCR with multiple replicates (e.g., eight replicates for the ease of experimental manipulations) to maintain a relatively high detection probability of the species contained in eDNA extracts (Miya and Sado 2019a). This practical recommendation is consistent with a previous simulation and empirical study (Ficetola et al. 2015), which advocated to perform at least eight PCR replicates if detection probability is not high.
New bioinformatics pipeline and reference database
MiFish metabarcoding uses a high-throughput NGS platform for massively parallel sequencing, resulting in the output of tens of millions to billions of DNA sequences overnight (Miya et al. 2015). Therefore, using a bioinformatics pipeline is indispensable for preprocessing and analyzing this large amount of the raw sequences (reads). Miya et al. (2015) constructed a custom bioinformatics pipeline by connecting various kinds of available software with a series of custom scripts. This pipeline has subsequently been implemented on the MitoFish server (https://mitofish.aori.u-tokyo.ac.jp/) with some additional functions for ecological analysis (Sato et al. 2018), and it is now available through a graphical user interface on the website.
However, it is well known that NGS data include numerous erroneous sequences that differ from the true biological sequences by one or few bases at random positions (Coissac et al. 2012; Edgar 2016). These errors may arise when Taq polymerase incorporates incorrect bases during eDNA amplification (1st and 2nd PCR) and/or MiSeq sequencing (cluster amplification and sequencing reaction). Additionally, image analysis during the MiSeq sequencing system also results in approximately 0.1–1% of bases being called incorrectly (Fox et al. 2014). The bioinformatics pipeline available on the web (Sato et al. 2018) does not explicitly remove such erroneous sequences. Instead, it performs clustering of sequences that are more similar to each other at a fixed threshold (e.g., 97% similarity) (Callahan et al. 2016). This sequence clustering into operational taxonomic units (OTUs) is called the OTU method, and its outputs have been demonstrated to lack reusability, reproducibility, and comprehensiveness (Callahan et al. 2017).
Recently, amplicon sequence variant (ASV) methods have been developed in the field of microbiology, replacing OTU methods due to their more accurate estimation of biological sequences. ASV methods infer the biological sequences in the sample prior to the introduction of amplification and sequencing errors, and distinguish sequence variants differing by as little as one nucleotide (Callahan et al. 2017). The core process of ASV methods is called “denoising,” which distinguishes the biological sequences from erroneous sequences on the basis of the expectation that the former are more likely to be repeatedly observed than the latter (Tsuji et al. 2020). Our research group currently incorporates this denoising process in a custom bioinformatics pipeline, and we summarize the updated pipeline from preprocessing the raw data to taxonomic assignment based on a custom database (PMiFish ver. 2.4; the latest version is available from https://github.com/rogotoh/PMiFish.git) in the following paragraphs. Note that most parameters in the PMiFish pipeline (e.g., percentage sequence identity in taxonomic assignment) can be modified by users according to the study purpose.
Data preprocessing and analysis of MiSeq raw reads are performed using USEARCH v10.0.240 (Edgar 2010) according to the following steps: (1) assembly of quality-filtered forward (R1) and reverse (R2) reads; (2) removal of primer sequences; (3) quality filtering; (4) dereplication; (5) denoising; and (6) taxonomic assignment. Note that Edgar (2010) recommended removal of singletons, doubletons, and tripletons, which are major sources of erroneous sequences. After the taxonomic assignment, an optional function in PMiFish ver. 2.4 can generate family-level phylogenies consisting of detected MiFish sequences and reference sequences (contained in a custom database) from those families. By visually checking those family-level phylogenies, one can confirm the authenticity of the taxonomic assignment. For more details about the bioinformatics pipeline and phylogeny-based authentication of the taxonomic assignment, see Komai et al. (2019) or Oka et al. (2020).
Miya et al. (2015) pointed out that there were several methodological challenges that must be addressed before the eDNA metabarcoding approach is likely to become a mainstream technology in fish biodiversity research. One of these challenges is completeness and accuracy of the reference sequence database, which is currently far from satisfactory considering the enormous diversity of fish encompassing more than 32,000 species known from aquatic environments worldwide (Nelson et al. 2016). Indeed, the custom reference sequence database in our laboratory (named “MiFish DB”) included 5085 MiFish sequences, covering around 4230 fish species placed in 457 families and 1827 genera, as of 4 October 2014, when the original study was published (Miya et al. 2015). MiFish DB has subsequently steadily expanded its taxonomic coverage over the past 5 years, almost doubling the number of MiFish sequences to 9708, covering around 8375 fish species placed in 62 orders, 479 families, and 2766 genera, as 14 June 2020 (Fig. 13).

Temporal accumulation of MiFish sequence numbers in the customized database (MiFish DB ver. 38) over a 5-year period between 2015 and 2020 after publication of Miya et al. (2015). For taxonomic coverage, see text
According to the latest information on species diversity from FishBase (Froese and Pauly 2019), the number of fish species worldwide is around 34,152, which are classified into 62 orders, 515 families, and 5124 genera (numbers of order and family follow Nelson et al. 2016). Therefore, the global diversity of fish covered by the current version of MiFish DB (ver. 38) is at 100% for order, 90.3% for family, 54.0% for genus, and 24.5% for species. For Japanese fish, 4554 species have been recorded to date (Motomura 2020), and MiFish DB ver. 38 covers 50 orders (100%), 345 families (90.6%), 1302 genera (81.2%), and 3257 species (71.5%). Apparently, these taxonomic coverages are far from satisfactory for accurate taxonomic assignment, and database construction through international collaboration is eagerly awaited. In this regard, it is worth noting that a joint initiative among related universities and research institutes in the California Current region has built its own reference database for enhanced MiFish eDNA metabarcoding, assembling reference sequences from 712 species among the 864 known species from the region (Gold et al. 2020).
Empirical studies in various aquatic environments
After developing MiFish primers using eDNA from aquarium tanks, Miya et al. (2015) sampled natural seawater from a rocky coast around the coral reef nearby the aquarium. Based on four 2-L seawater samples, Miya et al. (2015) performed MiFish metabarcoding, detecting a total of 93 fish species from the coral reef. This preliminary attempt represented the first demonstration of the feasibility of MiFish eDNA metabarcoding using natural water samples. Subsequently, several studies using MiFish eDNA metabarcoding have been conducted in various aquatic environments, which are summarized in the following sections with representative case studies in marine, freshwater, and estuary ecosystems.
Marine fish community
Marine ecosystems comprise extremely diverse habitats, horizontally ranging from coastal to oceanic areas in terms of distance from land, geographically extending from polar to equatorial regions, and vertically ranging from the surface to deep waters exceeding a depth of 10 km. Here, we introduce those case studies using MiFish eDNA metabarcoding conducted in the inner bay along the coast of Sea of Japan (Yamamoto et al. 2017), oceanic sites in Monterey Bay, southern California (Andruszkiewicz et al. 2017b), various coastal habitats off Qatar, Arabian Gulf (Sigsgaard et al. 2019), and a coral-reef lagoon in Okinawa Island, southern Japan (Oka et al. 2020). Additionally, we introduce a case study using sponges (phylum Poriphera) as eDNA samplers, comparing results from MiFish metabarcoding between the Antarctic and Mediterranean waters (Mariani et al. 2019).
Yamamoto et al. (2017) was the first to conduct a study that applied the MiFish eDNA metabarcoding approach to investigate marine fish communities. Maizuru Bay, an inner bay located along the central coast of Sea of Japan, was chosen as study site because underwater visual censuses of fish communities had been conducted over a 14-year period and baseline data of the faunal composition were available (Masuda 2008). To evaluate the ability of eDNA metabarcoding to reveal fish community structures in species-rich coastal waters, Yamamoto et al. (2017) conducted a systematic grid survey at 47 stations and collected water samples from both surface and bottom waters at each station. A total of 94 samples were subjected to MiFish eDNA metabarcoding and 128 fish species were detected, of which 62.5% (40 species) were also observed by underwater visual censuses and at least 23 species were newly recorded by the eDNA metabarcoding. While recognizing the need to optimize sampling methods (surface vs. bottom waters) and experimental protocols (number of technical replicates in the 1st PCR), Yamamoto et al. (2017) concluded that eDNA metabarcoding will enhance marine ecosystem-related research and that the method will potentially become a standard tool for surveying fish communities.
At about the same time, Andruszkiewicz et al. (2017b) published results from oceanic biodiversity monitoring of marine vertebrates in Monterey Bay, California, using MiFish eDNA metabarcoding methods. This study expanded on a previous eDNA metabarcoding study conducted along a short, 2.5-km transect in the same bay (Port et al. 2015) by extending the spatial scale to 45 km. The study’s main objective was to investigate if eDNA metabarcoding could identify spatial differences between vertebrate communities in Monterey Bay. Andruszkiewicz et al. (2017b) set ten stations along two transects from bottom depths of 27–2430 m, and sampled surface and subsurface waters (depth 20 or 40 m) with three biological replicates (one station sampled twice). A total of 66 samples were subjected to MiFish eDNA metabarcoding, and 72 vertebrate species were detected (including 68 fish species), of which 52 species (72.2%) were known to occur within the bay. Andruszkiewicz et al. (2017b) found that there were differences in vertebrate community composition identified by eDNA metabarcoding between the surface and subsurface waters, and between neritic (< 200 m depth) and deep-water stations, concluding that eDNA metabarcoding offers enormous potential for identifying spatial structures of vertebrate communities. This paper was followed by a series of subsequent eDNA metabarcoding studies using MiFish primers off California (Andruszkiewicz et al. 2017a; Truelove et al. 2019; Closek et al. 2019; Djurhuus et al. 2020; Gold 2020).
To test if eDNA can be informative for marine biodiversity monitoring at large spatial scales (tens to hundreds of kilometers), Sigsgaard et al. (2019) investigated marine vertebrate diversity off Qatar, the Arabian Gulf, through eDNA metabarcoding of seawater samples. They collected three 1-L seawater samples from 21 sites around Qatar that covered five habitat types, i.e., seagrass beds, coral reefs, mangroves, inshore sand bottom, and offshore sand bottom. A total of 72 seawater samples were collected during summer 2016 and spring 2017, from which eDNA was captured and extracted; the eDNA extracts were subsequently subjected to MiFish eDNA metabarcoding (another primer pair was also used for non-fish vertebrate metabarcoding). Sigsgaard et al. (2019) detected 163 fish species, covering 36% of the bony fish genera previously recorded in the Gulf, and found that the species compositions at different sites clearly reflected habitat types. Species accumulation analyses showed that the number of sample replicates was insufficient for some sampling sites, but suggested that a few hundred eDNA samples could potentially capture > 90% of the marine vertebrate diversity, including fishes. Sigsgaard et al. (2019) concluded that seawater samples contain habitat-characteristic molecular signatures and that eDNA biodiversity monitoring can efficiently cover vertebrate diversity at scales relevant to national and regional conservation and management.
One of the big challenges in eDNA metabarcoding is the feasibility of eDNA metabarcoding for monitoring highly diverse fish communities in “diversity hotspots,” such as cichlid fishes of the Great Lakes of East Africa (Kocher 2004), riverine fishes of the Amazon basin of South America (Lundberg et al. 1998), and tropical fishes of the “Coral Triangle,” western Central Pacific (Sanciangco et al. 2013). To test the feasibility of MiFish eDNA metabarcoding, Oka et al. (2020) examined to which extent species richness could be estimated in a small coral reef lagoon (1500 × 900 m) near Okinawa Island, southern Japan (Fig. 14), where the surrounding waters are likely to harbor more than 1500 species of fish (Sanciangco et al. 2013), and tested if eDNA metabarcoding could detect differences between adjacent fish communities inhabiting the offshore reef edge and shore-side seagrass beds within the lagoon (Dorenbosch et al. 2005). Before the eDNA metabarcoding survey, Oka et al. (2020) conducted capture-based sampling for 2 years (2017–2018) and confirmed occurrences of a total of 217 fish species. Subsequent MiFish eDNA metabarcoding from 11 sites on 1 day in May 2019 detected 291 fish species, identifying a total of 410 species distributed across 119 families and 193 genera. Of these 410 species, only 96 (24% of the total) were commonly identified by both methods, indicating that capture-based surveys failed to collect a number of species detected by eDNA metabarcoding. Interestingly, two different approaches to estimate species richness based on eDNA data yielded values close to the 410 species, including one suggesting that an additional three or more eDNA surveys from 11 sites (36 samples) would detect 90% of the 410 species. Furthermore, non-metric multi-dimensional scaling (NMDS) for fish assemblages clearly distinguished between the fish communities of the offshore reef edge and those of the shore-side seagrass beds (Fig. 15). Oka et al. (2020) concluded that the eDNA metabarcoding is a rapid, cost-effective, and useful approach for estimating species richness in tropical and subtropical fish communities with remarkable species diversity, and would be useful for detecting micro-scale habitat segregation. The detection of micro-scale habitat segregations was concurrently confirmed in tropical fish communities at Palmyra Atoll, equatorial Central Pacific, where reef and sand-flat fish communities were clearly distinguished by MiFish metabarcoding using eDNA samples from the respective habitats (Lafferty et al. 2020).

