
Abstract
The breakthrough CRISPR (clustered regularly interspaced short palindromic repeat)/Cas9-mediated genome-editing technology has led to great progress in monocot research; however, several factors need to be considered for the efficient implementation of this technology. To generate genome-edited crops, single guide (sg)RNA and Cas9 DNA are delivered into plant cells and expressed, and the predicted position is targeted. Analyses of successful targeted mutations have revealed that the expression levels, expression timing, and variants of both sgRNA and Cas9 need to be sophisticatedly regulated; therefore, the promoters of these genes and the target site positions are the key factors for genome-editing efficiency. Currently, various vectors and online tools are available to aid sgRNA design. Furthermore, to reduce the sequence limitation of the protospacer adjacent motif (PAM) and for other purposes, many Cas protein variants and base editors can be used in plants. Before the stable transformation of a plant, the evaluation of vectors and target sites is therefore very important. Moreover, the delivery of Cas9-sgRNA ribonucleoproteins (RNPs) is one strategy that can be used to prevent transgene issues with the expression of sgRNA and Cas proteins. RNPs can be used to efficiently generate transgene-free genome-edited crops that can reduce transgene issues related to the generation of genetically modified organisms. In this review, we introduce new techniques for genome editing and identifying marker-free genome-edited mutants in monocot crops. Four topics are covered: the design and construction of plasmids for genome editing in monocots; alternatives to SpCas9; protoplasts and CRISPR; and screening for marker-free CRISPR/Cas9-induced mutants. We have aimed to encompass a full spectrum of information for genome editing in monocot crops.
Similar content being viewed by others
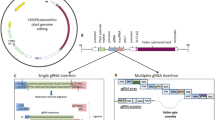
Design and Construction of Plasmids for Genome Editing in Monocots
The CRISPR/Cas9 system has been successfully used for genome editing in a variety of monocots, including rice (Oryza sativa), wheat (Triticum sp.), barley (Hordeum vulgare), and maize (Zea mays) (Feng et al. 2018; Gasparis et al. 2018; Hu et al. 2018; Kis et al. 2019; Okamoto et al. 2017; Wang et al. 2017; Zhang et al. 2016). To perform CRISPR/Cas9-mediated genome editing, the Cas9 endonuclease is guided by a single guide RNA (sgRNA) to recognize the complementary sequence and create double-strand breaks (DSBs), thereby generating a short deletion or insertion. Genome-edited plants can be generated either by stable or transient transformation. For stable transformations, the Agrobacterium-mediated transformation method is typically used to deliver transfer DNA (T-DNA) into the plant cell, where it is then inserted into the plant genome (Mikami et al. 2015a; Nandy et al. 2019). For transient transformations, particle bombardment and polyethylene glycol (PEG)-mediated methods are used to deliver plasmids or ribonucleoproteins (RNPs) into the plant cells (Woo et al. 2015; Svitashev et al. 2016; Zhang et al. 2016; Lin et al. 2018). Plasmids used for the stable genome editing of plants require a selection cassette, known as a sgRNA cassette, and a clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) cassette in the T-DNA region, while the selection cassette is not necessary for transient transformations (Fig. 1).
-
1.
Selection cassette: In monocots, several genes have served as useful selection markers for the efficient selection of transgenic plants, such as neomycin-phosphotransferase (NPTII), bar, mutated acetolactate synthase (ALS), plant phosphomannose isomerase, and hygromycin phosphotransferase (Hpt) (Miki and McHugh 2004; Hu et al. 2016). Among these genes, Hpt is the most widely used selection marker which confers tolerance to the herbicide hygromycin because several crops have a natural tolerance to kanamycin. Plant phosphomannose isomerase can also be used as a selectable marker for rice transformation (Hu et al. 2016). The expression of Hpt is usually driven by a strong constitutive promoter, such as maize Ubiquitin 1 (ZmUbi1), rice ACTIN 1, or Cauliflower mosaic virus (CaMV) 35S, for the ubiquitous expression of the antibiotic-tolerance gene (Mikami et al. 2015a).
-
2.
sgRNA cassette: sgRNA is a programmable 20-nucleotide (nt) sequence that recognizes the target DNA sequence and an invariant scaffold sequence (Ran et al. 2013), then directs the Cas nuclease to cleave the target sequence. Two factors are important for the function of sgRNA; promoter activity and the specificity of the sgRNA. A mixed dual promoter system is generally used in CRISPR/Cas9 system. In this system, Cas9 is directed by the RNA Polymerase II (Pol II) promoter while sgRNA expression is regulated by a Pol III promoter such as U6 or U3. To increase the transcription of the sgRNA, several monocot-specific U3 or U6 promoters have been cloned and used to direct the expression of the sgRNA, such as those from rice (Ma et al. 2015), maize (Qi et al. 2018), and wheat (Xing et al. 2014). In rice, the sgRNA driven by the OsU6 promoter produces more transcripts than when driven by the OsU3 promoter (Mikami et al. 2015a); however, several promoters, including OsU3, OsU6a, OsU6b, and OsU6c, have been used to direct the expression of sgRNA, and all of them could effectively direct genome editing with mutation rates of 81.4–90.0% (Ma et al. 2015; Shan et al. 2013; Xie and Yang 2013; Zhou et al. 2014). In addition to promoters, the specificity of the sgRNA for its target DNA sequence is another factor that affects the efficiency of genome editing. Several resources available on the web can be used to design highly specific sgRNAs for use with the CRISPR/Cas9 system (Table 1; modified from Zhang lab, https://zlab.bio/guide-design-resources). Nevertheless, even if these 20-nt sequences perfectly match the target gene, some sgRNAs do not work well. The online tools listed in Table 1 recommend targets with a low risk of an off-target match, but not all predicted target sequences may result in an efficient mutation. In addition, targeted DNA sequences with GC contents higher than 50% have higher genome-editing efficiencies (88.5–89.6%) than those with GC contents lower than 50% (77.2% efficiency) (Ma et al. 2015). Successive Ts in 20-nt target sequence is not good when sgRNA expression is driven by the U3 or U6 promoters (Wu et al. 2014).
-
3.
Cas9 cassette: Aspects of the Cas9 cassette that affect the mutation rate during genome editing include the expression level and codon usage of Cas9 (Ma et al. 2015; Xie et al. 2015). Several strategies have been conducted to improve the expression of Cas9, including the use of a strong constitutive promoter, the addition of a translational enhancer, and the addition of nuclear localization signals. Constitutive promoters have been used to direct the expression of Cas9, including the ZmUbi1 and 35S. The Cas9 nucleases from different bacteria may have variations in the protospacer adjacent motif (PAM) sequence that they require for cleavage; therefore, many Cas9 homologs with different PAM requirements have been isolated from different bacteria. These Cas proteins are introduced in ‘Alternatives to SpCas9’ section. Streptococcus pyogenes Cas9 (SpCas9) is the most common Cas used in CRISPR/Cas9-mediated genome editing. It has been codon-optimized for maize (Xing et al. 2014) and rice (Miao et al. 2013) to improve its expression levels in these monocots.

Schematic of the T-DNA region in a binary vector for genome editing in monocots. PPol III: Polymerase III promoter. PPol II: Polymerase II promoter. PUbi1: maize Ubiquitin 1 promoter and the first exon. Hpt: Hygromycin phosphotransferase. (Modified from Howells et al. 2018)
Strategies for Multiplex Genome Editing in Monocots
One advantage of the CRISPR/Cas9 system over other crop-breeding strategies is its flexibility for multiplex genome editing (Wang et al. 2017; Wang et al. 2018). The editing of multiple functional genes allows for the rapid improvement of multiple agronomic traits at one time, while editing the cis-acting elements of a promoter affects transcriptional regulation. The deletion of larger fragments between two sgRNA-targeted sites on the same chromosome following the generation of multiple DSBs has been reported in many species. The CRISPR/Cas9 system has been used to delete DNA fragments ranging from dozens of bases to greater than 1 Mb (Mali et al. 2013; Shan et al. 2013). In addition, targeted deletions of 10 bp to over 200 kb between two target sites have been reported in rice (Mikami et al. 2016; Zhou et al. 2014). The elimination of the Tos17 retrotransposon using CRISPR/Cas9 was reported in rice, providing a rapid breeding route for making reverting the agronomically important genes that have been inactivated by the insertion of transposable elements (Saika et al. 2019).
