
Abstract
Rice is a model system used for crop genomics studies. The completion of the rice genome draft sequences in 2002 not only accelerated functional genome studies, but also initiated a new era of resequencing rice genomes. Based on the reference genome in rice, next-generation sequencing (NGS) using the high-throughput sequencing system can efficiently accomplish whole genome resequencing of various genetic populations and diverse germplasm resources. Resequencing technology has been effectively utilized in evolutionary analysis, rice genomics and functional genomics studies. This technique is beneficial for both bridging the knowledge gap between genotype and phenotype and facilitating molecular breeding via gene design in rice. Here, we also discuss the limitation, application and future prospects of rice resequencing.
Similar content being viewed by others

Introduction
Rice is one of the most important staple crops worldwide as well as a model monocot used in genomics research. The world population has already exceeded seven billion and is still growing, while the amount of land suitable for agriculture is decreasing due to a variety of factors such as rapid climate change. To meet the global food demands of nine billion people by 2050, improvements in molecular genetics will be important to increase rice yield in the post-genomics era (Miura et al. [2011]; Huang et al. [2013]).
In 2002, draft genomic sequences of two rice subspecies, O. sativa ssp. japonica (Nipponbare) and O. sativa ssp. indica (93–11), were released (Yu et al. [2002]; Goff et al. [2002]) and subsequently, the International Rice Genome Sequencing Project ([2005]) completed the final genome sequence of Nipponbare. These achievements have not only greatly accelerated functional genomics research, but also provided a reference genome for resequencing rice genomes using high-throughput sequencing technologies (Gao et al. [2012]; Feuillet et al. [2011]). New sequencing technologies are also known as next-generation sequencing (NGS), which makes reference to the first generation of Sanger sequencing technology. Three mainstream NGS platforms, i.e., Illumina/Solexa, Roche/454 and ABI/SOLiD sequencing, which are known collectively as high-throughput sequencing, can generate large amounts of data in a single run and analyze more than 100 kb of DNA (Ansorge [2009]).
The advent of NGS technologies has greatly enhanced rice functional genomics and molecular breeding studies (Xie et al. [2010]; Gao et al. [2012]). The model system has been readily adapted to the new sequencing technologies, as rice is a self-fertilizing plant with the smallest completed high-quality genome among cereal crops. Furthermore, abundant diverse rice germplasm resources are available for genome-wide association studies (GWAS) and evolutionary analysis (Guo et al. [2004]; Huang et al. [2012a]). With the release of the complete bacterial artificial chromosome (BAC) physical map for the aus rice cultivar ‘Kasalath’ and an updated version of the whole genome sequence for the indica rice variety ’93-11’ (Kanamori et al. [2013]; Gao et al. [2013]), at least three references are now available together with the japonica ‘Nipponbare’ sequence. These advantages have enabled researchers to perform accurate alignments of short sequence reads produced by NGS with the reference genomes, as well as detailed genetic polymorphism analysis of rice in an efficient manner (Han and Huang [2013]). Using information from these studies, researchers have now succeeded in characterizing genomic variation, identifying QTLs (quantitative trait loci) by GWAS, investigating the origin of cultivated rice and performing molecular breeding studies.
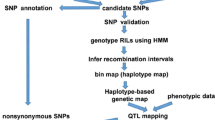
In this review, we briefly review the progress of the application of rice genome resequencing to studies of genome diversification, genotyping, gene identification and cultivated rice origin. We highlight the improved parental genome sequences, effective genetic mapping and genotyping techniques involving deep resequencing of an RIL (recombinant inbred line) population of the super hybrid rice Liang-You-Pei-Jiu (LYP9) (Gao et al. [2013]). Additionally, we discuss the future of rice resequencing.