(From Oka et al. (2020 with slight modifications)
Map (a) and photograph (b) showing locations of the 11 seawater sampling sites (stations, St.) in the coral-reef lagoon at Bise, Okinawa, southern Japan (Oka et al. 2020). The 11 samples were taken at a tide level of 180 cm above the lowest tide near high tide. Sts. 5–8 were located along the offshore reef edge, while Sts. 1–4 and 9–11 were located on the shore-side seagrass beds. The lower panels show coral assemblages around St. 8 (c), eelgrass beds around St. 9 (d) and an overview of the lagoon at the same tide level of the sampling day (e).

Non-metric multi-dimensional scaling (NMDS) for fish communities detected at four sites along the offshore reef edge (Sts. 5–8) and seven sites on shore-side seagrass beds (Sts. 1–4 and 9–11) in a coral-reef lagoon at Bise, Okinawa, southern Japan (Oka et al. 2020)
There are various types of water sampling equipment for eDNA analyses, such as buckets, disposable bottles, and Niskin or Van Dorn water samplers, although Mariani et al. (2019) may have been the first to use sponges (phylum Porifera) as natural eDNA samplers. Sponges are known as the planet’s most effective water-filterers, sifting up to 10,000 L of water in 1 day (Kahn et al. 2015). Mariani et al. (2019) collected five Antarctic and four Mediterranean sponge samples, from which eDNA was extracted and subjected to MiFish metabarcoding. The nine samples recovered at least 31 metazoan taxa, of which 22 could be identified at least to the family level or below. The Antarctic samples comprised five typical Antarctic notothenioid fishes (icefishes), while the Mediterranean samples comprised common fishes of that area, such as species of the families Carangidae, Serranidae, Clupeidae, and Sparidae. Additionally, chinstrap penguin, Weddell seal, and seven species of sea stars were detected (Mariani et al. 2019). Interestingly, sponges’ own DNA was not totally detected in the nine samples, and Mariani et al. (2019) speculated that their early-splitting phylogenetic position yielded primer/template mismatches, resulting in PCR dropouts from the supposedly predominant DNA of sponges in the eDNA extracts.
In addition to the above six case studies conducted in various marine environments, MiFish eDNA metabarcoding has been applied to biodiversity monitoring at two coastal areas of eastern Java and Sulawesi Sea, Indonesia (Masengi et al. 2019; Andriyono et al. 2019); port and urban canal area in Osaka Bay, central Japan (Kamimura et al. 2018); Innoshima, Seto Inland Sea, Japan (Sakamoto 2019); coastal waters off Zhoushan, China (Chen et al. 2019, 2020); a harbor at Sloehaven, the Netherlands (Hoorn and Gittenberger 2019); Gulf of St. Laurence Bay, Canada (Afzali et al. 2020); Caribbean bay in Panama (Nguyen et al. 2020); and coastal waters along the South Africa by members of the von der Heyden Lab (https://www.vonderheydenlab.com/environmental-dna-and-metabarcoding.html).
Freshwater fish community
MiFish eDNA metabarcoding has been conducted in both lotic (Nakagawa et al. 2018; Bylemans et al. 2018a, b, 2019; Sales et al. 2019; McDevitt et al. 2019) and lentic (Sato et al. 2017; Fujii et al. 2019; Hayami et al. 2020; Sakata et al. 2020) freshwater environments (flowing and still water, respectively). In addition to highlighting representative studies from these two different environments, in this section we briefly focuse on two topics concerning sampling strategies and sample media (water vs. sediment) for efficient biodiversity monitoring of freshwater fishes.
Nakagawa et al.’s study (2018) was the first to apply the MiFish eDNA metabarcoding approach to investigate freshwater fish communities. To evaluate the performance of MiFish eDNA metabarcoding to investigate local and regional diversities of stream fish species, these authors sampled 102 sites in 51 rivers around Lake Biwa in the central part of Honshu Island, Japan, within 10 person-days, and compared fish species compositions inferred from eDNA metabarcoding with existing observational data from a literature survey, hypothesizing that eDNA data are more consistent with fish assemblages observed upstream than to those downstream owing to a directional flow of river water. They found that when they used existing observational data from within 6 km upstream of the eDNA sampling sites, eDNA data were the most consistent with the observational data and also showed patterns consistent with known upstream–downstream turnover of related species and biogeographical assemblage patterns of certain species (Fig. 16). It appears that MiFish eDNA metabarcoding enabled them to obtain as much regional fish diversity data in a short period of time as the accumulated observations that have been assembled for a long period of time using significant amounts of resources.

(From Nakagawa et al. 2018 with slight modifications)
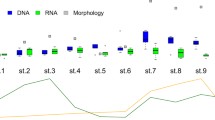
Comparisons of eDNA detections (a, b) and historical records (c, d) between two closely related congeners of the river sculpins (Cottus pollux and C. reinii) showing habitat segregation (Nakagawa et al. 2018). Red and yellow circles indicate sampling sites with and without eDNA detections (a, b) or historical records (c, d), respectively. Cottus pollux and C. reinii tend to occur upstream and near river mouths, respectively.
It is essential to choose the appropriate sampling strategies for biodiversity monitoring using eDNA metabarcoding (Shaw et al. 2016). To evaluate the impact of sampling strategies along an altitudinal and biodiversity gradient in the upper section of the Murrumbidgee River (Murray-Darling Basin, Australia), Bylemans et al. (2018b) chose five sampling sites ranging from high to low altitude (1303 and 520 m a.s.l., respectively) with a minimal distance of around 35 km between sampling sites. Twelve 2-L samples were collected at each site along four transects across the river width spanning a 100-m river section, and Bylemans et al. (2018b) performed MiFish eDNA metabarcoding to determine the local fish biodiversity with varying sampling intensity and spatial sampling replication. The results showed that optimal eDNA sampling strategies varied among sites and indicated that river morphology, species richness, and species abundance affected the optimal sampling intensity and spatial sampling replication needed to accurately assess fish biodiversity. These authors concluded that, while the generality of these patterns will need to be confirmed through future studies, these findings provided a basis to guide future eDNA metabarcoding surveys in river systems. The impact of sampling strategies was also evaluated by Sato et al. (2017), who performed MiFish eDNA metabarcoding in four satellite lakes of Lake Biwa, Japan, and compared the results from individual and pooled samples. In the individual samples from the four lakes, Sato et al. (2017) detected 31, 22, 33, and 31 fish species, while they detected only 30, 20, 29, and 27 fish species in the pooled samples from the respective lakes even after 15 PCR replicates. Based on the apparently lower detection rates from the pooled samples, they concluded that the pooling strategy is unsuitable for estimating fish species richness, although they acknowledged that this procedure is potentially useful for among-site comparison of representative fish communities. Recently, Hayami et al. (2020) compared results from MiFish metabarcoding based on seasonal sampling at multiple sites from the three dam reservoirs in central Honshu Island, Japan. They found that the efficiency of species detection by eDNA metabarcoding could be maximized by collecting water from shore locations during the breeding seasons of the inhabiting fish (Hayami et al. 2020). The results from these three case studies are useful for developing sampling strategies to maximize estimation efficiency of species richness and species composition in both lotic and lentic environments.
The impact of sampling medium and preservation methods on fish detection performance by eDNA metabarcoding is also important. Sales et al. (2019) performed MiFish eDNA metabarcoding using neotropical freshwater samples from Jequitinhonha River, Southeastern Brazil, and collected sediment and water samples from 11 sites with different preservation methods. Sales et al. (2019) found that sediment and water samples yielded a similar amount of fish species and that water stored in ice provided better results than those preserved with cationic surfactant benzalkonium chloride (also known as BAC), previously reported to be an effective preservative for eDNA (Yamanaka et al. 2016). Subsequently, Sakata et al. (2020) collected sedimentary and aqueous eDNA from a biotope (an artificial pond prepared with concrete) and a small satellite lake of Lake Biwa, central Honshu Island, Japan, and compared the decay rates and concentrations using quantitative PCR (qPCR), and species compositions using MiFish metabarcoding. They observed that the sedimentary eDNA exhibited lower decay rates and higher concentrations than those of aqueous eDNA, while species composition showed no significant difference between the two eDNA sources (Sakata et al. 2020). Sakata et al (2020) concluded that sedimentary eDNA analysis will expand future biomonitoring and ecological studies by providing a different timescale.
To validate the usefulness of the eDNA metabarcoding approach for evaluation of biodiversity in lakes, Fujii et al. (2019) compared results from seven capture-based sampling methods and MiFish eDNA metabarcoding in 31 backwater lakes in Hokkaido Island, northern Japan. The capture-based field surveys were often interrupted by macrophytes and muddy sediments in the 31 lakes, while the eDNA metabarcoding was interrupted by a lack of detectable eDNA at some sites, possibly because of PCR inhibitor in the eDNA extracts (Fujii et al. 2019). Comparisons of the results from eDNA metabarcoding and capture-based sampling showed that the detected fish communities were similar between methods, with a 70% overlap, leading the authors to conclude that the performance of eDNA metabarcoding using 1-L surface water sampling is similar to that of the capture-based sampling methods. Similarly, McDevitt et al. (2019) compared results from MiFish eDNA metabarcoding with those from two different types of electrofishing conducted along a stretch of the Huddersfield Narrow Canal in the UK, with the aim to validate the detection capability of eDNA metabarcoding. The canal has limited data available on fish assemblages and biodiversity. In addition to corroborating data obtained by electrofishing, eDNA provided a wider snapshot of fish assemblages. Given the semi-lotic nature of canals, McDevitt et al. (2019) encouraged the use of eDNA as a fast and cost-effective tool to detect and monitor whole fish communities. As these two case studies have concurrently performed capture-based sampling and MiFish eDNA metabarcoding, they provided strong proof-of-concepts for the eDNA approach being useful for fish biodiversity monitoring in both lentic and lotic environments.
In addition to the above ten case studies conducted in various freshwater environments, MiFish eDNA metabarcoding methods have been applied to biodiversity monitoring in the Duck and Clinch Rivers, Tennessee, USA (Paine 2019); Lake Michigan, USA (Jurecki 2020); Jequitinhonha River catchment, Brazil (Sales et al. 2020b); St. Laurence and Rupert Rivers, Canada (Boivin-Deslile et al. 2020; Berger et al. 2020); Lake Kasumigaura in Ibaraki, central Japan (Kondo et al. 2016); agricultural waterway in Saitama, central Japan (Kimochi et al. 2020); two rivers in Hokkaido and Hyogo, Japan (Akamatsu et al. 2018); 14 rivers in Sado Island, Niigata, Japan (Koseki 2019); a small river in Okinawa, southern Japan (Sato et al. 2018); an urban stream in Suwon City, Korea (Song et al. 2019); four Korean rivers (Alam et al. 2020); the middle and lower reaches of the Yangtze River, China (Nian et al. 2020); and irrigation water from two agro-ecological regions of Sri Lanka (Gamage et al. 2020). Furthermore, effects of seasonal thermal stratification and fish habitat preferences on the vertical distributions of eDNA were investigated inthe Experimental Lakes Area, north-western Ontario, Canada (Littlefair et al. 2020).
Estuarine fish community
Estuarine ecosystems play a significant ecological role at the boundary between freshwater and marine environments, providing foraging, breeding, and nursery habitats for a variety of fishes (Flitcroft et al. 2016). Despite their importance, estuarine ecosystems are susceptible to anthropogenic effects and are suffering from severe alterations, degradation, or losses (Wingard and Lorenz 2014). In light of this critical situation in estuarine ecosystems, three papers on biodiversity monitoring using the MiFish metabarcoding methods from East Asian countries have been consecutively published in the past 6 months (Zhang et al. 2019; Zou et al. 2020; Ahn et al. 2020).
In the Yangtze Estuary, near Shanghai, China, fisheries resources have been undergoing dramatic declines as a consequence of environmental changes and human activities (Zhang et al. 2015). To investigate the fish assemblage structure in the Yangtze Estuary and its adjacent waters, Zhang et al. (2019) collected 50 water samples from the estuary during three seasons in 2018 and subjected the eDNA samples to MiFish eDNA metabarcoding. They identified 41 species from the areas, and the fish assemblages differed significantly among seasons (Zhang et al. 2019). These authors concluded that eDNA metabarcoding can be an effective tool not only for monitoring for fisheries resources, but also for aquatic product processing, environmentally friendly development, and socioeconomic stability.
Subsequently, Zou et al. (2020) reported results from eDNA metabarcoding survey in a coastal wetland of the Pearl River Estuary, about 1220 km south of the Yangtze Estuary. These authors concurrently conducted eDNA sampling and bottom trawling at the same sampling sites and seasons and compared the results from eDNA metabarcoding and trawl catches. MiFish metabarcoding was performed on eDNA samples, and Zou et al. (2020) detected 57 fish species distributed across 24 families and 47 genera. In contrast, bottom trawling identified 32 fish species distributed across 19 families and 26 genera, a considerably lower detection sensitivity than that of the eDNA metabarcoding. Zou et al. (2020) concluded that the combined results from both methods provided a realistic picture of fish diversity of the Pearl River Estuary coastal wetland.