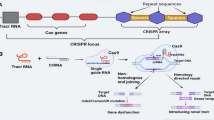
Multiplex genome editing can be achieved by the simultaneous delivery and expression of multiple sgRNAs; however, since most CRISPR/Cas9 components are transferred into plants via Agrobacterium-mediated transformations, an efficient plasmid construction strategy is required. Traditionally, multiple sgRNA expression cassettes (including a Pol III promoter, a sgRNA, and a terminator) can be stacked into one T-DNA (Fig. 2a); however, this may increase the cloning difficulties due to the limited restriction sites available, and the fact that large T-DNAs may decrease the transformation efficiency. Alternative strategies have therefore been developed to facilitate multiplexed genome editing in plants. These alternative strategies are based on the expression of multiple sgRNAs as a single transcript, after which multiple functional sgRNAs are generated following the processing of the transcripts by exogenous ribozymes (Gao and Zhao 2014), Csy-type ribonuclease 4 (Csy4) (Cermak et al. 2017), or the plant endogenous transfer RNA (tRNA)-processing system (Xie et al. 2015). The endogenous tRNA-processing system exists in almost all organisms and has been successfully used to perform multiplex genome editing in rice (Xie et al. 2015). In the tRNA-processing system, the Pol III promoter is used to direct the expression of a single synthetic gene containing multiple tRNA-sgRNAs or a polycistronic tRNA-sgRNA (PTG) gene. The PTG gene can be generated using the Golden Gate assembly method (Lowder et al. 2015) (Fig. 2b). After transcribing the PTG, the endogenous RNases specific to tRNA would recognize the tRNA components and cleave the individual sgRNAs from the PTG transcript. The resulting sgRNAs would then guide Cas9 to multiple target sites for genome editing. Many toolboxes are currently available in the public database Addgene (Cermak et al. 2017; Castel et al. 2019; Hahn et al. 2019), from where these vectors can be purchased by academic researchers.

Diagram of a plasmid construct used for multiplex genome editing. a Stacking of multiple sgRNA expression cassettes in one T-DNA. P Pol III: Polymerase III promoter. b Cloning of four tRNA-gRNAs into the CRISPR/Cas9 binary vector using a single Golden Gate ligation. (Modified from Kurata et al. 2018)
Reducing off-Target Mutations and Lethality in Monocots
Several reports have indicated that constitutively expressed Cas9 produces an excess of sgRNA-Cas9, which may increase the incidence of genome-wide off-target mutations (Hsu et al. 2013; Hu et al. 2018; Pattanayak et al. 2013; Svitashev et al. 2016). By contrast, conditionally or transiently expressing Cas9 significantly reduces the frequency of off-target mutations (Srivastava et al. 2017; Zhang et al. 2014). Increasing evidence has indicated that conditionally expressing Cas9 at the plant regeneration phase can markedly improve genome-editing efficiency, and this kind of conditional targeting could avoid the lethal phenotype caused by the cleavage of genes essential for development (Srivastava et al. 2017; Zhang et al. 2014). The promoter of the gene encoding heat-shock protein 17.5E (Hsp17.5E) from soybean (Glycine max) has been used to direct the expression of Cas9 for genome editing in rice. The mutation frequency was 16% and 50–63% among the transgenic lines before and after a heat treatment, respectively (Nandy et al. 2019). In maize, Cas9 driven by the meiosis-specific Disrupted Meiotic cDNA 1 (ZmDMC1) promoter was able to generate up to 66% homozygous or bi-allelic mutants, and no off-target mutations were detected using whole-genome sequencing (Feng et al. 2018). The vector delivery of preassembled Cas9-sgRNA RNPs instead of DNA has been reported to significantly reduce the frequency of off-site cleavage in both protoplast and zygote systems (Toda et al. 2019). In maize embryo cells, the delivery of DNA vectors containing Cas9 and sgRNA showed a high frequency of off-site mutations (50%) when compared with the Cas9-sgRNA RNP complex (0%) (Svitashev et al. 2016). In most plant species, the isolation, cultivation, and regeneration of protoplasts remains a challenge; however, recent work demonstrated the direct delivery of a Cas9-sgRNA RNP into rice zygotes using the in vitro fertilization of isolated gametes, resulting in a targeted mutation rate of 14–64%. This protoplast-free zygote system makes the RNP-mediated genome-editing system much easier to perform, which could be a potential avenue for crop improvement in many monocot species (Toda et al. 2019).
Alternatives to SpCas9
The CRISPR/Cas systems are divided into two classes. The Class 1 systems possess multiple Cas protein subunits, whereas the Class 2 systems utilize a single, multifunctional protein effector (Shmakov et al. 2015). Class 2 CRISPR systems are further divided into types II, V, and VI. Along with the commonly used type II effector SpCas9, other orthologs with RNA-guided site-specific nuclease (SSN) activity have been engineered for use as tools in the genome editing of eukaryotic cells. These orthologs include proteins from Staphylococcus aureus (SaCas9), Streptococcus thermophilus (St1Cas9), Francisella novicida (FnCas9), Neisseria meningitidis (NmCas9), Brevibacillus laterosporus (BlCas9), and Campylobacter jejuni (CjCas9) (Cong et al. 2013; Hou et al. 2013; Sampson et al. 2013; Ran et al. 2015; Karvelis et al. 2015; Kim et al. 2017a; Chatterjee et al. 2018; Edraki et al. 2019). These Cas9s can be a welcome complement to the editing ability of SpCas9 in plants. Here, we describe plant genome-editing tools developed from these orthologs and discuss their advantages for research in botany.
Type II Cas9 Systems
Type II CRISPR systems are abundant in prokaryotic organisms and are the principal resource used to develop gene-editing tools. In addition to the first-reported SpCas9, several studies have described the Cas9 ortholog-mediated genetic engineering of plants. SaCas9 is most frequently used in place of SpCas9 and provides comparable editing efficiency for eukaryotic genomes. Compared with SpCas9, SaCas9 may reduce the delivery barrier of the CRISPR system because of its smaller size (Friedland et al. 2015; Ran et al. 2015). SaCas9 and its engineered variant SaKKH (E782K/N968K/R1015H) (Kleinstiver et al. 2015), which relaxes the canonical NNGRRT PAM of SaCas9 to NNNRRT, have been used to achieve the efficient targeted mutagenesis of Arabidopsis thaliana, tobacco (Nicotiana tabacum), rice, and citrus (Citrus sp.) (Kaya et al. 2016; Steinert et al. 2015; Jia et al. 2017; Qin et al. 2019).
SaCas9 and SaKKH have also been used to develop plant base editors (BEs), including the cytosine BE (CBE) responsible for a C·G to T·A conversion and the adenine BE (ABE) responsible for the reverse substitution of A·T to G·C. Similar to the BEs derived from SpCas9, the SaCas9 BEs have been successfully used to induce specific base conversions in the rice genome (Hua et al. 2018; Hua et al. 2019; Qin et al. 2019). Notably, the editing windows of the SaCas9 BEs are much broader than those of the SpCas9 BEs, possibly due to differences in the formation of the R-loop complex (Kim et al. 2017d). Compared the conventional CRISPR/Cas9-targeted mutagenesis system, BEs developed from SpCas9 tend to be less efficient at editing certain targets and less flexible for specific nucleotides. Because of their recognition of different PAMs and the enlargement of the editing window, the SaCas9 BEs have provided alternative tools for precise genome editing in plants.
Another Cas9 ortholog, St1Cas9, has been used to induce mutations in the Arabidopsis gene ALCOHOL DEHYDROGENASE 1 (ADH1) (Steinert et al. 2015), while FnCas9 was used to confer molecular immunity against RNA viruses in tobacco and Arabidopsis (Zhang et al. 2018).
Type V Cas Systems
The CRISPR RNA (crRNA)-guided SSN activity of the type V Cas system, which includes Cas12a and Cas12b, also generates DSBs. Several Cas12a (formerly Cpf1) orthologs, such as F. novicida Cas12a (FnCas12a), Acidaminococcus sp. Cas12a (AsCas12a), and Lachnospiraceae sp. Cas12a (LbCas12a), have been engineered as a class of genome-editing tools distinct from the Cas9 system (Zetsche et al. 2015). The Cas12a proteins induce staggered DSBs at sites distal to a 5′ T-rich PAM, generating relatively longer deletions via the non-homologous end joining repair pathway in various plant species, including rice (Endo et al. 2016a; Begemann et al. 2017; Hu et al. 2017; Kim et al. 2017c; Tang et al. 2017; Xu et al. 2017). Similar to the SpCas9 systems, transgene-free Cas12a-mediated mutants can be generated by simply segregating the T-DNA fragment in the transition from the T0 to T1 generations (Xu et al. 2017). In addition, mixing RNP with Cas12a and crRNA has allowed transgene-free genome editing in soybean and tobacco (Kim et al. 2017b). Interestingly, the Cas12a proteins have different sensitivities to temperature in plants (Malzahn et al. 2019); therefore, the optimization of the incubation temperature used in crop culture systems may facilitate a higher editing efficiency when using Cas12a proteins.