More recently, Ahn et al. (2020) conducted biodiversity monitoring using MiFish eDNA metabarcoding at five estuaries with a broader geographic scale along the Japanese archipelago. Water samples were collected from river mouths and adjacent coastal areas of two and three estuaries with high and low degrees of development, respectively (Ahn et al. 2020). A total of 182 fish species across 67 families were detected, 11 of which occurred in all of the rivers. Species richness apparently to decrease with increasing the degree of human development along each river, while species composition varied among rivers, reflecting the distribution and migration of fishes. Ahn et al. (2020) concluded that further development of the eDNA technique will offer an alternative method for accurate and non-invasive monitoring of aquatic life.
In addition to the above three case studies, eDNA collected from four estuaries around the English Channel and North Sea were used to compare primer performance in eDNA metabarcoding (Collins et al. 2019). Also, Siegenthaler et al. (2019) used MiFish metabarcoding to analyze the gut contents of a generalist predator/scavenger, the European brown shrimp, Crangon crangon, from the intertidal zone at 21 sites distributed over six estuaries in the Netherlands, Portugal, and the UK to monitor fish biodiversity (for details, see below).
Developments of new techniques
The results from eDNA metabarcoding analysis provide a taxonomic table with read abundances. However, read abundances are greatly affected by the amplification efficiency of the PCR primers and by PCR cycles, the latter being an exponential process that increases the very low concentration of eDNA to make sufficient amounts for further analysis (Kelly et al. 2019). Therefore, comparing read abundances between different samples and studies is not a straightforward procedure.
Ushio et al (2018a) attempted to quantify fish eDNA copy numbers in weekly samples collected from a coastal marine ecosystem in Maizuru Bay, Japan, using MiFish metabarcoding methods. The key to this technique is the addition of internal standard DNAs at several levels of concentration (i.e., known amounts of short DNA fragments from fish species that have never been observed in a sampling area) to the eDNA samples. A correlation equation was obtained for each sample using the relationship between the number of sequence reads and the added amount of standard DNAs, and this equation was used to estimate the copy numbers from the sequence reads of non-standard fish eDNA. The calculated copy numbers showed a significant and positive correlation with those determined by qPCR, suggesting that eDNA metabarcoding with standard DNA enabled useful quantifications of eDNA. Using this method, Ushio et al. (2018a) demonstrated the temporal dynamics of the ten most abundant fish species in the bay (Fig. 17), while acknowledging that the method requires that (fish) species-specific PCR amplification biases need to be corrected for between-species comparisons. Nevertheless, this method (called qMiSeq) appears to provide ecologically meaningful information on the natural fish community dynamics. From an experimental point of view, continued use of the same set of the standards should be avoided; otherwise the laboratory will be contaminated with amplicons of the standards, compromising the quantification.

The versatility of MiFish primers for gene amplification across diverse taxa has led to the development of two new techniques for single-species detections (Stoeckle et al. 2018; Truelove et al. 2019). In addition, two new software tools have been developed for bioinformatics pipeline (Curd et al. 2019) and ecological analyses based on an output taxonomic table (Kandlikar et al. 2018), with their own analyzed data from MiFish eDNA metabarcoding as examples to demonstrate software performance. Below we briefly introduce these four papers.
Stoeckle et al. (2018) developed a strategy for single-species eDNA presence/absence assays using nested PCR techniques, naming these assays “GoFish”. The assays first amplify a mitochondrial 12S rRNA gene fragment with MiFish primers, followed by nested PCR with species-specific primers. The species-specific primer set was designed on the hypervariable regions in the MiFish amplicon for the amplification of the target species. Stoeckle et al. (2018) designed GoFish assays for 11 bony fish species common in the lower Hudson River estuary and then tested their performance using eDNA samples from two sites. These authors argue that GoFish does not require tissues of target and related species for assay development, that a basic thermal cycler is sufficient, and that indexing and batching samples are unnecessary and advanced bioinformatics expertise is not needed, unlike for qPCR and Illumina metabarcoding, respectively. They concluded that this approach will be a useful addition to current eDNA methods for analyzing the presence/absence of known species, when turnaround time is important, and in educational settings.
Truelove et al. (2019) recently developed a rapid eDNA sequencing methodology using a portable the Oxford Nanopore MinIon Sequencer (Oxford Nanopore Technologies Ltd., Oxford, UK) to identify the presence of white sharks (Carcharodon carcharias), a threatened top oceanic predator. The methodology consisted of filtration of 3-L seawater from a Niskin bottle, eDNA collection from the filter, library preparation using MiFish primers, and sequencing, all occurring while on board the research vessel (Truelove et al. 2019). This rapid eDNA protocol accurately identified white sharks and cookie cutter sharks in the water column over 1800 km from shore, providing a first step toward using eDNA techniques in the high seas as an oceanic census technique. Truelove et al. (2019) argued that future eDNA studies using MinIon sequencing in the field can follow their protocol to detect the presence of other fish species of research or conservation interest using the MiFish universal fish primers.
Curd et al. (2019) developed “Anapaca Toolkit”, a bioinformatic pipeline with modules for (1) creating custom reference databases, (2) executing equality control and multilocus read parsing, and (3) generating taxonomic assignments for all quality reads produced by MiSeq. The Anacapa Toolkit is modular and its parameters are easily modifiable, making it easily adapted to user-specific needs in several important ways. To test the toolkit on field-collected eDNA metabarcoding datasets, MiFish primers were used for library preparation, and the raw reads from MiSeq were analyzed by the toolkit. Curd et al (2019) found that the identified taxa included several of interest for natural resource managers, including species of special concern (e.g., basking shark Cetorhinus maximus, and giant black sea bass Stereolepis gigas) and species that are the subject of focused monitoring efforts (California sheepshead Semicossyphus pulcher). Output data from Anapaca Toolkit can be subjected to ecological analysis using “ranacapa,” the R package that helps perform exploratory biodiversity analyses and visualization (Kandlikar et al. 2018).
Application of MiFish eDNA metabarcoding to other organisms
MiFish metabarcoding has occasionally detected non-fish vertebrates, such as dolphins, sea birds, and turtles, from marine environments (Miya et al. 2015). Indeed, a research group in California has continuously used MiFish primers to census marine vertebrates (Andruszkiewicz et al. 2017a, 2017b; Closek et al. 2019; Djurhuus et al. 2020), suggesting that optimization of the priming sites in MiFish primers to accommodate variations of non-fish vertebrates would be effective for monitoring these animals. Additionally, terrestrial animals must have frequent contact with water to survive, implying that eDNA originating from those animals should be detectable from places containing water in terrestrial ecosystems (Ushio et al. 2017).
To validate the possible detection of terrestrial mammals from eDNA in their habitats, Ushio et al. (2017) developed a universal primer pair (MiMammal, a modified version of MiFish primers) for metabarcoding mammalian eDNA and then tested its performance using water samples from zoo cages and natural water samples from forest ponds. Using MiMammal primers, these authors successfully detected mammals reared in the zoo and representative mammals in the forest from those samples, and subsequent studies demonstrated that MiMammal primers were useful for detecting tropical forest mammals from a natural saltlick (Ishige et al. 2017), Neotropical mammals from the Amazon and Atlantic Forest in Brazil (Sales et al. 2020a), forest mammals from Jasper Ridge Biological Preserve, California (Leempoel et al. 2020), and mammal species in snow tracks during winter in Kyoto, Japan (Kinoshita et al. 2019). To the best of our knowledge, MiMammal primers have not been used to detect marine mammals, partly because most of the species identified by the marine mammal visual surveys have been detected with MiFish primers (Closek et al. 2019), and Closek et al. (2019) stated that MiMammal primers can be used in future studies.
Regarding mammals, Ushio et al. (2018c) developed a universal primer pair (MiBird, a modified version of MiFish/MiMammal primers) for metabarcoding bird eDNA and tested its performance using water samples from zoo cages and from a natural pond. MiBird primers successfully detected known species from the zoo cages and representative species from the natural pond (Ushio et al. 2018c).
In addition to vertebrates, our research group recently developed a set of universal primers for metabarcoding eDNA from a representative invertebrate group, i.e., decapod crustaceans comprising approximately 14,000 extant species and including several commercially important species (Komai et al. 2019). The new primers (named “MiDeca”) were designed for two conservative regions of the mitochondrial 16S rRNA gene that amplify a short hypervariable region (average 164 bp, range 153–184 bp) with sufficient interspecific variations. After confirmation of the amplification from tissue-derived DNA extracts for 250 species distributed across 65 families and 186 genera, Komai et al. (2019) performed preliminary eDNA metabarcoding from natural seawater collected at a rocky shore in Tateyama, the Pacific coast of central Japan, and detected 42 decapod species.
Both mammals and birds play significant roles in marine, freshwater, and estuarine ecosystems as top predators (Valenzuela et al. 2013), and decapod crustaceans are one of the most important fishery resources in several countries worldwide (Pan et al. 2007). Therefore, non-invasive, rapid, and cost-effective detection of those animals using eDNA metabarcoding approaches would further contribute to our understanding of aquatic ecosystems, ultimately leading to sound ecosystem conservation and fishery resource management.
Applications of MiFish metabarcoding to bulk DNA samples
In addition to fish eDNA suspended in aquatic environments or deposited on the bottom sediments, the MiFish metabarcoding approach is applicable to bulk DNA samples (total DNA) containing multiple fish species (Miya et al. 2015). These include samples from (1) net collections containing early life history stages or damaged specimens with no diagnostic characters for species identification (Miya et al. 2015; Kacev et al. 2018), (2) digested stomach contents of the piscivorous or generalist/scavenger animals (Devloo-Delva et al. 2019; Siegenthaler et al. 2019), and (3) processed foods (Marín et al. 2018; Calosso et al. 2020). Here, we highlight the use of bulk samples from net collections as the most promising application of the MiFish metabarcoding approach.
Ichthyoplankton refers to the early life history stages of fish (eggs and larvae), and research in this field is an important tool to understand trophic dynamics, recruitment processes, and the association between environmental fluctuations and productivity of recreational, commercially, and ecologically important fish stocks (Auth and Brodeur 2013). A long-term sampling program (> 60 years) using plankton nets has been conducted in the southern California Current since 1949 (McClatchie et al. 2014), and net collections have been sorted into major taxonomic groups, followed by morphological identification based on highly specialized taxonomic expertise, which is laborious, time consuming, and costly (Kacev et al. 2018). To develop a framework for cost-effective and informative ichthyoplankton monitoring in the southern California Current, Kacev et al. (2018) compared results from traditional approaches to those of metabarcoding to determine whether metabarcoding is a viable alternative. A total of 74 ichthyoplankton samples were collected from 37 stations. Fish samples were sorted and identified to lowest taxonomic level by expert taxonomists. Subsequently, fish samples were homogenized, from which total DNA was extracted and subjected to MiFish metabarcoding. The metabarcoding results closely matched those of traditional methods regarding family-level composition within a sample, but at finer taxonomic levels, they did not agree as closely to those of traditional methodologies, requiring well-curated reference libraries for as many species as possible. Kacev et al. (2018) concluded that continued refinement of this new method (in particular that of reference libraries) will increase the utility of this high-throughput genetic approach for routine monitoring and assessment. Actually, subsequent efforts to assemble the MiFish sequences from fishes of the California Current region resulted in 717 reference sequences, which cover 84.7% of the total fauna, with 864 fish species currently included (Gold et al. 2020).
Other than the net collections, MiFish metabarcoding has been used to analyze the gut contents of a generalist predator/scavenger, the European brown shrimp Crangon crangon (Siegenthaler et al. 2019). Siegenthaler et al. (2019) used this shrimp species as a sampler that consumes a wide variety of food items, ranging from meiofauna to fish. To evaluate the suitability of trophic contents to assess fish diversity, adult brown shrimps (total length 20–50 mm) were collected from the intertidal zone at 21 sites distributed over six estuaries in the Netherlands, Portugal, and the UK. Siegenthaler et al. (2019) performed metabarcoding analysis using both MiFish and COI primers, identifying a total of 32 teleost fish species from those estuaries, which was twice the number of species recovered from the traditional capture-based sampling. MiFish primers consistently outperformed COI primers in terms of mean number of detected fish taxa per sample (Siegenthaler et al. 2019). In another study, MiFish primers were used as supplementary to an analysis of stomach contents of piscivorous tropical-reef fish (coral trout genus Plectropomus spp.) (Devloo-Delva et al. 2019). However, their utility was limited owing to incomplete reference libraries compared with those of the COI and 16S rRNA genes.
MiFish primers (not metabarcoding) have been also used to identify food samples being sold at wholesalers, retailers, and restaurants (Marín et al. 2018; Calosso et al. 2020). To increase knowledge about species diversity available across the Peruvian supply chains (from landing sites to markets and restaurants), Marín et al. (2018) applied barcoding approaches targeting three mitochondria genes (COI, 12S, and 16S) in 131 samples. Among these, 35 samples (26.7%) were mislabeled, including tilapia labeled as wild marine fish, dolphinfish and hake labeled as grouper, and different shark species sold as “smooth-hounds”. Such seafood mislabeling was also reported from the Turks and Caicos Islands, a small island nation in the Wider Caribbean Region. Calosso et al. (2020) collected market samples of groupers (Epinephelinae spp.), which are frequent targets for fraudulent substitution on the world’s major seafood markets and performed barcoding with the COI and 12S genes. These authors found that only 18% of samples from restaurants and stores were confirmed as groupers, while 96% were mislabeled in some way. The results from these two case studies indicate that MiFish primers are useful as supplementary to barcoding when amplification fails or sub-optimal reads are obtained with the full COI fish-barcoding primer cocktail (Calosso et al. 2020). In the case of processed foods (e.g., surimi-based products or fish cakes), where multiple raw materials may be used, a metabarcoding approach would be useful for identification of the source materials (Giusti et al. 2017). Similarly, MiFish metabarcoding can be applied to fish silage used for animal feed or fish-oil production, which consists of minced fish from multiple species dissolved in acid upon capture (Hansen et al. 2018).
Concluding remarks
In this review article, we first introduced three recent independent studies on performance evaluation of eDNA metabarcoding primers including MiFish (Bylemans et al. 2018a; Collins et al. 2019; Zhang et al. 2020). Then, we provided an overview of the latest developments in the MiFish eDNA metabarcoding methods and related techniques (from water filtration to library preparation and data analysis) and their applications to ecological studies on fish communities in various aquatic environments (marine, freshwater, and estuarine). In addition, we have covered topics on other applications of MiFish primers and MiFish metabarcoding methods, including developments of new techniques in eDNA studies (i.e., single-species detections and quantitative MiSeq), eDNA metabarcoding of other animal groups (mammals, birds, and decapod crustaceans), and metabarcoding of bulk DNA samples (net collections and food samples). Although we have skipped some of more general topics, such as the “ecology” of eDNA (from production and persistence/degradation, to transport of eDNA) (Barnes and Turner 2016; Seymour et al. 2018; Harrison et al. 2019; Beng and Corlett 2020), it is essential to consider the effects of environmental factors on those processes for a more appropriate interpretation of eDNA detections (Yamamoto et al. 2016; Sassoubre et al. 2016; Murakami et al. 2019).
Throughout the review, we have attempted to highlight not only the strengths, but also the limitations of MiFish primers and metabarcoding methods, and have suggested alternative strategies to mitigate those limitations. Indeed, although recent studies (Bylemans et al. 2018a; Collins et al. 2019; Zhang et al. 2020) have demonstrated that MiFish primers outperformed other competing primers, primer/template mismatches in MiFish primers occasionally cause underrepresentation of some fish groups (Fig. 7), and MiFish sequences often lack interspecific variations among some closely related congeners, most notably among commercially important groups, such as tunas (Miya et al. 2015), salmons (Morita et al. 2019), and freshwater eels (Takeuchi et al. 2019). However, these technical problems can be alleviated by the optimization of MiFish primers to local fauna and by simultaneous use of universal MiFish primers and newly developed group-specific primers amplifying more variable regions. Revised laboratory settings and experimental protocols consistently enable the preparation of high-quality libraries for massively parallel sequencing that are less susceptible to carryover contamination. The new bioinformatics pipeline implementing the newly developed denoising process has greatly improved taxonomic assignment accuracy and its reproducibility, although taxonomic coverage of the reference database is still far from satisfactory, resulting in a number of detections with ambiguous species identity. Quantitative MiFish metabarcoding (Ushio et al. 2018a) would be a promising approach to capture spatiotemporal dynamics of fish communities that have never been easily monitored by conventional methods based on capture-based sampling and/or underwater visual censuses. Analysis of bulk DNA samples of fish eggs and larvae from plankton net collections (Kacev et al. 2018) in combination with concurrent analysis of eDNA samples would be another promising approach toward a comprehensive understanding of oceanic fish communities.
Nevertheless, eDNA simply represents indirect, extraorganismal genetic markers shed from macroorganisms. Thus, even if eDNA copy number per single unit volume of water (eDNA concentration) can be accurately estimated through quantitative PCR (Takahara et al. 2012) or droplet digital PCR (Doi et al. 2015), too many challenges need to be overcome to correlate the eDNA concentration with absolute abundance or biomass of the multiple target fish species (Rees et al. 2014). This is partly because eDNA itself does not contain any information on the size and number of individuals, age, weight, life-history stages, and (in most cases) sex, while all of this information is available from specimens collected by traditional capture-based sampling (Hansen et al. 2018). Therefore, for fisheries purposes, MiFish eDNA metabarcoding demonstrates its strength when a concept of “ecosystem-based fishery management” (Pikitch et al. 2014) is applied and continuous biodiversity monitoring is performed at multiple sites. By applying MiFish eDNA metabarcoding in this way, we could quickly detect spatial and temporal changes in ecosystems, such as biodiversity loss or invasion of alien species, thereby taking immediate measures to resolve such environmental issues.
Indeed, in August 2017 our research group started biweekly eDNA surveys at 11 stations along the southernmost tip of the Boso Peninsula, central Japan (Fig. 18), in an attempt to monitor the spatiotemporal dynamics of coastal fish communities (M. Miya et al. unpublished). In this project, two personnel have conducted a single survey along the 130-km-long coastline, while performing a bucket water sampling and on-site filtration using a Sterivex filter cartridge combined with a 50-ml syringe at each station (Miya and Sado 2019b), then bringing the filtered cartridges back to the laboratory. The eDNA was then extracted from the cartridges following the methods developed by Miya et al. (2016), and the eDNA extracts subjected to library preparation and MiSeq sequencing. The initial 50 surveys of this project were completed by August 2019, with a total of 550 samples, and the project has been continued with eDNA surveys carried out at monthly intervals. All molecular experiments on the initial 550 samples have been completed successfully, and a large amounts of raw sequence data (approx. 500 million reads) are ready for data analysis in the near future. Surprisingly, there have been no missing data during the initial 50 surveys despite the occasional field sampling under unfavorable weather and sea conditions. Apparently, field sampling for traditional capture-based sampling or underwater visual censuses could not be conducted at this frequency with this spatial scale, indicating the excellent feasibility of eDNA sampling coupled with MiFish metabarcoding methods.

Stations along the 130-km coastline of the southern tip of Boso Peninsula for biweekly eDNA sampling from August 2017 (central map). A bucket water sampling and on-site filtration of 1-L seawater using a combination of the Sterivex filter cartridge and a 50-ml syringe was performed at each station, and 50 samplings have been completed by August 2019, resulting in 550 eDNA samples without missing data
However, to perform eDNA biodiversity monitoring on a scale comparable to that of physical attributes, several technical challenges are yet to be overcome. For example, it is essential to develop an autonomous sampling instrument, called an “environmental sample processor” (ESP) (Scholin et al. 2017), which is a robotic device that can be programmed to automate water sample filtration and preservation of the captured material for immediate analyses in situ (Yamahara et al. 2019; Fukuba et al. 2019). Furthermore, such instrumentation should be cost-effective to be installed in large numbers over a wide area. By combining on-site eDNA sampling and sample processing automation with rapid developments in molecular analysis technology and quantitative MiFish metabarcoding (Ushio et al. 2018a), a large quantitative dataset could be generated to capture fluctuating interspecific interactions among component species in fish communities (e.g., Ushio et al. 2018c). If such complex and dynamic interspecific interactions can be modeled using a non-linear method (e.g., empirical dynamic modeling [EDM]), it may be possible to forecast fish community dynamics from time series data (Chang et al. 2017). Such a forecast should be useful for healthy ecosystem conservation and the sustainable use of fisheries resources, which meet the goals of SDGs 14 and 15 (conservation and sustainable use of marine and terrestrial [including inland freshwater] ecosystems, respectively; https://sdgs.un.org/goals).
References
Afzali SF, Bourdages H, Laporte M, Mérot C, Audet C, Bernatchez L (2020) Comparing environmental metabarcoding and trawling survey of demersal fish communities in the Gulf of St. Lawrence, Canada. Environ DNA (in press)
Ahn H, Kume M, Terashima Y, Ye F, Kameyama S, Miya M, Yamashita Y, Kasai A (2020) Evaluation of biodiversity in estuaries using environmental DNA metabarcoding. BioRxiv. https://doi.org/10.1101/2020.03.22.997809
Akamatsu Y, Tsuzuki T, Yokoyama R, Funabashi Y, Ohta M, Azegami M, Naito D, Inui R (2018) Comparative study between fish collection by national census on river environment and environmental DNA metabarcoding. Ann J Hydr Engineer 74:415–420. https://doi.org/10.2208/jscejhe.74.5_I_415
Alam MJ, Kim N, Andriyono S, Choi H, Lee J, Kim H (2020) Assessment of fish biodiversity in four Korean rivers using environmental DNA metabarcoding. PeerJ 8:e9508. https://doi.org/10.7717/peerj.9508
Alexander JB, Bunce M, White N, Wilkinson SP, Adam AAS, Berry T, Stat M, Thomas L, Newman SJ, Dugal L, Richards ZT (2019) Development of a multi-assay approach for monitoring coral diversity using eDNA metabarcoding. Coral Reefs 39:1–13. https://doi.org/10.1007/s00338-019-01875-9
Andriyono S, Alam MJ, Kim H-W (2019) Environmental DNA (eDNA) metabarcoding: diversity study around the Pondok Dadap fish landing station, Malang, Indonesia. Biodiversitas 20:3772–3781. https://doi.org/10.13057/biodiv/d201241
Andruszkiewicz EA, Sassoubre LM, Boehm AB (2017a) Persistence of marine fish environmental DNA and the influence of sunlight. PLoS ONE 12:e0185043. https://doi.org/10.1371/journal.pone.0185043
Andruszkiewicz EA, Starks HA, Chavez FP, Sassoubre LM, Block BA, Boehm AB (2017b) Biomonitoring of marine vertebrates in Monterey Bay using eDNA metabarcoding. PLoS ONE 12:e0176343. https://doi.org/10.1371/journal.pone.0176343
Auth TD, Brodeur RD (2013) An overview of ichthyoplankton research in the northern California Current region: contributions to ecosystem assessments and management. CalCOFI Rep 54:107–126
Barbier EB, Hacker SD, Kennedy C, Koch EW, Stier AC, Silliman BR (2011) The value of estuarine and coastal ecosystem services. Ecol Monogr 81:169–193. https://doi.org/10.1890/10-1510.1
Barnes MA, Turner CR (2016) The ecology of environmental DNA and implications for conservation genetics. Conserv Genet 17:1–17. https://doi.org/10.1007/s10592-015-0775-4
Beng KC, Corlett RT (2020) Applications of environmental DNA (eDNA) in ecology and conservation: opportunities, challenges and prospects. Biodivers Conserv 29:2089–2121. https://doi.org/10.1007/s10531-020-01980-0
Berger C, Hernandez C, Laporte M, Côté G, Paradis Y, Kameni T, Dominique W, Normandeau E, Bernatchez L (2020) Fine-scale spatial structuration of lotic fish communities revealed by environmental DNA metabarcoding. Environ DNA (in press). https://doi.org/10.1002/edn3.129
Bohmann K, Evans A, Gilbert MTP, Carvalho GR, Creer S, Knapp M, Yu DW, de Bruyn M (2014) Environmental DNA for wildlife biology and biodiversity monitoring. Trends Ecol Evol 29:358–367. https://doi.org/10.1016/j.tree.2014.04.003
Boivin-Deslile D, Laporte M, Burton F, Dion R, Normandeau E, Bernatchez L (2020) Using environmental DNA (eDNA) for evaluation of freshwater fish communities: comparison with established gillnet surveys. Environ DNA (in press)
Brinson A, Wallmo K (2015) Stakeholder attitudes toward ecosystem-based fisheries management. Mar Fish Rev 77:17–30. https://doi.org/10.7755/mfr.77.3.2
Bylemans J, Gleeson DM, Hardy CM, Furlan E (2018a) Toward an ecoregion scale evaluation of eDNA metabarcoding primers: a case study for the freshwater fish biodiversity of the Murray-Darling Basin (Australia). Ecol Evol 8:8697–8712. https://doi.org/10.1002/ece3.4387