Cas12a proteins not only induce DSBs, but also process their own pre-crRNA for maturation. This RNase activity provides a simple pattern by which single or multiplex crRNA(s) can be expressed from the same transcription unit of Cas12a, facilitating the highly efficient genome editing of plants (Wang et al. 2017; Ding et al. 2018; Wang et al. 2018; Xu et al. 2019). In the genome editing mediated by the CRISPR/Cas9 system, RNA could be used as a repair template for homology-directed repair (Butt et al. 2017). The self-processing activity of Cas12a means that homologous recombination-mediated gene replacements can be generated using the FnCas12a system (Li et al. 2019). In mammalian cells, CBEs were also developed using Cas12a proteins to facilitate precise C-to-T conversions in A/T-rich regions (Li et al. 2018); however, similar systems have not yet been reported in plants. The engineered Alicyclobacillus acidiphilus Cas12b (AaCas12b) has been used to edit mammalian genomes under a wide range of temperatures (31–59 °C; Teng et al. 2018), but it is still unclear whether the high-temperature-preferring Cas12b systems function in plants.
Type VI Cas Systems
The type VI CRISPR proteins, such as Cas13a, can target and cleave the target RNA under the guidance of crRNA. The heterologous expression of Leptotrichia wadei Cas13a (LwaCas13a) causes the knockdown of target RNAs in mammalian and plant cells (Abudayyeh et al. 2017). Similarly, in both dicot and monocot plants, the transient or stable expression of a Cas13a-crRNA complex results in the targeting and degradation of the RNA transcripts of endogenous genes and foreign reporters (Aman et al. 2018; Zhang et al. 2019a, 2019b). To precisely edit nucleotides in RNA, the ADAR2 adenosine deaminase or an evolved ADAR2 cytidine deaminase was fused to a catalytically inactive Cas13a (dCas13a), generating C-to-U and A-to-I RNA editors in mammalian cells (Cox et al. 2017; Abudayyeh et al. 2019). RNA editing, especially organellar RNA editing, plays an irreplaceable role in plant growth and development; therefore, similar Cas13a tools are highly anticipated to facilitate related research in plants.
Protoplasts and CRISPR
Protoplast Isolation and Validation
Many different vectors are available for the expression of Cas proteins using different promoters. Researchers should choose a suitable vector based on their needs, and importantly should evaluate the target sites and constructs using a transient expression system before performing the stable transformation to reduce the time and labor required. Protoplasts are often used for plant science investigations (Marx 2016), and the convenience and speed of their transfection means they are an attractive model in which to assess the mutagenesis efficiency of a CRISPR/Cas system, including the validation of Cas protein codon optimizations or modifications, sgRNA target sites, the promoters used for sgRNA and Cas9 proteins, and different vector designs (Andersson et al. 2017; Butt et al. 2017; Cermak et al. 2017; Endo et al. 2019; Hsu et al. 2019; Hsu et al. in preparation; Li et al. 2013; Li et al. 2018; Liang et al. 2014; Lowder et al. 2015; Shan et al. 2013; Sun et al. 2015; Zong et al. 2017, 2018). Rice, tobacco, and soybean protoplasts have been used to analyze Cas9 and Cas12a (Kim et al. 2017; Tang et al. 2017), while Cas13a was examined in rice protoplasts (Abudayyeh et al. 2017). Protoplast transfection can also be used to evaluate the efficiency of the use of RNPs (Andersson et al. 2018; Hsu et al. in preparation; Kim et al. 2017; Malnoy et al. 2016; Woo et al. 2015).
In comparison with the number of articles on stable rice CRISPR transformations, very few have been published using protoplasts for validation. One of the reasons for this is the efficiency of protoplast transfection and isolation. We have evaluated the different methods of protoplast transfection in rice, including PEG, electroporation, and liposome delivery, which revealed that the PEG method has highest transfection efficiency (Lin and Hsu, personal comm.). The main bottleneck restricting the application of rice protoplasts in the evaluation of genome-editing reagents is thus considered to be protoplast isolation. We previously improved the protocol for isolating Arabidopsis protoplasts (Tape Arabidopsis Sandwich; Wu et al. 2009) to facilitate their use for various purposes, and we are currently trying to develop a convenient rice protoplast isolation method, modifying two of the steps reported in previous rice mesophyll protoplast isolation protocols (Chen et al. 2006; Zhang et al. 2011).
First, based on our observations, seedlings cut cross-sectionally retain more cells within the leaf sheath after digestion, meaning the mesophyll cells can be digested but not released. The veins in rice run parallel to each other; therefore, we changed the cut direction from a random or cross-sectional cut to a longitudinal cut parallel to the veins, which allows the enzyme solution to more easily access the cells and provides more surface area from which the protoplasts are released. To increase the efficiency of this process, multiple blades were fixed by a holder, creating a tool that can increase the speed of cutting (Lin et al. 2018). Second, we also assessed the enzyme components required for the digestion solution when using longitudinal cuts, revealing that the less expensive Cellulase R10 (Yakult, Japan) could be used in place of the more expensive Cellulase RS (Yakult, Japan) solution typically used for rice (Zhang et al. 2011). This protocol can also be applied to other Poaceae species, including wheat, bamboo (Bambusa oldhamii), millet (Setaria italica), and maize (Lin et al. 2018). We believe this convenient method will be of benefit not only in rice, but also for Poaceae crop research in general. We have also established protoplast isolation protocols for use with the Solanaceae and Cruciferae (Hsu et al. submitted). The protoplasts isolated using these methods can be used to more rapidly evaluate the genome-editing efficiency in crops (Lin et al. 2018).
Single Protoplast Analysis
When using protoplasts to validate CRISPR efficiency, more than 100,000 protoplasts are typically used in each transfection experiment. DNA is extracted from the pooled protoplasts to enable the amplification of the target region using PCR. The pooled protoplasts also contain unedited DNA, making mutations difficult to detect if the mutagenesis efficiency is low (Lin et al. 2018). Mutagenesis efficiency can also be assessed using next-generation sequencing, from which the density ratio of the target fragments or the editing percentage can be determined. This method is relatively accurate, but it is expensive and time consuming. Recently, a convenient and reliable protocol for evaluating CRISPR mutagenesis efficiency from a single cell was established (Lin et al. 2018). In this approach, single cells can be isolated from various species and subjected to two rounds of PCR amplification and enzyme digestion without DNA purification to identify successful mutants. The mutated sequences and the mutation efficiency could thus be analyzed directly, allowing even low-efficiency mutation events to be detected in maize. This single-cell analysis technique could be used to improve the precision and application range of CRISPR gene editing using protoplasts.
Although these convenient methods could be used for mesophyll isolation to provide the materials for the evaluation of CRISPR editing efficiency and accuracy, it is important to consider the correlation between CRISPR efficiencies using mesophyll protoplasts and stable transformation, particularly in rice, for which a callus is typically used as the material for stable transformation (Kaya et al. 2016). Certain target sites were found to have a high CRISPR efficiency in stable transformation experiments, but their use did not result in mutations in the mesophyll protoplasts (Toki, Endo, and Lin, personal comm.). We are therefore working on developing a rice protoplast isolation protocol using callus materials, which will enable the assessment of the gene-editing relationship between these protoplasts and the stably transformed calli.
CRISPR-Edited Protoplast Regeneration
In addition to the validation of transformation efficiency, mutated protoplasts have the advantage of being able to regenerate into entire mutant plants. Protoplasts isolated from meristematic tissues or totipotent cells were first used for plant regeneration in the early 1970s (Takebe et al. 1971), and just a few years later, researchers used protoplasts as materials for plant transformation (Marton et al. 1979). The progeny of N. tabacum regenerated from transformed protoplasts displayed the mutant phenotype, indicating that the transgenic tumor markers (octopine and nopaline) were inherited through meiosis (Wullems et al. 1981a; Wullems et al. 1981b). Monocot protoplast regeneration (Abdullah et al. 1986; Fujimura et al. 1985; Rhodes et al. 1988a) and transformation (Rhodes et al. 1988b; Shimamoto et al. 1989; Toriyama et al. 1988) protocols have also been established.
Cas proteins and sgRNAs are sufficient for CRISPR/Cas genome editing and are no longer required once the genes have been edited. Transient expression or the direct delivery of sgRNAs and Cas proteins into the cells is sufficient for editing; therefore, the DNA encoding the Cas proteins and sgRNAs does not need to be integrated into the genome for their continued expression, making the plants regenerated from these edited cells transgene-free. In 2015, Prof. Jin-Soo Kim’s group published a milestone article using protoplasts (Woo et al. 2015), in which RNP was used as the genome-editing reagent to edit lettuce (Lactuca sativa) protoplasts, which were subsequently regenerated into transgene-free plants.