Bylemans J, Gleeson DM, Lintermans M, Gilligan DM, Hardy CM, Beitzel M, Gilligan DM, Furlan EM, Gilligan DM, Lintermans M (2018b) Monitoring riverine fish communities through eDNA metabarcoding: determining optimal sampling strategies along an altitudinal and biodiversity gradient. Metabarcoding Metagenom 2:1361–1412. https://doi.org/10.3897/mbmg.2.30457
Bylemans J, Gleeson DM, Duncan RP, Hardy CM, Furlan EM (2019) A performance evaluation of targeted eDNA and eDNA metabarcoding analyses for freshwater fishes. Environ DNA 1:402–414. https://doi.org/10.1002/edn3.41
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:1–7. https://doi.org/10.1038/nmeth.3869
Callahan BJ, McMurdie PJ, Holmes SP (2017) Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J 11:2639–2643. https://doi.org/10.1038/ismej.2017.119
Calosso MC, Claydon JAB, Mariani S, Cawthorn D-M (2020) Global footprint of mislabeled seafood on a small island nation. Biol Conserv 245:108557. https://doi.org/10.1016/j.biocon.2020.108557
Capo E, Spong G, Königsson H, Byström P (2020) Effects of filtration methods and water volume on the quantification of brown trout (Salmo trutta) and Arctic char (Salvelinus alpinus) eDNA concentrations via droplet digital PCR. Environ DNA 2:152–160. https://doi.org/10.1002/edn3.52
Chen Z, Chen J-W, Wang X-Y, Gao T-X, Liu Y (2019) Establishment and optimization of the eDNA preservation method for Zhoushan coastal waters. Oceanol Limnol Sinica 50:1098–1107
Chen Z, Minamoto T, Lin L, Gao T (2020) An optional low-cost method of extracting environmental DNA of macro-organisms from filter membranes in large scale eDNA surveys. Pak J Zool. https://doi.org/10.17582/journal.pjz/20190118100108
Cheung WWL, Reygondeau G, Frölicher TL (2016) Large benefits to marine fisheries of meeting the 1.5°C global warming target. Science 354:1591–1594. https://doi.org/10.1126/science.aag2331
Cilleros K, Valentini A, Allard L, Dejean T, Etienne R, Grenouillet G, Iribar A, Taberlet P, Vigouroux R, Brosse S (2019) Unlocking biodiversity and conservation studies in high-diversity environments using environmental DNA (eDNA): a test with Guianese freshwater fishes. Mol Ecol Resour 19:27–46. https://doi.org/10.1111/1755-0998.12900
Closek CJ, Santora JA, Starks HA, Schroeder ID, Andruszkiewicz EA, Sakuma KM, Bograd SJ, Hazen EL, Field JC, Boehm AB (2019) Marine vertebrate biodiversity and distribution within the central California Current using environmental DNA (eDNA) metabarcoding and ecosystem surveys. Front Mar Sci 6:1–17. https://doi.org/10.3389/fmars.2019.00732
Coissac E, Riaz T, Puillandre N (2012) Bioinformatic challenges for DNA metabarcoding of plants and animals. Mol Ecol 21:1834–1847. https://doi.org/10.1111/j.1365-294x.2012.05550.x
Collins RA, Bakker J, Wangensteen OS, Soto AZ, Corrigan L, Sims DW, Genner MJ, Mariani S (2019) Non-specific amplification compromises environmental DNA metabarcoding with COI. Methods Ecol Evol 10:1985–2001. https://doi.org/10.1111/2041-210x.13276
Common MS, Norton TW (1994) Biodiversity, natural resource accounting and ecological monitoring. Environ Resour Econ 4:29–53. https://doi.org/10.1007/bf00691931
Costanza R, d’Arge R, de Groot R, Farber S, Grasso M, Hannon B, Limburg K, Naeem S, O’Neill RV, Paruelo J, Raskin RG, Sutton P, van den Belt M (1997) The value of the world’s ecosystem services and natural capital. Nature 387:253–260. https://doi.org/10.1038/387253a0
Cristescu ME (2014) From barcoding single individuals to metabarcoding biological communities: towards an integrative approach to the study of global biodiversity. Trends Ecol Evol 29:566–571. https://doi.org/10.1016/j.tree.2014.08.001
Curd EE, Gold Z, Kandlikar GS, Gomer J, Ogden M, O’Connell T, Pipes L, Schweizer TM, Rabichow L, Lin M, Shi B, Barber PH, Kraft N, Wayne R, Meyer RS (2019) Anacapa Toolkit: an environmental DNA toolkit for processing multilocus metabarcode datasets. Methods Ecol Evol 10:1469–1475. https://doi.org/10.1111/2041-210x.13214
Darling JA, Galil BS, Carvalho GR, Rius M, Viard F, Piraino S (2017) Recommendations for developing and applying genetic tools to assess and manage biological invasions in marine ecosystems. Mar Policy 85:54–64. https://doi.org/10.1016/j.marpol.2017.08.014
Deiner K, Walser J-C, Mächler E, Altermatt F (2015) Choice of capture and extraction methods affect detection of freshwater biodiversity from environmental DNA. Biol Conserv 183:53–63. https://doi.org/10.1016/j.biocon.2014.11.018
Deiner K, Bik HM, Mächler E, Seymour M, Roussel AL, Altermatt F, Creer S, Bista I, Lodge DM, Vere N, Pfrender MEL, BERNATCHEZ (2017) Environmental DNA metabarcoding: transforming how we survey animal and plant communities. Mol Ecol 26:5872–5895. https://doi.org/10.1111/mec.14350
Devloo-Delva F, Huerlimann R, Chua G, Matley JK, Heupel MR, Simpfendorfer CA, Maes GE (2019) How does marker choice affect your diet analysis: comparing genetic markers and digestion levels for diet metabarcoding of tropical-reef piscivores. Mar Freshw Res 70:8–11. https://doi.org/10.1071/mf17209
DiBattista JD, Coker DJ, Sinclair-Taylor TH, Stat M, Berumen ML, Bunce M (2017) Assessing the utility of eDNA as a tool to survey reef-fish communities in the Red Sea. Coral Reefs 36:1245–1252. https://doi.org/10.1007/s00338-017-1618-1
DiBattista JD, Reimer JD, Stat M, Masucci GD, Biondi P, Brauwer MD, Wilkinson SP, Chariton AA, Bunce M (2020) Environmental DNA can act as a biodiversity barometer of anthropogenic pressures in coastal ecosystems. Sci Rep 10:8365. https://doi.org/10.1038/s41598-020-64858-9
Djurhuus A, Closek CJ, Kelly RP, Pitz KJ, Michisaki RP, Starks HA, Walz KR, Andruszkiewicz EA, Olesin E, Hubbard K, Montes E, Otis D, Muller-Karger FE, Chavez FP, Boehm AB, Breitbart M (2020) Environmental DNA reveals seasonal shifts and potential interactions in a marine community. Nat Commun 11:254. https://doi.org/10.1038/s41467-019-14105-1
Doble CJ, Hipperson H, Salzburger W, Horsburgh GJ, Mwita C, Murrell DJ, Day JJ (2019) Testing the performance of environmental DNA metabarcoding for surveying highly diverse tropical fish communities: a case study from Lake Tanganyika. Environ DNA 2:24–41. https://doi.org/10.1002/edn3.43
Doi H, Takahara T, Minamoto T, Matsuhashi S, Uchii K, Yamanaka H (2015) Droplet digital polymerase chain reaction (PCR) outperforms real-time PCR in the detection of environmental DNA from an invasive fish species. Environ Sci Tech 49:5601–5608. https://doi.org/10.1021/acs.est.5b00253
Doi H, Fukaya K, Oka S, Sato K, Kondoh M, Miya M (2019) Evaluation of detection probabilities at the water-filtering and initial PCR steps in environmental DNA metabarcoding using a multispecies site occupancy model. Sci Rep 9:1–8. https://doi.org/10.1038/s41598-019-40233-1
Dorenbosch M, Grol M, Nagelkerken I, van der Velde G (2005) Distribution of coral reef fishes along a coral reef-seagrass gradient: edge effects and habitat segregation. Mar Ecol Prog Ser 299:277–288. https://doi.org/10.3354/meps299277
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. https://doi.org/10.1093/bioinformatics/btq461
Edgar RC (2016) UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. BioRxiv. https://doi.org/10.1101/081257
Elbrecht V, Leese F (2017) PrimerMiner: an r package for development and in silico validation of DNA metabarcoding primers. Methods Ecol Evol 8:622–626. https://doi.org/10.1111/2041-210x.12687
Evans NT, Li Y, Renshaw MA, Olds BP, Deiner K, Turner CR, Jerde CL, Lodge DM, Lamberti GA, Pfrender ME (2017) Fish community assessment with eDNA metabarcoding: effects of sampling design and bioinformatic filtering. Can J Fish Aquat Sci 74:1362–1374. https://doi.org/10.1139/cjfas-2016-0306
Ficetola GF, Coissac E, Zundel S, Riaz T, Shehzad W, Bessière J, Taberlet P, Pompanon F (2010) An In silico approach for the evaluation of DNA barcodes. BMC Genomics 11:434. https://doi.org/10.1186/1471-2164-11-434
Ficetola GF, Pansu J, Bonin A, Coissac E, Giguet-Covex C, Barba MD, Gielly L, Lopes CM, Boyer F, Pompanon F, Rayé G, Taberlet P (2015) Replication levels, false presences and the estimation of the presence/absence from eDNA metabarcoding data. Mol Eco Res 15:543–556. https://doi.org/10.1111/1755-0998.12338
Flitcroft RL, Bottom DL, Haberman KL, Bierly KF, Jones KK, Simenstad CA, Gray A, Ellingson KS, Baumgartner E, Cornwell TJ, Campbell LA (2016) Expect the unexpected: place-based protections can lead to unforeseen benefits. Aquat Conser Mar Freshw Ecosyst 26:39–59. https://doi.org/10.1002/aqc.2660
Fogarty M (2014) The art of ecosystem-based fishery management. Can J Fish Aquat Sci 71:479–490. https://doi.org/10.1139/cjfas-2013-0203
Fox EJ, Reid-Bayliss KS, Emond MJ, Loeb LA (2014) Accuracy of next generation sequencing platforms. Next Gener Seq Appl 1:1000106. https://doi.org/10.4172/jngsa.1000106
Froese R, Pauly D (2019) FishBase. www.fishbase.org
Fujii K, Doi H, Matsuoka S, Nagano M, Sato H, Yamanaka H (2019) Environmental DNA metabarcoding for fish community analysis in backwater lakes: a comparison of capture methods. PLoS ONE 14:0210357. https://doi.org/10.1371/journal.pone.0210357
Fujikura K, Lindsay D, Kitazato H, Nishida S, Shirayama Y, Schnur J (2010) Marine biodiversity in Japanese waters. PLoS ONE 5:e11836. https://doi.org/10.1371/journal.pone.0011836
Fukuba T, Sano Y, Yamamoto H, Miwa T, Fujii T (2019) Development, deployment and improvement of miniaturized in situ bio/biochemical analysis systems towards multi-modal ocean sensing. IEEE Underw Technol (UT). https://doi.org/10.1109/UT.2019.8734416
Gamage CD, Sato Y, Kimura R, Yamashiro T, Toma C (2020) Understanding leptospirosis eco-epidemiology by environmental DNA metabarcoding of irrigation water from two agro-ecological regions of Sri Lanka. PLoS Neglect Trop Dis 14:e0008437. https://doi.org/10.1371/journal.pntd.0008437
Giusti A, Armani A, Sotelo CG (2017) Advances in the analysis of complex food matrices: species identification in surimi-based products using next generation sequencing technologies. PLoS ONE 12:e0185586. https://doi.org/10.1371/journal.pone.0185586
Gold ZJ (2020) Design and implementation of environmental DNA metabarcoding methods for monitoring the Southern California marine protected area network. Ph.D. thesis, University of California, Los Angeles
Gold Z, Choi E, Kacev D, Frable B, Burton R, Goodwin K, Thompson A, Barber P (2020) FishCARD: Fish 12S California Current specific reference database for enhanced metabarcoding efforts. Authorea. https://doi.org/10.22541/au.159136805.55528691
Goodwin KD, Muller-Karger FE, Djurhuus A, Allen LZ, Allen AE, McCrow JP, Hyde GC (2018) Molecular approaches for an operational marine biodiversity observation network. World seas: an environmental evaluation. Academic Press, New York, pp 613–631
Grizzetti B, Lanzanova D, Liquete C, Reynaud A, Cardoso AC (2016) Assessing water ecosystem services for water resource management. Environ Sci Policy 61:194–203. https://doi.org/10.1016/j.envsci.2016.04.008
Halpern BS, Longo C, Lowndes JSS, Best BD, Frazier M, Katona SK, Kleisner KM, Rosenberg AA, Scarborough C, Selig ER (2015) Patterns and emerging trends in global ocean health. PLoS ONE 10:e0117863–e117921. https://doi.org/10.1371/journal.pone.0117863
Hansen BK, Bekkevold D, Clausen LW, Nielsen EE (2018) The sceptical optimist: challenges and perspectives for the application of environmental DNA in marine fisheries. Fish Fish 19:751–768. https://doi.org/10.1111/faf.12286
Harrison JB, Sunday JM, Rogers SM (2019) Predicting the fate of eDNA in the environment and implications for studying biodiversity. Proc Roy Soc B 286:20191409–20191419. https://doi.org/10.1098/rspb.2019.1409
Hayami K, Sakata MK, Inagawa T, Okitsu J, Katano I, Doi H, Nakai K, Ichiyanagi H, Gotoh RO, Miya M, Sato H, Yamanaka H, Minamoto T (2020) Effects of sampling seasons and locations on fish environmental DNA metabarcoding in dam reservoirs. Ecol Evol. https://doi.org/10.1002/ece3.6279
Hellström M, Blomstrand CE (2019) Environmental DNA inventory of fish in the fishing port in Lövsta. AquaBiota Report 8. AquaBiota Water Research, Stockholm, Sweden
Hill SL, Hinke J, Bertrand S, Fritz L, Furness RW, Ianelli JN, Murphy M, Oliveros-Ramos R, Pichegru L, Sharp R, Stillman RA, Wright PJ, Ratcliffe N (2020) Reference points for predators will progress ecosystem-based management of fisheries. Fish Fish 21:368–378. https://doi.org/10.1111/faf.12434
Hoegh-Guldberg O, Mumby PJ, Hooten AJ, Steneck RS, Greenfield P, Gomez E, Harvell CD, Sale PF, Edwards AJ, Caldeira K, Knowlton N, Eakin CM, Iglesias-Prieto R, Muthiga N, Bradbury RH, Dubi A, Hatziolos ME (2007) Coral reefs under rapid climate change and ocean acidification. Science 318:1737–1742. https://doi.org/10.1126/science.1152509
Hoffmann C, Schubert G, Calvignac-Spencer S (2016) Aquatic biodiversity assessment for the lazy. Mol Ecol 25:846–848. https://doi.org/10.1111/mec.13535
Hopkins GW, Freckleton RP (2002) Declines in the numbers of amateur and professional taxonomists: implications for conservation. Anim Conserv 5:245–249. https://doi.org/10.1017/s1367943002002299
Ishige T, Miya M, Ushio M, Sado T, Ushioda M, Maebashi K, Yonechi R, Lagan P, Matsubayashi H (2017) Tropical-forest mammals as detected by environmental DNA at natural saltlicks in Borneo. Biol Conserv 210:281–285. https://doi.org/10.1016/j.biocon.2017.04.023
Jerde CL, Wilson EA, Dressler TL (2019) Measuring global fish species richness with eDNA metabarcoding. Mol Ecol Res 19:19–22. https://doi.org/10.1111/1755-0998.12929
Johns GC, Avise JC (1998) Tests for ancient species flocks based on molecular phylogenetic appraisals of Sebastes rockfishes and other marine fishes. Evolution 52:1135–1146. https://doi.org/10.1111/j.1558-5646.1998.tb01840.x
Jurecki S (2020) Application and validation of the eDNA-metabarcoded MiFish/MitoFish pipeline for assessment of native and non-native fish communities of Lake Michigan. Ph.D. thesis, Purdue University.