There are several advantages to generating transgene-free edited crops using protoplasts:
-
1.
Protoplast transformation can be applied to edit hybrid and long-juvenile-phase crops, which are typically propagated using vegetative methods. Traditionally, the use of Agrobacterium tumefaciens-mediated or other stable transformation techniques means the transgene (selection markers, sgRNA, and Cas9) must be integrated into the genome. In inbred crops, such as rice, this transgene can be removed through crossing; however, this causes the segregation of the desired traits in the offspring. This separation issue also occurs in systems with a low editing efficiency, in which the transformants must be crossed in order to obtain the homozygous genotype. By contrast, homozygous edited crops can be achieved from heterozygous edited protoplasts using a second transfection (Hsu et al. 2019). Several methods are available for achieving this goal, including the delivery of RNP into the callus using a biolistic approach (Liang et al. 2017, 2019) or the transient expression of Cas9 and sgRNA using an Agrobacterium-mediated transfection (Chen et al. 2018); however, the gene-editing/mutation efficiencies of these methods are low (less than 10%). The protoplast transformation strategy had a higher efficiency; for example, over 50% of tobacco protoplasts were mutagenized using this technique, which was similar to the numbers transformed during the efficiency evaluation without antibiotic selection.
-
2.
Protoplast transformation can deliver more than one reagent. The current Agrobacterium vectors used for stable transformations are multiplex sgRNAs. The genes encoding Cas proteins are large, and no multiplex Cas protein vectors are currently available. Three different Cas protein vectors (SaCas9, FnCas12a, and nCas9-Target-AID) can be delivered into a single protoplast to edit three different target sites without interference (Hsu et al. 2019). The introduction of these three Cas protein vectors requires three subsequent Agrobacterium-mediated transformations, the crossing of individual mutants, or the co-transformation of Agrobacterium harboring independent Cas9s and different selection markers.. In addition to sgRNA and Cas proteins, donor DNA fragments are also required for knock-in genome editing, and must be delivered using Agrobacterium (Endo et al. 2016b; Miki et al. 2018;, Wolter and Puchta 2019). Using these protocols, only a few copies of donor DNA can be used in genome editing, which may be one of the reasons for the low efficiency of knock-in genome editing. Although multiple T-DNAs can be delivered into plant nuclei to increase the editing efficiency, this also increases the difficulty of removing the T-DNAs to produce marker-free plants. In protoplasts, however, the donor DNA can be delivered in microgram quantities, which may help increase the knock-in efficiency. Not only can the sgRNAs, Cas proteins, and donor DNAs be more easily delivered into protoplasts, but the application time and amounts of genome-editing reagents can be controlled to improve the editing efficiency.
-
3.
Using RNPs, the issues of promoter and codon modification in different species can be solved. The original Cas proteins were obtained from microorganisms; therefore, their codons must be modified for vector construction in plant species, and their expression must be driven by plant promoters. These issues have been investigated in Arabidopsis, resulting in the identification of a T-DNA architecture causing homozygous mutations in the first generation after transformation (Castel et al. 2019); however, the transcription and translation of these genes can still be problematic in different target crops. The resulting amount of Cas proteins in the cell can cause a low editing efficiency; however, in protoplasts, RNPs can be used to solve this problem and provide a higher delivery efficiency for transfection.
-
4.
With the exception of Arabidopsis, most Agrobacterium-mediated transformation protocols were performed using a tissue culture platform. In dicots, the regenerated plants were derived using organogenesis, meaning they were derived from multiple cells. Many edited transformants are therefore chimeric (Kaya et al2016). To validate their edited sequences, the transformant DNAs were sequenced. Diploids contain two alleles in each cell; therefore, if the transformants contained more than two alleles they were determined to be chimeric (Kaya et al. 2016). If the edited alleles are not present in the reproductive organs, the alleles cannot be passed on to the progeny. In contrast, protoplasts are single cells that are edited before the first cell division is completed. The regenerates are then derived from a single edited protoplast, meaning all cells have same genomic background, which enables the edited alleles to be transmitted to the next generation. In our previous studies, non-chimeric regenerates were derived from protoplasts edited using the Cas proteins Cas9, Cas12a, and Target-AID, and the genotypes were inherited in a Mendelian manner (Hsu et al. 2019). This phenomenon was also reported in lettuce (Woo et al. 2015).
RNPs (Andersson et al. 2018; Woo et al. 2015) and plasmids (Andersson et al. 2017; Zong et al. 2018) have been delivered into lettuce and potato (Solanum tuberosum) protoplasts, which were subsequently regenerated into transgene-free genome-edited plants. In our lab, we established N. tabacum (Hsu et al. 2019; Lin et al. 2018), rapid cycle brassica (Brassica oleracea), wild tomato (Solanum peruvianum), and N. benthamiana (Lin et al. in preparation) protoplast regenerations, and used these systems to establish RNP and plasmid DNA gene-editing platforms (Hsu et al. in preparation). Some issues are yet to be resolved in protoplast regeneration during genome editing, however:
-
1.
The regeneration protocol is difficult to establish. Only a few protocols for genome editing and protoplast regeneration have been developed, all of which were achieved in dicots (Andersson et al. 2017, 2018; Hsu et al. 2019; Jin et al. 2019; Lin et al. 2018; Tuncel et al. 2019; Woo et al. 2015). Many protoplast regeneration protocols are available in other species, including rice (Shimamoto et al. 1989; Toriyama et al. 1988) and other important Poaceae species (Rhodes et al. 1988a, 1988b). Recent discoveries have elucidated the mechanisms by which plants can regenerate, which could be applied to improve protoplast regeneration in the future (Lowe et al. 2016). In rice, it is possible to solve the lack of an efficient protoplast regeneration protocol using an alternative method; Cas9-sgRNA RNPs could be directly delivered into rice zygotes, which can then be cultured into mature plants (Toda et al. 2019). A total of 14–64% of the transgene-free plants obtained using this method were found to contain the target mutations.
-
2.
Unexpected mutations can occur during protoplast regeneration. When 15 protoplast potato regenerants were sequenced, they were found to include a variety of mutations, including insertions/deletions, chromosome rearrangements, and aneuploidy (Fossi et al. 2019). Indeed, somaclonal mutations occur in all tissue culture strategies, including micropropagation and somatic embryogenesis. In our experience using bamboo, somaclonal mutations occurred after a long-term subculture (Lin and Chang 1998; Lin et al. 2006, 2007; Liu et al. 2007). Using a shorter period for the protoplast regeneration or reducing the amount of supplemental plant growth regulators provided may reduce this mutation rate (Lin and Hsu, personal comm.). CRISPR/Cas can also introduce off-target mutations, with the mutation rate dependent on the CRISPR/Cas system; for example, in rice, CBE but not ABE induces genome-wide off-target mutations (Jin et al. 2019). Fortunately, off-target mutations do not have a major impact on crop breeding like they do in medical applications (Tang et al. 2019); rather, they simply result in more than one edited transformant for each transformation. In addition, mutation breeding is a strategy used in traditional breeding. We can select the transformants with good traits and use these edited lines as parental lines during crop production. In our opinion, although mutations can occur during regeneration, the protoplast regeneration techniques currently available are useful tools for transgene-free genome editing.
Screening for Marker-Free CRISPR/Cas9-Induced Mutants
Genome editing is widely used produce new genetic variants in plants. Several approaches for creating genome-edited crops have been developed, including the CRISPR/Cas system, which can be used with various tissue types including protoplasts (as described in the previous section), callus, leaf discs, and germline cells. Agrobacterium-mediated, PEG-mediated, particle bombardment, and virus infection transformation methods are commonly used for the delivery of the CRISPR/Cas system into plants, resulting in stable or transient expression patterns.
The transient expression of the CRISPR/Cas system can deliver DNA-based sgRNA and Cas9 RNP sequences or the proteins themselves (Chen et al. 2018; Zhang et al. 2016). In a DNA-based CRISPR/Cas9 system, recombinant DNA can be transferred to the plant using Agrobacterium- or PEG-mediated transformations or particle bombardment, eliminating the need for herbicide or antibiotic selection steps. This method allows the expression of the CRISPR/Cas9 plasmid without requiring its integration into the plant genome. This approach can reduce regeneration time via tissue culture while producing mutation frequencies similar to the stable expression of the CRISPR/Cas9 vector at the target site. More than 86% of wheat T0 mutants generated using this technique were transgene free (Zhang et al. 2016). In the Transgene-free CRISPR/Cas9 system, RNPs are assembled in vitro and directly delivered into the protoplasts using a PEG fusion approach. The RNP complex directly targets the recognized sequences and induces DSBs (Park and Choe 2019; Woo et al. 2015). The binding of the RNP complex to the target DNA is tight, and the half-life for dissociation is slow (more than 6 hours in vitro) (Didovyk et al. 2016). After dissociation, the RNP complex is degraded quickly in the cell.