Kacev D, Gillett D, Carvalho AF de, Cash C, Walther S, Thompson A, Thompson L, Bowlin N, Goodwin K, Stein ED (2018) Assessment of Ichthyoplankton Metabarcoding for Routine Monitoring. Southern California Coastal Water Research Project Technical Report 1031. https://ftp.sccwrp.org/pub/download/DOCUMENTS/TechnicalReports/1031_IchthyoplanktonMetabarcoding.pdf
Kahn AS, Yahel G, Chu JWF, Tunnicliffe V, Leys SP (2015) Benthic grazing and carbon sequestration by deep-water glass sponge reefs. Limnol Oceanogr 60:78–88. https://doi.org/10.1002/lno.10002
Kamimura S, Kozuki Y, Otani S, Hirakawa R, Iwami K, Takeyama K, Yamanaka R (2018) Fish diversity detection at port and urban canal area using environmental DNA metabarcoding. J JSCEB3 74:I_474–I_479. https://doi.org/10.2208/jscejoe.74.i_474
Kandlikar GS, Gold ZJ, Cowen MC, Meyer RS, Freise AC, Kraft NJB, Moberg-Parker J, Sprague J, Kushner DJ, Curd EE (2018) ranacapa: an R package and Shiny web app to explore environmental DNA data with exploratory statistics and interactive visualizations. F1000Res 7:1734–1818. https://doi.org/10.12688/f1000research.16680.1
Kelly RP, Port JA, Yamahara KM, Crowder LB (2014a) Using environmental DNA to census marine fishes in a large mesocosm. PLoS ONE 9:e86175. https://doi.org/10.1371/journal.pone.0086175.s007
Kelly RP, Port JA, Yamahara KM, Martone RG, Lowell N, Thomsen PF, Mach ME, Bennett M, Prahler E, Caldwell MR, Crowder LB (2014b) Harnessing DNA to improve environmental management. Science 344:1455–1456. https://doi.org/10.1126/science.1251156
Kelly RP, Shelton AO, Gallego R (2019) Understanding PCR processes to draw meaningful conclusions from environmental DNA studies. Sci Rep 9:1–14. https://doi.org/10.1038/s41598-019-48546-x
Kimochi Y, Watanabe K, Tanaka H, Suzuki K, Tamura K, Takahashi Y, Saito Y, Mizushima K, Oota M, Koizumi N (2020) Evaluation of fish diversity in domestic wastewater polluted rivers using environmental DNA metabarcoding analysis. J Water Waste 62:47–58
Kinoshita G, Yonezawa S, Murakami S, Isagi Y (2019) Environmental DNA collected from snow tracks is useful for identification of mammalian species. Zool Sci 36:198–207. https://doi.org/10.2108/zs180172
Kitagawa T, Muraoka K, Yamada T, Nakamura K (2020) Analysis for trial cases of environmental DNA metabarcoding to fish survey in the National Census on River Environments. Adv River Eng 26:319–324
Kocher TD (2004) Adaptive evolution and explosive speciation: the cichlid fish model. Nat Rev Genet 5:288–298. https://doi.org/10.1038/nrg1316
Komai T, Gotoh RO, Sado T, Miya M (2019) Development of a new set of PCR primers for eDNA metabarcoding decapod crustaceans. Metabarcoding Metagenom 3:343–419. https://doi.org/10.3897/mbmg.3.33835
Kondo NI, Nakagawa M, Matsuzaki S, Ueno R, Takamura K, Ito H (2016) DNA barcoding, environmental DNA and an ongoing attempt of detecting biodiversity in Lake Kasumigaura. J Integr Field Sci 13:21–29
Koseki Y (2019) An evaluation of environmental DNA metabarcoding approach for monitoring stream fish species. J Human Cult Stud 184–187
Koslow J (2009) The role of acoustics in ecosystem-based fishery management. ICES J Mar Sci 66:966–973. https://doi.org/10.1093/icesjms/fsp082
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Lafferty KD, Garcia-Vedrenne AE, McLaughlin JP, Childress JN, Morse MF, Jerde CL (2020) At Palmyra Atoll, the fish-community environmental DNA signal changes across habitats but not with tides. J Fish Biol (in press). https://doi.org/10.1111/jfb.14403
Leempoel K, Hebert T, Hadly EA (2020) A comparison of eDNA to camera trapping for assessment of terrestrial mammal diversity. Proc R Soc B 287:20192353. https://doi.org/10.1098/rspb.2019.2353
Leese F, Altermatt F, Bouchez A, Ekrem T, Hering D, Meissner K, Mergen P, Pawlowski J, Piggott J, Rimet F, Steinke D, Taberlet P, Weigand A, Abarenkov K, Beja P, Bervoets L, Björnsdóttir S, Boets P, Boggero A, Bones A, Borja A, Bruce K, Bursić V, Carlsson J, Čiampor F, Čiamporová-Zatovičová Z, Coissac E, Costa F, Costache M, Creer S, Csabai Z, Deiner K, DelValls Á, Drakare S, Duarte S, Eleršek T, Fazi S, Fišer C, Flot J-F, Fonseca V, Fontaneto D, Grabowski M, Graf W, Guðbrandsson J, Hellström M, Hershkovitz Y, Hollingsworth P, Japoshvili B, Jones J, Kahlert M, Stroil BK, Kasapidis P, Kelly M, Kelly-Quinn M, Keskin E, Kõljalg U, Ljubešić Z, Maček I, Mächler E, Mahon A, Marečková M, Mejdandzic M, Mircheva G, Montagna M, Moritz C, Mulk V, Naumoski A, Navodaru I, Padisák J, Pálsson S, Panksep K, Penev L, Petrusek A, Pfannkuchen M, Primmer C, Rinkevich B, Rotter A, Schmidt-Kloiber A, Segurado P, Speksnijder A, Stoev P, Strand M, Šulčius S, Sundberg P, Traugott M, Tsigenopoulos C, Turon X, Valentini A, van der Hoorn B, Várbíró G, Hadjilyra MV, Viguri J, Vitonytė I, Vogler A, Vrålstad T, Wägele W, Wenne R, Winding A, Woodward G, Zegura B, Zimmermann J (2016) DNAqua-Net: developing new genetic tools for bioassessment and monitoring of aquatic ecosystems in Europe. Res Ideas Outcomes 2:e11321–e11325. https://doi.org/10.3897/rio.2.e11321
Leese F, Bouchez A, Abarenkov K, Altermatt F, Borja Á, Bruce K, Ekrem T, Čiampor F, Čiamporová-Zaťovičová Z, Costa FO, Duarte S, Elbrecht V, Fontaneto D, Franc A, Geiger MF, Hering D, Kahlert M, Stroil BK, Kelly M, Keskin E, Liska I, Mergen P, Meissner K, Pawlowski J, Penev L, Reyjol Y, Rotter A, Steinke D, van der Wal B, Vitecek S, Zimmermann J, Weigand AM (2018) Why we need sustainable networks bridging countries, disciplines, cultures and generations for aquatic biomonitoring 2.0: a perspective derived from the DNAqua-Net COST action. Adv Ecol Res 58:63–99. https://doi.org/10.1016/bs.aecr.2018.01.001
Lepetz V, Massot M, Schmeller DS, Clobert J (2009) Biodiversity monitoring: some proposals to adequately study species’ responses to climate change. Biodivers Conserv 18:3185. https://doi.org/10.1007/s10531-009-9636-0
Li J, Handley LL, Read DS, Hänfling B (2018) The effect of filtration method on the efficiency of environmental DNA capture and quantification via metabarcoding. Mol Eco Resour 18:1102–1114. https://doi.org/10.1111/1755-0998.12899
Littlefair JE, Hrenchuk LE, Blanchfield PJ, Rennie MD, Cristescu ME (2020) Thermal stratification and fish thermal preference explain vertical eDNA distributions in lakes. BioRxiv. https://doi.org/10.1101/2020.04.21.042820
Long R, Charles A, Stephenson R (2017) Key principles of ecosystem-based management: the fishermen’s perspective. Fish Fish 18:244–253. https://doi.org/10.1111/faf.12175
Lundberg JG, Marshall LG, Guerrero J, Horton B, Malabarba MCSL, Wesselingh F (1998) The stage for Neotropical fish diversification: a history of tropical South American rivers. In: Malabarba LR, et al. (eds) Phylogeny and classification of neotropical fishes. Editora Universitaria da PUCRS, Rio Grande Sul, Brazil, pp 14–48
Mariani S, Baillie C, Colosimo G, Riesgo A (2019) Sponges as natural environmental DNA samplers. Curr Biol 29:R401–R402. https://doi.org/10.1016/j.cub.2019.04.031
Marín A, Serna J, Robles C, Ramírez B, Reyes-Flores LE, Zelada-Mázmela E, Sotil G, Alfaro R (2018) A glimpse into the genetic diversity of the Peruvian seafood sector: unveiling species substitution, mislabeling and trade of threatened species. PLoS ONE 13:e0206596. https://doi.org/10.1371/journal.pone.0206596
Masengi KWA, Mandagi IF, Manu L, Silooy F, Labaro IL, Masengi AWR, Sebua N, Masengi EIKG, Pinontoan B, Hutabarat Y, Hukom F, Iwata M, Abe Y, Sato Y, Kimura R, Yamahira K (2019) Study on existence of the fisheries resources abundance by using environmental deoxyribonucleic acid (e-DNA) approach at fishing grounds in the Sulawesi Sea, Indonesia. IOP Conf Series: Mater Sci Eng 567:012026. https://doi.org/10.1088/1757-899x/567/1/012026
Masuda R (2008) Seasonal and interannual variation of subtidal fish assemblages in Wakasa Bay with reference to the warming trend in the Sea of Japan. Environ Biol Fish 82:387–399. https://doi.org/10.1007/s10641-007-9300-z
McClatchie S, Duffy-Anderson J, Field J, Goericke R, Griffith D, Hanisko D, Hare J, Lyczkowski-Shultz J, Peterson W, Watson W, Weber E, Zapfe G (2014) Long time series in US fisheries oceanography. Oceanography (Wash DC) 27:48–67. https://doi.org/10.5670/oceanog.2014.86
McCulloch M, Falter J, Trotter J, Montagna P (2012) Coral resilience to ocean acidification and global warming through pH up-regulation. Nat Clim Change 2:623–627. https://doi.org/10.1038/nclimate1473
McDevitt AD, Sales NG, Browett SS, Sparnenn AO, Mariani S, Wangensteen OS, Coscia I, Benvenuto C (2019) Environmental DNA metabarcoding as an effective and rapid tool for fish monitoring in canals. J Fish Biol 6:25–35. https://doi.org/10.1111/jfb.14053
Minamoto T, Miya M, Sado T, Seino S, Doi H, Kondoh M, Nakamura K, Takahara T, Yamamoto S, Yamanaka H, Araki H, Iwasaki W, Kasai A, Masuda R, Uchii K (2020) An illustrated manual for environmental DNA research: Water sampling guidelines and experimental protocols. Environ DNA (in press)
Miya M, Nishida M (2015) The mitogenomic contributions to molecular phylogenetics and evolution of fishes: a 15-year retrospect. Ichthyol Res 62:29–71. https://doi.org/10.1007/s10228-014-0440-9
Miya M, Sado T (2019a) Multiple species detection using MiFish primers. In: eDNA Method Standardization Committee (ed) Environmental DNA Sampling and Experiment Manual Version 2.1. The eDNA Society, Otsu, Japan, pp 55–92
Miya M, Sado T (2019b) Water sampling and on-site filtration using a filter cartridge. In: eDNA Method Standardization Committee (ed) Environmental DNA Sampling and Experiment Manual Version 2.1. The eDNA Society, Otsu, Japan, pp 12–22
Miya M, Sato Y, Fukunaga T, Sado T, Poulsen JY, Sato K, Minamoto T, Yamamoto S, Yamanaka H, Araki H, Kondoh M, Iwasaki W (2015) MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: detection of more than 230 subtropical marine species. R Soc Open Sci 2:150088. https://doi.org/10.1098/rsos.150088
Miya M, Minamoto T, Yamanaka H, Oka S, Sato K, Yamamoto S, Sado T, Doi H (2016) Use of a filter cartridge for filtration of water samples and extraction of environmental DNA. J Visual Exper. https://doi.org/10.3791/54741
Morita K, Sahashi G, Miya M, Kamada S, Kanbe T, Araki H (2019) Ongoing localized extinctions of stream-dwelling white-spotted charr populations in small dammed-off habitats of Hokkaido Island, Japan. Hydrobiologia 840:207–213. https://doi.org/10.1007/s10750-019-3891-1