The majority of genome-edited plants involve the stable integration of the CRISPR/Cas9 system into the plant genome. CRISPR/Cas9 DNA is delivered by methods similar to those employed for transient expression, followed by a herbicide or antibiotic selection of successful transformants containing a marker gene. Antibiotic and herbicide resistance markers have been widely used in plant biotechnology (Wilmink and Dons 1993); however, genome-integrated CRISPR/Cas9 carries a risk of increased off-target effects and requires researchers to follow the current regulations for typical genetically modified crops. To bypass these strict biosafety regulations, CRISPR/Cas binary vectors containing selection markers or foreign DNA can be segregated in the progeny by self-pollinating or crossing the transformants (Gao et al. 2016). The PCR amplification of vector sequences can be used to verify the presence of foreign DNA in the genome. A rapid method using antibiotics to identify marker-free genome-edited plants was also reported recently (Wu et al. 2019). Leaf sections from the T1 progeny of genome-edited rice were incubated with hygromycin B, an antibiotic commonly used for the positive selection of transgenic plants. In the presence of hygromycin B, the genome-edited rice plants that did not retain CRISPR/Cas vectors in their genome could produce reactive oxygen species (e.g., H2O2) in their mitochondria and chloroplasts (Oung et al. 2015). These elevated H2O2 levels can be visualized directly using 3,3’diaminobenzidine (DAB) staining (Wu et al. 2019). This approach can be easily applied to most monocot species.
Identification of CRISPR/Cas9-Induced Mutations
Following the delivery of the CRISPR/Cas system into monocot calli or other tissues, a T0 generation of plants harboring edited genes is regenerated. Three categories of site-directed nuclease systems (SDN1–3) are employed in genome-editing techniques (Podevin et al. 2013). SDN1 relies on the most common endogenous processes of non-homologous end-joining to repair DSBs in the plant DNA. This process is error prone, and may result in random mutations at the break site (Bortesi and Fischer 2015). SDN2 involves homology-directed repair using one or a few nucleotides as a template (EFSA Panel on Genetically Modified Organisms 2012). SDN3 uses the same repair mechanism as SDN2 but with a longer nucleotide template. Unlike SDN1, the repair processes used by SDN2 and SDN3 is not random, and does not cause substitutions, insertions, or deletions at the repair sites.
Researchers have developed several methods to increase the efficiency of screening large numbers of mutants (Table 2). These methods can detect on-target or off-target variants and include the restriction enzyme (RE) cleaved amplified polymorphic sequences (CAPS) assay (Shan et al. 2014), RE site created assays (Hodgens et al. 2017), T7 endonuclease I assays (T7E1) (Vouillot et al. 2015), polyacrylamide gel electrophoresis (PAGE)-based genotyping assay (Zhu et al. 2014), high-resolution melting analysis (HRM) (Thomas et al. 2014), PCR- and labeling-based method (Biswas et al. 2019), and annealing at critical temperature PCR (ACT-PCR) (Hua et al. 2017).
The CAPS, indCAPS, and T7E1 cleavage assays for identifying gene-edited mutants are based on an enzymatic approach. Wild-type and mutant sequences are amplified using PCR then subjected to enzyme digestion. A typical CAPS assay can be used if a RE cutting site is present at the CRISPR target site, which is disrupted once the sequences are mutated; however, RE sites are not always present within target regions. A similar dCAPS assay has been developed for genome regions not possessing different RE sites between the wild type and the mutant. This dCAPS assay introduces or disrupts a RE site near the mutation site by amplifying several mismatched nucleotides, which greatly increases the flexibility for selecting the sgRNA target sites. T7 endonuclease 1 can be useful for digesting the mismatched heteroduplexes formed between the wild-type and mutated strands; however, this method lacks the sensitivity to distinguish between homozygous mutants and the wild type.
Other PCR-based analyses include PAGE, HRM, ACT-PCR, and PCR−/labeling-based assays. To distinguish genome-edited mutations from wild-type target sequences, target region amplicons are migrated on a native polyacrylamide gel. Homoduplex DNA migrates faster than heteroduplex DNA. To distinguish homozygous mutants from the wild type, unknown amplicons can be pre-mixed with wild-type amplicons before the assay. The migration pattern of the pre-mixed homozygous mutant and wild type will be similar to that of the heterozygous mutant. HRM is a fluorescence-based technique for determining the differences in the melting temperatures of heteroduplex and homoduplex DNA fragments. This technique can detect differences as small as 0.1 °C; however, its sensitivity is influenced by the amplicon length and variation of the mutated sequence. ACT-PCR assay can also distinguish homozygous mutant sequences at the target site using optimal annealing temperature and specific primers; however, the assay requires the design of specific primers and is time consuming and labor intensive. A simple PCR- and amplicon labeling-based method was recently used to identify the CRISPR/Cas9-generated mutants in rice (Biswas et al. 2019). This approach requires two pairs of primers or a FAM-labeled allele-specific primer. The sensitivity, precision, and reliability of the FAM-labeled method allows for the detection of indels with a high sensitivity (down to ±1 bp).
Although all these PCR-based analyses enable the effective, accurate, and economical screening of CRISPR/Cas9-generated mutants, the identification of sequence changes resulting from SDN1-generated mutations requires the Sanger sequencing of amplicons generated from the target region. In addition, whole-genome sequencing is a powerful tool for identifying not only on-target and off-target mutations, but also transgene-free plants produced by genome editing. The only drawback of whole-genome sequencing is its cost and time requirement.
Availability of data and materials
Not applicable.
References
Abdullah R, Cocking EC, Thompson AJ (1986) Efficient plant regeneration from rice protoplasts through somatic embryogenesis. Nat Biotechnol 4:1087–1090
Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A (2017) RNA targeting with CRISPR-Cas13. Nature 550:280
Abudayyeh OO, Gootenberg JS, Franklin B, Koob J, Kellner MJ, Ladha A, Joung J, Kirchgatterer P, Cox DBT, Zhang F (2019) A cytosine deaminase for programmable single-base RNA editing. Science 365:382–386
Aman R, Ali Z, Butt H, Mahas A, Aljedaani F, Khan MZ, Ding S, Mahfouz M (2018) RNA virus interference via CRISPR/Cas13a system in plants. Genome Biol 19:1
Bao A, Burritt DJ, Chen H, Zhou X, Cao D, Tran LP (2019) The CRISPR/Cas9 system and its applications in crop genome editing. Crit Rev Biotechnol 39:321–336
Begemann MB, Gray BN, January E, Gordon GC, He Y, Liu H, Wu X, Brutnell TP, Mockler TC, Oufattole M (2017) Precise insertion and guided editing of higher plant genomes using Cpf1 CRISPR nucleases. Sci Rep 7:11606
Biswas S, Li R, Yuan Z, Zhang D, Zhao X, Shi J (2019) Development of methods for effective identification of CRISPR/Cas9-induced indels in rice. Plant Cell Rep 38:503–510
Bortesi L, Fischer R (2015) The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol Adv 33:41–52
Butt H, Eid A, Ali Z, Atia MAM, Mokhtar MM, Hassan N, Lee CM, Bao G, Mahfouz MM (2017) Efficient CRISPR/Cas9-mediated genome editing using a chimeric single-guide RNA molecule. Front Plant Sci 8:1441
Castel B, Tomlinson L, Locci F, Yang Y, Jones JDG (2019) Optimization of T-DNA architecture for Cas9-mediated mutagenesis in Arabidopsis. PLoS One 14:e0204778
Cermak T, Curtin SJ, Gil-Humanes J, Cegan R, Kono TJY, Konecna E, Belanto JJ, Starker CG, Mathre JW, Greenstein RL, Voytas DF (2017) A multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell 29:1196–1217
Chatterjee P, Jakimo N, Jacobson JM (2018) Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci Adv 4:eaau0766
Chen, S., Tao, L., Zeng, L., Vega‐Sanchez, M.E., Umemura, K. and Wang, G.L. (2006) A highly efficient transient protoplast system for analyzing defense gene expression and protein‐protein interactions in rice. Mol. Plant Pathol. 7, 417– 427.