Motomura H (2020) List of Japan’s all fish species. Current standard Japanese and scientific names of all fish species recorded from Japanese waters. The Kagoshima University Museum, Kagoshima, Japan
Murakami H, Yoon S, Kasai A, Minamoto T, Yamamoto S, Sakata MK, Horiuchi T, Sawada H, Kondoh M, Yamashita Y, Masuda R (2019) Dispersion and degradation of environmental DNA from caged fish in a marine environment. Fish Sci 85:327–337. https://doi.org/10.1007/s12562-018-1282-6
Nakagawa H, Yamamoto S, Sato Y, Sado T, Minamoto T, Miya M (2018) Comparing local- and regional-scale estimations of the diversity of stream fish using eDNA metabarcoding and conventional observation methods. Freshw Biol 63:569–580. https://doi.org/10.1111/fwb.13094
Näslund J, Didrikas T, Hellström P, Hellström M (2019) Inventory of fish at Gåsefjärden in Karlskrona archipelago using capture-based sampling and eDNA metabarcoding AquaBiota Report 15. AquaBiota Water Research, Stockholm, Sweden
Nelson JS, Grande TC, Wilson MVH (2016) Fishes of the world, 5th edn. Wiley & Sons, Hoboken, New Jersey
Nester GM, Brauwer MD, Koziol A, West KM, DiBattista JD, White NE, Power M, Heydenrych MJ, Harvey E, Bunce M (2020) Development and evaluation of fish eDNA metabarcoding assays facilitate the detection of cryptic seahorse taxa (family: Syngnathidae). Environ DNA (in press). https://doi.org/10.1002/edn3.93
Nguyen BN, Shen EW, Seemann J, Correa AMS, O’Donnell JL, Altieri AH, Knowlton N, Crandall KA, Egan SP, McMillan WO, Leray M (2020) Environmental DNA survey captures patterns of fish and invertebrate diversity across a tropical seascape. Sci Rep 10:6729. https://doi.org/10.1038/s41598-020-63565-9
Nian XU, Xiong M, Shao K, Que Y, Li J (2020) Preliminary study on environmental DNA metabarcoding for detecting biodiversity in the middle and lower reaches of the Yangtze River. Res Environ Sci 33:1187–1196. https://doi.org/10.13198/j.issn.1001-6929.2020.03.06
Oka S, Doi H, Miyamoto K, Hanahara N, Sado T, Miya M (2020) Environmental DNA metabarcoding for biodiversity monitoring of a highly-diverse tropical fish community in a coral-reef lagoon: Estimation of species richness and detection of habitat segregation. Environmental DNA (in press)
Paine RT (2019) Environmental DNA: A molecular approach to delineating the distribution and community composition of fishes in the Duck and Clinch Rivers, Tennessee. Ph.D. thesis, Tennessee Technological University
Palumbi S (1996) Nucleic acids II: the polymerase chain reaction. In: Hillis D, Moritz C, Mable B (eds) Molecular systematics. Sinauer Associates, Sunderland, MA, pp 205–247
Pan M, Gallego A, Hay S, Ieno EN, Pierce GJ, Zuur AF, Smith GM (2007) Univariate methods to analyse abundance of decapod larvae. Analyzing ecological data. Springer, New York, NY, pp 373–388
Pandolfi JM, Connolly SR, Marshall DJ, Cohen AL (2011) Projecting coral reef futures under global warming and ocean acidification. Science 333:418–422. https://doi.org/10.1126/science.1204794
Pedersen MW, Overballe-Petersen S, Ermini L, Sarkissian CD, Haile J, Hellstrom M, Spens J, Thomsen PF, Bohmann K, Cappellini E, Schnell IB, Wales NA, Caroe C, Campos PF, Schmidt AMZ, Gilbert MTP, Hansen AJ, Orlando L, Willerslev E (2014) Ancient and modern environmental DNA. Phil Trans R Soc B 370:20130383–20130383. https://doi.org/10.1098/rstb.2013.0383
Pikitch EK, Santora C, Babcock EA, Bakun A, Bonfil R, Conover DO, Dayton P, Doukakis P, Fluharty D, Heneman B, Houde ED, Link J, Livingston PA, Mangel M, McAllister MK, Pope J, Sainsbury KJ (2014) Ecosystem-based fishery management. Science 305:346–347. https://doi.org/10.1126/science.1098222
Pitz K, Truelove N, Nye C, P R, Chavez F (2020) Environmental DNA (eDNA) 12S Metabarcoding Illumina MiSeq NGS PCR Protocol (Touchdown) v2. protocol.io. dx.doi.org/10.17504/protocols.io.bcppivmn
Port JA, O’Donnell JL, Romero-Maraccini OC, Leary PR, Litvin SY, Nickols KJ, Yamahara KM, Kelly RP (2015) Assessing vertebrate biodiversity in a kelp forest ecosystem using environmental DNA. Mol Ecol 25:527–541. https://doi.org/10.1111/mec.13481
Pörtner H (2008) Ecosystem effects of ocean acidification in times of ocean warming: a physiologist’s view. Mar Ecol Prog Ser 373:203–217. https://doi.org/10.3354/meps07768
Quesne WJFL, Pinnegar JK (2012) The potential impacts of ocean acidification: scaling from physiology to fisheries. Fish Fish 13:333–344. https://doi.org/10.1111/j.1467-2979.2011.00423.x
Rapport DJ, Costanza R, McMichael AJ (1998) Assessing ecosystem health. Trends Ecol Evol 13:397–402. https://doi.org/10.1016/s0169-5347(98)01449-9
Rees HC, Maddison BC, Middleditch DJ, Patmore JRM, Gough KC (2014) The detection of aquatic animal species using environmental DNA—a review of eDNA as a survey tool in ecology. J Appl Ecol 51:1450–1459. https://doi.org/10.1111/1365-2664.12306
Riaz T, Shehzad W, Viari A, Pompanon F, Taberlet P, Coissac E (2011) ecoPrimers: inference of new DNA barcode markers from whole genome sequence analysis. Nucleic Acids Res 39:e145–e145. https://doi.org/10.1093/nar/gkr732
Riser SC, Freeland HJ, Roemmich D, Wijffels S, Troisi A, Belbéoch M, Gilbert D, Xu J, Pouliquen S, Thresher A, Traon P-YL, Maze G, Klein B, Ravichandran M, Grant F, Poulain P-M, Suga T, Lim B, Sterl A, Sutton P, Mork K-A, Vélez-Belchí PJ, Ansorge I, King B, Turton J, Baringer M, Jayne SR (2016) Fifteen years of ocean observations with the global Argo array. Nat Clim Change 6:145–153. https://doi.org/10.1038/nclimate2872
Robison BH (2009) Conservation of deep pelagic biodiversity. Conserv Biol 23:847–858. https://doi.org/10.1111/j.1523-1739.2009.01219.x
Sakamoto K (2019) Detection of fish species in seagrass and macroalgae beds at Innoshima, Seto Inland Sea, using environmental DNA. Bull Biogeogr Soc Japan 74:3–7
Sakata MK, Yamamoto S, Gotoh RO, Miya M, Yamanaka H, Minamoto T (2020) Sedimentary eDNA provides different information on timescale and fish species composition compared with aqueous eDNA. Environ DNA (in press). https://doi.org/10.1002/edn3.75
Sales NG, Wangensteen OS, Carvalho DC, Mariani S (2019) Influence of preservation methods, sample medium and sampling time on eDNA recovery in a neotropical river. Environ DNA 2:1–12. https://doi.org/10.1002/edn3.14
Sales NG, da Kaizer MC, Coscia I, Perkins JC, Highlands A, Boubli JP, Magnusson WE, Silva MNFD, Benvenuto C, McDevitt AD (2020a) Assessing the potential of environmental DNA metabarcoding for monitoring Neotropical mammals: a case study in the Amazon and Atlantic Forest, Brazil. Mamm Rev 82:400–405. https://doi.org/10.1111/mam.12183
Sales NG, Wangensteen O, Carvalho DC, Deiner K, Praebel K, Coscia I, McDevitt A, Mariani S (2020b) Space-time dynamics in monitoring neotropical fish communities using eDNA metabarcoding. BioRxiv. https://doi.org/10.1101/2020.02.04.933366
Sanciangco JC, Carpenter KE, Etnoyer PJ, Moretzsohn F (2013) Habitat availability and heterogeneity and the Indo-Pacific warm pool as predictors of marine species richness in the tropical Indo-Pacific. PLoS ONE 8:e56245. https://doi.org/10.1371/journal.pone.0056245
Sassoubre LM, Yamahara KM, Gardner LD, Block BA, Boehm AB (2016) Quantification of environmental DNA (eDNA) shedding and decay rates for three marine fish. Environ Sci Technol 50:10456–10464. https://doi.org/10.1021/acs.est.6b03114
Sato H, Sogo Y, Doi H, Yamanaka H (2017) Usefulness and limitations of sample pooling for environmental DNA metabarcoding of freshwater fish communities. Sci Rep 7:1–12. https://doi.org/10.1038/s41598-017-14978-6
Sato Y, Miya M, Fukunaga T, Sado T, Iwasaki W (2018) MitoFish and MiFish pipeline: a mitochondrial genome database of fish with an analysis pipeline for environmental DNA metabarcoding. Mol Biol Evol 10:421–423. https://doi.org/10.1093/molbev/msy074
Scholin C, Birch J, JensenMarin SR III, Massion E, Pargett D, Preston C, Roman B, Ussler W III (2017) The quest to develop ecogenomic sensors: a 25-year history of the environmental sample processor (ESP) as a case study. Oceanography (Wash DC) 30:100–113. https://doi.org/10.5670/oceanog.2017.427
Sefers S, Schmitz JE (2018) Molecular contamination and amplification product inactivation. Advanced techniques in diagnostic microbiology. Springer, Cham, Switzerland, pp 505–526
Seymour M, Durance I, Cosby BJ, Ransom-Jones E, Deiner K, Ormerod SJ, Colbourne JK, Wilgar G, Carvalho GR, de Bruyn M, Edwards F, Emmett BA, Bik HM, Creer S (2018) Acidity promotes degradation of multi-species environmental DNA in lotic mesocosms. Comm Biol 1:4. https://doi.org/10.1038/s42003-017-0005-3
Shaw JLA, Clarke LJ, Wedderburn SD, Barnes TC, Weyrich LS, Cooper A (2016) Comparison of environmental DNA metabarcoding and conventional fish survey methods in a river system. Biol Conserv 197:131–138. https://doi.org/10.1016/j.biocon.2016.03.010
Siegenthaler A, Wangensteen OS, Soto AZ, Benvenuto C, Corrigan L, Mariani S (2019) Metabarcoding of shrimp stomach content: Harnessing a natural sampler for fish biodiversity monitoring. Mol Ecol Resour 19:206–220. https://doi.org/10.1111/1755-0998.12956
Sigsgaard EE, Torquato F, Frøslev TG, Moore ABM, Sørensen JM, Range P, Hamadou RB, Bach SS, Møller PR, Thomsen PF (2019) Using vertebrate environmental DNA from seawater in biomonitoring of marine habitats. Conserv Biol 21:3–14. https://doi.org/10.1111/cobi.13437
Six KD, Kloster S, Ilyina T, Archer SD, Zhang K, Maier-Reimer E (2013) Global warming amplified by reduced sulphur fluxes as a result of ocean acidification. Nat Clim Change 3:975–978. https://doi.org/10.1038/nclimate1981
Song Y-K, Kim J-H, Won S-Y, Park C (2019) Possibility in identifying species composition of fish communities using the environmental DNA metabarcoding technique—with the preliminary results at urban ecological streams. J Korean Env Res Tech 22:125–138. https://doi.org/10.13087/kosert.2019.22.6.125
Spens J, Evans AR, Halfmaerten D, Knudsen SW, Sengupta ME, Mak SST, Sigsgaard EE, Hellström M (2016) Comparison of capture and storage methods for aqueous macrobial eDNA using an optimized extraction protocol: advantage of enclosed filter. Methods Ecol Evol. https://doi.org/10.1111/2041-210x.12683