Chen L, Li W, Katin-Grazzini L, Ding J, Gu X, Li Y, Gu T, Wang R, Lin X, Deng Z, McAvoy RJ, Gmitter FG Jr, Deng Z, Zhao Y, Li Y (2018) A method for the production and expedient screening of CRISPR/Cas9-mediated non-transgenic mutant plants. Hortic Res 5:13
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, Zhang F (2017) RNA editing with CRISPR-Cas13. Science 358:1019–1027
Didovyk A, Borek B, Tsimring L, Hasty J (2016) Transcriptional regulation with CRISPR-Cas9: principles, advances, and applications. Curr Opin Biotechnol 40:177–184
Ding D, Chen K, Chen Y, Li H, Xie K (2018) Engineering introns to express RNA guides for Cas9- and Cpf1-mediated multiplex genome editing. Mol Plant 11:542–552
Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, Root DE. (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 34:184-191.
Edraki A, Mir A, Ibraheim R, Gainetdinov I, Yoon Y, Song CQ, Cao Y, Gallant J, Xue W, Rivera-Pérez JA, Sontheimer EJ (2019) A compact, high-accuracy Cas9 with a dinucleotide PAM for in vivo genome editing. Mol Cell 73:714–726.e4
EFSA Panel on Genetically Modified Organisms (2012) Scientific opinion addressing the safety assessment of plants developed using zinc finger nuclease 3 and other site-directed nucleases with similar function. EFSA J 10:2943
Endo A, Masafumi M, Kaya H, Toki S (2016a) Efficient targeted mutagenesis of rice and tobacco genomes using Cpf1 from Francisella novicida. Sci Rep 6:38169
Endo M, Mikami M, Endo A, Kaya H, Itoh T, Nishimasu H, Nureki O, Toki S (2019) Genome editing in plants by engineered CRISPR-Cas9 recognizing NG PAM. Nat Plants 5:14–17
Endo M, Mikami M, Toki S (2016b) Biallelic gene targeting in Rice. Plant Physiol 170:667–677
Feng C, Su H, Bai H, Wang R, Liu Y, Guo X, Liu C, Zhang J, Yuan J, Birchler JA, Han F (2018) High-efficiency genome editing using a dmc1 promoter-controlled CRISPR/Cas9 system in maize. Plant Biotechnol J 16:1848–1857
Fossi M, Amundson K, Kuppu S, Britt A, Comai L (2019) Regeneration of Solanum tuberosum plants from protoplasts induces widespread genome instability. Plant Physiol 180:78–86
Friedland AE, Baral R, Singhal P, Loveluck K, Shen S, Sanchez M, Marco E, Gotta GM, Maeder ML, Kennedy EM (2015) Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol 16:257
Fujimura T, Sakurai M, Akagi H, Negishi T, Hirose A (1985) Regeneration of Rice plants from protoplasts. Plant Tiss Cult Letters 2:74–75
Gao X, Chen J, Dai X, Zhang D, Zhao Y (2016) An effective strategy for reliably isolating heritable and Cas9-free Arabidopsis mutants generated by CRISPR/Cas9-mediated genome editing. Plant Physiol 171:1794–1800
Gao Y, Zhao Y (2014) Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J Integr Plant Biol 56:343–349
Gasparis S, Kala M, Przyborowski M, Lyznik LA, Orczyk W, Nadolska-Orczyk A (2018) A simple and efficient CRISPR/Cas9 platform for induction of single and multiple, heritable mutations in barley (Hordeum vulgare L.). Plant Methods 14:111
Hahn F, Korolev A, Loures LS, Nekrasov V (2019) A modular cloning toolkit for genome editing in plants. bioRxiv. https://doi.org/10.1101/738021
Hodgens C, Nimchuk ZL, Kieber JJ (2017) indCAPS: a tool for designing screening primers for CRISPR/Cas9 mutagenesis events. PLoS one 12:e0188406
Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, Thomson JA (2013) Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci U S A 110:15644–15649
Howells RM, Craze M, Bowden S, Wallington EJ (2018) Efficient generation of stable, heritable gene edits in wheat using CRISPR/Cas9. BMC Plant Biol 18:215
Hsu CT, Cheng YJ, Yuan YH, Hung WF, Cheng QW, Wu FH, Lee LY, Gelvin SB, Lin CS (2019) Application of Cas12a and nCas9-activation-induced cytidine deaminase for genome editing and as a non-sexual strategy to generate homozygous/multiplex edited plants in the allotetraploid genome of tobacco. Plant Mol Biol. https://doi.org/10.1007/s11103-019-00907-w
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31:827–832
Hu L, Li H, Qin R, Xu R, Li J, Li L, Wei P, Yang J (2016) Plant phosphomannose isomerase as a selectable marker for rice transformation. Sci Rep 6:25921
Hu X, Meng X, Liu Q, Li J, Wang K (2018) Increasing the efficiency of CRISPR-Cas9-VQR precise genome editing in rice. Plant Biotechnol J 16:292–297
Hu X, Wang C, Liu Q, Fu Y, Wang K (2017) Targeted mutagenesis in rice using CRISPR-Cpf1 system. J Genet Genomics 44:71–73
Hua K, Tao X, Yuan F, Wang D, Zhu JK (2018) Precise a· T to G· C base editing in the rice genome. Mol Plant 11:627–630
Hua K, Tao X, Zhu JK (2019) Expanding the base editing scope in rice by using Cas9 variants. Plant Biotechnol J 17:499–504
Hua Y, Wang C, Huang J, Wang K (2017) A simple and efficient method for CRISPR/Cas9-induced mutant screening. J Genet Genomics 44:207–213
Jia H, Xu J, Orbović V, Zhang Y, Wang N (2017) Editing citrus genome via SaCas9/sgRNA system. Front Plant Sci 8:2135
Jin S, Zong Y, Gao Q, Zhu Z, Wang Y, Qin P, Liang C, Wang D, Qiu JL, Zhang F, Gao C (2019) Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 364:292–295
Karvelis T, Gasiunas G, Young J, Bigelyte G, Silanskas A, Cigan M, Siksnys V (2015) Rapid characterization of CRISPR-Cas9 protospacer adjacent motif sequence elements. Genome Biol 16:253
Kaya H, Mikami M, Endo A, Endo M, Toki S (2016) Highly specific targeted mutagenesis in plants using Staphylococcus aureus Cas9. Sci Rep 6:26871
Kim E, Koo T, Park SW, Kim D, Kim K, Cho HY, Song DW, Lee KJ, Jung MH, Kim S, Kim JH, Kim JH, Kim JS (2017a) In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun 8:14500
Kim H, Kim ST, Ryu J, Kang BC, Kim JS, Kim SG (2017b) CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat Commun 8:14406
Kim HK, Song M, Lee J, Menon AV, Kim H (2017c) In vivo highthroughput profiling of CRISPRCpf1 activity. Nat Methods 14:153
Kim YB, Komor AC, Levy JM, Packer MS, Zhao KT, Liu DR (2017d) Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol 35:371–376
Kis A, Hamar E, Tholt G, Ban R, Havelda Z (2019) Creating highly efficient resistance against wheat dwarf virus in barley by employing CRISPR/Cas9 system. Plant Biotechnol J 17:1004–1006
Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z, Joung JK (2015) Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol 33:1293–1298
Kurata M, Wolf NK, Lahr WS, Weg MT, Kluesner MG, Lee S, Hui K, Shiraiwa M, Webber BR, Moriarity BS (2018) Highly multiplexed genome engineering using CRISPR/Cas9 gRNA arrays. PLoS One 13:e0198714
Li S, Li J, He Y, Xu M, Zhang J, Du W, Zhao Y, Xia L (2019) Precise gene replacement in rice by RNA transcript-templated homologous recombination. Nat Biotechnol 37:445
Li X, Wang Y, Liu Y, Yang B, Wang X, Wei J, Lu Z, Zhang Y, Wu J, Huang X (2018) Base editing with a Cpf1–cytidine deaminase fusion. Nat Biotechnol 36:324–327
Liang Z, Chen K, Gao C (2019) Biolistic delivery of CRISPR/Cas9 with ribonucleoprotein complex in wheat. Methods Mol Biol 1917:327–335
Liang Z, Chen K, Li T, Zhang Y, Wang Y, Zhao Q, Liu J, Zhang H, Liu C, Ran Y, Gao C (2017) Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nat Commun 8:14261
Lin CS, Chang WC (1998) Micropropagation of Bambusa edulis through nodal explants of field-grown culms and flowering of regenerated plantlets. Plant Cell Rep 17:617–620
Lin CS, Lai YH, Sun CW, Liu NT, Tsay HS, Chang WC, Chen JJW (2006) Identification of ESTs differentially expressed in green and albino mutant bamboo (Bambusa edulis) by suppressive subtractive hybridization (SSH) and microarray analysis. Plant Cell Tissue Organ Cult 86:169–175
Lin CS, Liang CJ, Hsaio HW, Lin MJ, Chang WC (2007) In vitro flowering of green and albino Dendrocalamus latiflorus. New For 34:177–186
Liu NT, Jane WN, Tsay HS, Wu H, Chang WC, Lin CS (2007) Chloroplast genome aberration in micropropagation-derived albino Bambusa edulis mutants, ab1 and ab2. Plant Cell Tissue Organ Cult 88:147–156
Lowder LG, Zhang D, Baltes NJ, Paul JW 3rd, Tang X, Zheng X, Voytas DF, Hsieh TF, Zhang Y, Qi Y (2015) A CRISPR/Cas9 toolbox for multiplexed plant genome editing and transcriptional regulation. Plant Physiol 169:971–985
Lowe K, Wu E, Wang N, Hoerster G, Hastings C, Cho MJ, Scelonge C, Lenderts B, Chamberlin M, Cushatt J, Wang L, Ryan L, Khan T, Chow-Yiu J, Hua W, Yu M, Banh J, Bao Z, Brink K, Igo E, Rudrappa B, Shamseer PM, Bruce W, Newman L, Shen B, Zheng P, Bidney D, Falco C, Register J, Zhao ZY, Xu D, Jones T, Gordon-Kamm W (2016) Morphogenic regulators Baby boom and Wuschel improve monocot transformation. Plant Cell 28:1998–2015
Ma X, Zhang Q, Zhu Q, Liu W, Chen Y, Qiu R, Wang B, Yang Z, Li H, Lin Y, Xie Y, Shen R, Chen S, Wang Z, Chen Y, Guo J, Chen L, Zhao X, Dong Z, Liu YG (2015) A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol Plant 8:1274–1284
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826
Malzahn AA, Tang X, Lee K, Ren Q, Sretenovic S, Zhang Y, Chen H, Kang M, Bao Y, Zheng X, Deng K, Zhang T, Salcedo V, Wang K, Zhang Y, Qi Y (2019) Application of CRISPR-Cas12a temperature sensitivity for improved genome editing in rice, maize, and Arabidopsis. BMC Biol 17:9
Marton L, Wullems GJ, Molendijk L, Schilperoort RA (1979) In vitro transformation of cultured cells from Nicotiana tabacum by Agrobacterium tumefaciens. Nature 277:129–131
Marx V (2016) Plants: a tool box of cell-based assays. Nat Methods 13:551–554
Miao J, Guo D, Zhang J, Huang Q, Qin G, Zhang X, Wan J, Gu H, Qu LJ (2013) Targeted mutagenesis in rice using CRISPRCas system. Cell Res 23:1233–1236
Mikami M, Toki S, Endo M (2015) Comparison of CRISPR/Cas9 expression constructs for efficient targeted mutagenesis in rice. Plant Mol Biol 88:561–572
Mikami M, Toki S, Endo M (2016) Precision targeted mutagenesis via Cas9 paired nickases in rice. Plant Cell Physiol 57:1058–1068
Miki B, McHugh S (2004) Selectable marker genes in transgenic plants: applications, alternatives and biosafety. J Biotechnol 107:193–232
Miki D, Zhang W, Zeng W, Feng Z, Zhu JK (2018) CRISPR/Cas9-mediated gene targeting in Arabidopsis using sequential transformation. Nat Commun 9:1967
Nandy S, Pathak B, Zhao S, Srivastava V (2019) Heat-shock-inducible CRISPR/Cas9 system generates heritable mutations in rice. Plant Direct 3:e00145
Okamoto T, Ohnishi Y, Toda E (2017) Development of polyspermic zygote and possible contribution of polyspermy to polyploid formation in angiosperms. J Plant Res 130:485–490
Oung HM, Lin KC, Wu TM, Chandrika NNP, Hong CY (2015) Hygromycin B-induced cell death is partly mediated by reactive oxygen species in rice (Oryza sativa L.). Plant Mol Biol 89:577–588
Park J, Choe S (2019) DNA-free genome editing with preassembled CRISPR/Cas9 ribonucleoproteins in plants. Transgenic Res 28(Suppl 2):61–64
Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR (2013) High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31:839–843
Podevin N, Davies HV, Hartung F, Nogue F, Casacuberta JM (2013) Site-directed nucleases: a paradigm shift in predictable, knowledge-based plant breeding. Trends Biotechnol 3:375–383
Qi X, Dong L, Liu C, Mao L, Liu F, Zhang X, Cheng B, Xie C (2018) Systematic identification of endogenous RNA polymerase III promoters for efficient RNA guide-based genome editing technologies in maize. The Crop Journal 6:314–320
Qin R, Li J, Li H, Zhang Y, Liu X, Miao Y, Zhang X, Wei P (2019) Developing a highly efficient and wildly adaptive CRISPR-SaCas9 toolset for plant genome editing. Plant Biotechnol J 17:706–708
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F (2015) In vivo genome editing using Staphylococcus aureus Cas9. Nature 520:186
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308
Rhodes CA, Lowe KS, Ruby KL (1988a) Plant regeneration from protoplasts isolated from embryogenic maize cell cultures. Nat Biotechnol 6:56–60
Rhodes CA, Pierce DA, Mettler IJ, Mascarenhas D, Detmer JJ (1988b) Genetically transformed maize plants from protoplasts. Science 240:204–207
Saika H, Mori A, Endo M, Toki S (2019) Targeted deletion of rice retrotransposon Tos17 via CRISPR/Cas9. Plant Cell Rep 38:455–458
Sampson TR, Saroj SD, Llewellyn AC, Tzeng YL, Weiss DS (2013) A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 497:254–257
Shan Q, Wang Y, Chen K, Liang Z, Li J, Zhang Y, Zhang K, Liu J, Voytas DF, Zheng X, Zhang Y, Gao C (2013) Rapid and efficient gene modification in rice and Brachypodium using TALENs. Mol Plant 6:1365–1368
Shan Q, Wang Y, Li J, Gao C (2014) Genome editing in rice and wheat using the CRISPR/Cas system. Nat Protoc 9:2395–2410
Shimamoto K, Terada R, Izawa T, Fujimoto H (1989) Fertile transgenic rice plants regenerated from transformed protoplasts. Nature 338:274–276
Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, Zhang F, Koonin EV (2015) Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell 60:385–397
Srivastava V, Underwood JL, Zhao S (2017) Dual-targeting by CRISPR/Cas9 for precise excision of transgene from rice genome. Plant Cell Tissue Organ Cult 129:153–160
Steinert J, Schiml S, Fauser F, Puchta H (2015) Highly efficient heritable plant genome engineering using Cas9 orthologues from Streptococcus thermophilus and Staphylococcus aureus. Plant J 84:1295–1305
Svitashev S, Schwartz C, Lenderts B, Young JK, Mark Cigan A (2016) Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nat Commun 7:13274
Takebe I, Labib G, Melchers G (1971) Regeneration of whole plants from isolated mesophyll protoplasts of tobacco. Naturwissenschaften 58:318–320
Tang J, Chen L, Liu YG (2019) Off-target effects and the solution. Nat Plants 5:341–342
Tang X, Liu G, Zhou J, Ren Q, You Q, Tian L, Xin X, Zhong Z, Liu B, Zheng X, Zhang D, Malzahn A, Gong Z, Qi Y, Zhang T, Zhang Y (2018) A large-scale whole-genome sequencing analysis reveals highly specific genome editing by both Cas9 and Cpf1 (Cas12a) nucleases in rice. Genome Biol 19:84
Tang X, Lowder LG, Zhang T, Malzahn AA, Zheng X, Voytas DF, Zhong Z, Chen Y, Ren Q, Li Q, Kirkland ER, Zhang Y, Qi Y (2017) A CRISPR-Cpf1 system for efficient genome editing and transcriptional repression in plants. Nat Plants 3:17013
Teng F, Cui T, Feng G, Guo L, Xu K, Gao Q, Li T, Li J, Zhou Q, Li W (2018) Repurposing CRISPR-Cas12b for mammalian genome engineering. Cell Discov 4:63
Thomas HR, Percival SM, Yoder BK, Parant JM (2014) High-throughput genome editing and phenotyping facilitated by high resolution melting curve analysis. PLoS One 9:e114632
Toda E, Koiso N, Takebayashi A, Ichikawa M, Kiba T, Osakabe K, Osakabe Y, Sakakibara H, Kato N, Okamoto T (2019) An efficient DNA- and selectable-marker-free genome-editing system using zygotes in rice. Nat Plants 5:363–368