Stiasny MH, Mittermayer FH, Sswat M, Voss R, Jutfelt F, Chierici M, Puvanendran V, Mortensen A, Reusch TBH, Clemmesen C (2016) Ocean acidification effects on Atlantic cod larval survival and recruitment to the fished population. PLoS ONE 11:e0155448. https://doi.org/10.1371/journal.pone.0155448
Stoeckle MY, Mishu MD, Charlop-Powers Z (2018) GoFish: a versatile nested PCR strategy for environmental DNA assays for marine vertebrates. PLoS ONE 13:e0198717–e198817. https://doi.org/10.1371/journal.pone.0198717
Sumaila UR, Cheung WWL, Lam VWY, Pauly D, Herrick S (2011) Climate change impacts on the biophysics and economics of world fisheries. Nat Clim Change 1:449–456. https://doi.org/10.1038/nclimate1301
Taberlet P, Coissac E, Pompanon F, Brochmann C, Wllerslev E (2012) Towards next-generation biodiversity assessment using DNA metabarcoding. Mol Ecol 21:2045–2050. https://doi.org/10.1111/j.1365-294x.2012.05470.x
Taberlet P, Bonin A, Coissac E, Zinger L (2018) Environmental DNA: for biodiversity research and monitoring. Oxford University Press, London
Takahara T, Minamoto T, Yamanaka H, Doi H, Kawabata Z (2012) Estimation of fish biomass using environmental DNA. PLoS ONE 7:e35868. https://doi.org/10.1371/journal.pone.0035868.s002
Takeuchi A, Sado T, Gotoh RO, Watanabe S, Tsukamoto K, Miya M (2019) New PCR primers for metabarcoding environmental DNA from freshwater eels, genus Anguilla. Sci Rep 9:1–11. https://doi.org/10.1038/s41598-019-44402-0
Tang CQ (2020) eDNA monitoring for migratory fish assemblages. Natural England Commissioned Report, Number 290
Tang CQ, Crampton-Platt A, Townsend S, Bruce K, Bista I, Creer S (2018) Development of DNA applications in Natural England 2016/2017. Natural England Comissioned Reports, Number 252 The eDNA Society (2019) Environmental DNA Sampling and Experiment Manual Version 2.1. The eDNA Society, Otsu, Japan
Thomsen PF, Willerslev E (2015) Environmental DNA —an emerging tool in conservation for monitoring past and present biodiversity. Biol Conserv 183:4–18. https://doi.org/10.1016/j.biocon.2014.11.019
Thomsen PF, Kielgast J, Iversen LL, Møller PR, Rasmussen M, Willerslev E (2012) Detection of a diverse marine fish fauna using environmental DNA from seawater samples. PLoS ONE 7:e41732. https://doi.org/10.1371/journal.pone.0041732.s001
Truelove NK, Andruszkiewicz EA, Block BA (2019) A rapid environmental DNA method for detecting white sharks in the open ocean. Methods Ecol Evol 10:1128–1135. https://doi.org/10.1111/2041-210x.13201
Tsuji S, Miya M, Ushio M, Sato H, Minamoto T, Yamanaka H (2020) Evaluating intraspecific genetic diversity using environmental DNA and denoising approach: a case study using tank water. Environ DNA 2:42–52. https://doi.org/10.1002/edn3.44
Turner CR, Uy KL, Everhart RC (2015) Fish environmental DNA is more concentrated in aquatic sediments than surface water. Biol Conserv 183:93–102. https://doi.org/10.1016/j.biocon.2014.11.017
Ushio M, Fukuda H, Inoue T, Makoto K, Kishida O, Sato K, Murata K, Nikaido M, Sado T, Sato Y, Takeshita M, Iwasaki W, Yamanaka H, Kondoh M, Miya M (2017) Environmental DNA enables detection of terrestrial mammals from forest pond water. Mol Eco Resour 17:e63–e75. https://doi.org/10.1111/1755-0998.12690
Ushio M, Hiroaki M, Masuda R, Sado T, Miya M, Sakurai S, Yamanaka H, Minamoto T, Kondoh M (2018a) Quantitative monitoring of multispecies fish environmental DNA using high-throughput sequencing. Metabarcoding Metagenom 2:e23297. https://doi.org/10.3897/mbmg.2.23297
Ushio M, Hsieh C-h, Masuda R, Deyle ER, Ye H, Chang C-W, Sugihara G, Kondoh M (2018b) Fluctuating interaction network and time-varying stability of a natural fish community. Nature 554:360–363. https://doi.org/10.1038/nature25504
Ushio M, Murata K, Sado T, Nishiumi I, Takeshita M, Iwasaki W, Miya M (2018c) Demonstration of the potential of environmental DNA as a tool for the detection of avian species. Sci Rep 8:1–10. https://doi.org/10.1038/s41598-018-22817-5
Valenzuela AEJ, Rey AR, Fasola L, Samaniego RAS, Schiavini A (2013) Trophic ecology of a top predator colonizing the southern extreme of South America: feeding habits of invasive American mink (Neovison vison) in Tierra del Fuego. Mamm Biol 78:104–110. https://doi.org/10.1016/j.mambio.2012.11.007
Hoorn B van der, Gittenberger A (2019) A multi-substrate metabarcoding approach for detecting non-indigenous species in a Dutch port. 43 pages. Naturalis Biodiversity Center, Leiden, the Netherlands
Walsh DA, Zaikova E, Hallam SJ (2009) Large volume (20L+) filtration of coastal seawater samples. J Vis Expdoi. https://doi.org/10.3791/1161
Weigand H, Beermann AJ, Čiampor F, Costa FO, Csabai Z, Duarte S, Geiger MF, Grabowski M, Rimet F, Rulik B, Strand M, Szucsich N, Weigand AM, Willassen E, Wyler SA, Bouchez A, Borja A, Čiamporová-Zaťovičová Z, Ferreira S, Dijkstra KD, Eisendle U, Freyhof J, Gadawski P, Graf W, Haegerbaeumer A, van der Hoorn BB, Japoshvili B, Keresztes L, Keskin E, Leese F, Macher JN, Mamos T, Paz G, Pešić V, Pfannkuchen DM, Pfannkuchen MA, Price BW, Rinkevich B, Teixeira MAL, Várbíró G, Ekrem T (2019) DNA barcode reference libraries for the monitoring of aquatic biota in Europe: Gap-analysis and recommendations for future work. Sci Total Environ 678:499–524. https://doi.org/10.1016/j.scitotenv.2019.04.247
Wingard GL, Lorenz JJ (2014) Integrated conceptual ecological model and habitat indices for the southwest Florida coastal wetlands. Ecol Indic 44:92–107. https://doi.org/10.1016/j.ecolind.2014.01.007
Yamahara KM, Preston CM, Birch J, Walz K, Marin R, Jensen S, Pargett D, Roman B, Ussler W, Zhang Y, Ryan J, Hobson B, Kieft B, Raanan B, Goodwin KD, Chavez FP, Scholin C (2019) In situ autonomous acquisition and preservation of marine environmental DNA using an autonomous underwater vehicle. Front Mar Sci 6:373. https://doi.org/10.3389/fmars.2019.00373
Yamamoto S, Minami K, Fukaya K, Takahashi K, Sawada H, Murakami H, Tsuji S, Hashizume H, Kubonaga S, Horiuchi T, Hongo M, Nishida J, Okugawa Y, Fujiwara A, Fukuda M, Hidaka S, Suzuki KW, Miya M, Araki H, Yamanaka H, Maruyama A, Miyashita K, Masuda R, Minamoto T, Kondoh M (2016) Environmental DNA as a ‘snapshot’ of fish distribution: a case study of Japanese jack mackerel in Maizuru Bay, Sea of Japan. PLoS ONE 11:e0149786–e149818. https://doi.org/10.1371/journal.pone.0149786
Yamamoto S, Masuda R, Sato Y, Sado T, Araki H, Kondoh M, Minamoto T, Miya M (2017) Environmental DNA metabarcoding reveals local fish communities in a species-rich coastal sea. Sci Rep 6:40368. https://doi.org/10.1038/srep40368
Yamanaka H, Minamoto T, Matsuura J, Sakurai S, Tsuji S, Motozawa H, Hongo M, Sogo Y, Kakimi N, Teramura I, Sugita M, Baba M, Kondo A (2016) A simple method for preserving environmental DNA in water samples at ambient temperature by addition of cationic surfactant. Limnology 18:233–241. https://doi.org/10.1007/s10201-016-0508-5
Yamanoue Y, Miya M, Matsuura K, Miyazawa S, Tsukamoto N, Doi H, Takahashi H, Mabuchi K, Nishida M, Sakai H (2009) Explosive speciation of Takifugu: another use of fugu as a model system for evolutionary biology. Mol Biol Evol 26:623–629. https://doi.org/10.1093/molbev/msn283
Zhang H, Xian W, Liu S (2015) Ichthyoplankton assemblage structure of springs in the Yangtze Estuary revealed by biological and environmental visions. PeerJ 3:e1186. https://doi.org/10.7717/peerj.1186
Zhang H, Yoshizawa S, Iwasaki W, Xian W (2019) Seasonal fish assemblage structure using environmental DNA in the Yangtze Estuary and Its adjacent waters. Front Mar Sci 6:671–710. https://doi.org/10.3389/fmars.2019.00515
Zhang S, Zhao J, Yao M (2020) A comprehensive and comparative evaluation of primers for metabarcoding eDNA from fish. Methods Ecol Evol (in press)
Zou K, Chen J, Ruan H, Li Z, Guo W, Li M, Liu L (2020) eDNA metabarcoding as a promising conservation tool for monitoring fish diversity in a coastal wetland of the Pearl River Estuary compared to bottom trawling. Sci Total Env 702:134704. https://doi.org/10.1016/j.scitotenv.2019.134704
Acknowledgements
We sincerely appreciate the Editorial Board of Fisheries Science for generously inviting one of us (MM) to write this review article. Yumi Terashima kindly gave us editorial advice throughout manuscript writing process. Comments from two anonymous reviewers were helpful for improving the manuscript. Masayuki Ushio kindly provided us with the latest information on empirical dynamic modeling (EDM) and its potential application to spatiotemporal analysis of fish communities based on eDNA metabarcoding data. Developments of the MiFish eDNA metabarcoding methods would not have been possible without tremendous support and guidance from Yukuto Sato during an earlier stage of the eDNA metabarcoding projects. A series of our studies on the MiFish eDNA metabarcoding methods has been supported as basic research by CREST (JPMJCR13A2) from the Japan Science and Technology Agency (JST); the Canon Foundation; the Environment Research and Technology Development Fund (4-1602) of the Ministry of the Environment, Japan; Japan Society for the Promotion of Science KAKENHI (JP19H03291), and Ministry of Education, Culture, Sports, Science and Technology; and the Ocean Resource Use Promotion Technology Development Program (JPMXD0618068274). Members of the first three projects (in particular those of CREST) have greatly contributed to the successful developments of MiFish eDNA metabarcoding technologies and their applications. Louis Bernachez, Stefano Mariani, Owen S. Wangensteen, and Meng Yao kindly provided us with information on their papers using MiFish primers. Rupert A. Collins, Hideyuki Doi, Kei-ichi Fukaya, Stefano Mariani, Shin-ichiro Oka, Hikaru Nakagawa kindly provided us with their original figures. We would like to thank Editage (www.editage.com) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Published with support by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant no. JP19HP2002.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miya, M., Gotoh, R.O. & Sado, T. MiFish metabarcoding: a high-throughput approach for simultaneous detection of multiple fish species from environmental DNA and other samples. Fish Sci 86, 939–970 (2020). https://doi.org/10.1007/s12562-020-01461-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12562-020-01461-x