Toriyama K, Arimoto Y, Uchimiya H, Hinata K (1988) Transgenic rice plants after direct gene transfer into protoplasts. Nat Biotechnol 6:1072–1074
Tuncel A, Corbin KR, Ahn-Jarvis J, Harris S, Hawkins E, Smedley MA, Harwood W, Warren FJ, Patron NJ, Smith AM (2019) Cas9-mediated mutagenesis of potato starch-branching enzymes generates a range of tuber starch phenotypes. Plant Biotechnol J. https://doi.org/10.1111/pbi.13137
Vouillot L, Thelie A, Pollet N (2015) Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda) 5:407–415
Wang M, Mao Y, Lu Y, Tao X, Zhu JK (2017) Multiplex gene editing in rice using the CRISPR-Cpf1 system. Mol Plant 10:1011–1013
Wang M, Mao Y, Lu Y, Wang Z, Tao X, Zhu JK (2018) Multiplex gene editing in rice with simplified CRISPR-Cpf1 and CRISPR-Cas9 systems. J Integr Plant Biol 60:626–631
Wilmink A, Dons JJM (1993) Selective agents and marker genes for use in transformation of monocotyledonous plants. Plant Mol Biol Rep 11:165–185
Wolter F, Puchta H (2019) In planta gene targeting can be enhanced by the use of CRISPR/Cas12a. Plant J. https://doi.org/10.1111/tpj.14488
Woo JW, Kim J, Kwon SI, Corvalan C, Cho SW, Kim H, Kim SG, Kim ST, Choe S, Kim JS (2015) DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat Biotechnol 33:1162–1164
Wu TM, Huang JZ, Oung HM, Hsu YT, Tsai YC, Hong CY (2019) H2O2-based method for rapid detection of transgene-free rice plants from segregating CRISPR/Cas9 genome-edited progenies. Int J Mol Sci 20:3885
Xie K, Minkenberg B, Yang Y (2015) Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci U S A 112:3570–3575
Xie K, Yang Y (2013) RNA-guided genome editing in plants using a CRISPR-Cas system. Mol Plant 6:1975–1983
Xing HL, Dong L, Wang ZP, Zhang HY, Han CY, Liu B, Wang XC, Chen QJ (2014) A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol 14:327
Xu R, Qin R, Li H, Li D, Li L, Wei P, Yang J (2017) Generation of targeted mutant rice using a CRISPR-Cpf1 system. Plant Biotechnol J 15:713–717
Xu R, Qin R, Li H, Li J, Yang J, Wei P (2019) Enhanced genome editing in rice using single transcript unit CRISPR-LbCpf1 systems. Plant Biotechnol J 17:553–555
Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F (2015) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163:759–771
Zhang H, Zhang J, Wei P, Zhang B, Gou F, Feng Z, Mao Y, Yang L, Zhang H, Xu N, Zhu JK (2014) The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol J 12:797–807
Zhang T, Zhao Y, Ye J, Cao X, Xu C, Chen B, An H, Jiao Y, Zhang F, Yang X, Zhou G (2019b) Establishing CRISPR/Cas13a immune system conferring RNA virus resistance in both dicot and monocot plants. Plant Biotechnol J 17:1185–1187
Zhang T, Zheng Q, Yi X, An H, Ma S, Zhou G (2018) Establishing RNA virus resistance in plants by harnessing CRISPR immune system. Plant Biotechnol J 16:1415–1423
Zhang Y, Liang Z, Zong Y, Wang Y, Liu J, Chen K, Qiu JL, Gao C (2016) Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA. Nat Commun 7:12617
Zhang Y, Malzahn AA, Sretenovic S, Qi Y (2019a) The emerging and uncultivated potential of CRISPR technology in plant science. Nat Plants 5:778–794
Zhou H, Liu B, Weeks DP, Spalding MH, Yang B (2014) Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res 42:10903–10914
Zhu X, Xu Y, Yu S, Lu L, Ding M, Cheng J, Song G, Gao X, Yao L, Fan D, Meng S, Zhang X, Hu S, Tian Y (2014) An efficient genotyping method for genome-modified animals and human cells generated with CRISPR/Cas9 system. Sci Rep 4:6420
Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, Cheng AW, Trevino AE, Konermann S, Chen S, et al. (2014) Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol 32, 670–676.
Lin CS, Hsu CT, Yang LH, Lee LY, Fu JY, Cheng QW, Wu FH, Hsiao HC, Zhang Y, Zhang R, Chang WJ, Yu CT, Wang W, Liao LJ, Gelvin SB, Shih MC (2018) Application of protoplast technology to CRISPR/Cas9 mutagenesis: from single-cell mutation detection to mutant plant regeneration. Plant Biotechnol J 16:1295-1310
Andersson M, Turesson H, Nicolia A, Falt AS, Samuelsson M, Hofvander P (2017) Efficient targeted multiallelic mutagenesis in tetraploid potato ( Solanum tuberosum) by transient CRISPR-Cas9 expression in protoplasts. Plant Cell Rep 36:117-128.
Andersson M, Turesson H, Olsson N, Falt AS, Ohlsson P, Gonzalez MN, Samuelsson M, Hofvander P (2018) Genome editing in potato via CRISPR-Cas9 ribonucleoprotein delivery. Physiol Plant 64:378-384
Li JF, Norville JE, Aach J, McCormack M, Zhang D, Bush J, Church GM, Sheen J (2013) Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nature Biotechnol 31:688-691.
Liang Z, Zhang K, Chen K, Gao C (2014) Targeted mutagenesis in Zea mays using TALENs and the CRISPR/Cas system. J Genet Gen 41:63-68
Sun X, Hu Z, Chen R, Jiang Q, Song G, Zhang H, Xi Y (2015) Targeted mutagenesis in soybean using the CRISPR-Cas9 system. Sci Rep 5:10342
Zong Y, Wang Y, Li C, Zhang R, Chen K, Ran Y, Qiu JL, Wang D, Gao C (2017) Precise base editing in rice, wheat and maize with a Cas9-cytidine deaminase fusion. Nature Biotechnol 35:438-440.
Zong Y, Song Q, Li C, Jin S, Zhang D, Wang Y, Qiu JL, Gao C. 2018 Efficient C-to-T base editing in plants using a fusion of nCas9 and human APOBEC3A. Nature Biotechnol 36, 950–953.
Kim H, Kim ST, Ryu J, Kang BC, Kim JS, Kim SG (2017) CRISPR/Cpf1-mediated DNA-free plant genome editing. Nature Commun 8:14406.
Malnoy M, Viola R, Jung MH, Koo OJ, Kim S, Kim JS, Velasco R, Nagamangala Kanchiswamy C (2016) DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers Plant Sci 7:1904.
Wu FH, Shen SC, Lee LY, Lee SH, Chan MT, Lin CS (2009) Tape- Arabidopsis Sandwich - a simpler Arabidopsis protoplast isolation method. Plant Methods 5:16
Zhang Y, Su J, Duan S, Ao Y, Dai J, Liu J, Wang P, Li Y, Liu B, Feng D, Wang J, Wang H (2011) A highly efficient rice green tissue protoplast system for transient gene expression and studying light/chloroplast‐related processes. Plant Methods 7: 30
Wullems GJ, Molendijk L, Ooms G, Schilperoort RA (1981a) Differential expression of crown gall tumor markers in transformants obtained after in vitro Agrobacterium tumefaciens-induced transformation of cell wall regenerating protoplasts derived from Nicotiana tabacum. Proc Natl Acad Sci USA 78:4344-4348
Wullems GJ, Molendijk L, Ooms G, Schilperoort RA (1981b) Retention of tumor markers in F1 progeny plants from in vitro induced octopine and nopaline tumor tissues. Cell 24:719-727
Acknowledgments
We thank Miranda Loney and Fu-Hui Wu for editing.
Funding
This research was supported by Academia Sinica, the Innovative Translational Agricultural Research Administrative Office (AS-KPQ-107-ITAR-10), and the Ministry of Science and Technology (105–2313-B-001 -007 -MY3 and 108–2313-B-001-11 to CSL; 105-2628-B-036-MY3 and 108-2313-B-002-055-MY3 to CYH; 108-2313-B-002 -051 and107-2313-B-002 -023 to YCT), Taiwan.
Author information
Authors and Affiliations
Contributions
JJY, CYH, PW, YCT, and CSL contributed to the writing. CSL organized and prepared this manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yue, JJ., Hong, CY., Wei, P. et al. How to start your monocot CRISPR/Cas project: plasmid design, efficiency detection, and offspring analysis. Rice 13, 9 (2020). https://doi.org/10.1186/s12284-019-0354-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-019-0354-2