Review
Genetic diversity and genome variation analysis of rice germplasm
Rice has more than 100,000 accession germplasm resources including Asian cultivated rice, African cultivated rice and 22 wild rice species (The International Rice Genebank: http://irri.org/our-work/research/genetic-diversity), which are indispensable genetic resources for further improvement of cultivated rice varieties. Recently, rice molecular biology research has been involved in exploring genetic diversity and exploiting genome variation in rice germplasm. Genetic variation can be assayed using a variety of molecular markers, including structure variation (SV) markers such as insertions/deletions (InDels) and copy number variations (CNV). Resequencing can be used to identify genetic variation within a species and to assess the population structure and the pattern of linkage disequilibrium. Resequencing a key germplasm subset representing various geographically distributed populations can provide scientists with in-depth knowledge of the range of genome variation and genetic diversity within the population based on sequence databases.
Numerous diverse rice germplasm resources that have been used to detect genome variation are shown in Table 1 (Subbaiyan et al. [2012]; Jeong et al. [2013]). Kojima et al. ([2005]) detected 554 alleles from 332 accessions of cultivated rice based on a genome-wide RFLP survey, and developed a rice diversity research set of 69 accessions of germplasm, including two reference varieties, Nipponbare and Kasalath. McNally et al. ([2009]) identified 160,000 genome-wide SNPs in 20 diverse rice varieties via microarray-based resequencing and discovered their introgression patterns and pedigree relationships. Zhao et al. ([2011]) genotyped 44,100 SNP variants across 413 accessions of O. sativa collected from 82 countries for genetic structure analysis and cross-population-based mapping. Huang et al. ([2010]) resequenced 517 indica subspecies of Chinese rice landraces with approximately one-fold-coverage Illumina sequencing. A total of 3,625,200 non-redundant SNPs were identified, resulting in an average of 9.32 SNPs per kb, with 167,514 SNPs located in the coding regions of 25,409 annotated genes. A high-density SNP map and haplotype map (HapMap) of the rice genome was constructed using a novel data-imputation method. Subsequently, Huang et al. ([2012b]) extended this methodology to a larger, and more diverse, sample of 950 worldwide rice varieties, including indica and japonica subspecies. In the non-repeated regions, 4,109,366 non-singleton SNPs and 191,476 non-redundant InDels ranging from 1 bp to 376 bp in size were identified in genic regions. The authors investigated the worldwide rice population structure and constructed a neighbor-joining tree involving five divergent groups: indica, aus, temperate japonica, tropical japonica and intermediate, which were consistent with the five-distinct-groups detected by Garries et al. ([2005]). The authors resequenced an elite Japanese rice cultivar, Koshihikari, corresponding to 80.1% identity with the Nipponbare sequence, and leading to the identification of 67,051 SNPs. They also genotyped 151 representative Japanese cultivars using 1,917 SNPs to clarify the dynamics of the pedigree haplotypes (Yamamoto et al. [2010]). Based on genome-wide SNP analysis, these studies revealed relationships among landraces and modern varieties of rice, and genetic diversity that can be used for breeding programs in rice.
Genome diversity has increasingly been identified in wild rice. Wing et al. ([2005]) constructed bacterial artificial chromosome and/or sequence tag connector (BAC/STC)-based physical maps of 11 wild and one cultivated rice species for alignment with the rice reference genome. Resequencing the wild species of the genus Oryza has enormous potential in identifying approaches to increase agricultural productivity of the cultivated rice species O. sativa and O. glaberrima. Xu et al. ([2012]) directly resequenced 50 accessions of cultivated and wild rice, and identified genome-wide variation patterns including the identification of more than 6.5 million high-quality SNPs, 808,000 InDels, 94,700 SVs (>100 bp) and 1,676 CNVs. In another study, 66 accessions from three taxa (22 each from O. sativa indica, O. sativa japonica and O. rufipogon) were chosen for whole genome sequencing (He et al. [2011]). Huang et al. ([2012a]) also generated genome sequences from 446 geographically diverse accessions of the wild rice species O. rufipogon, the immediate ancestral progenitor of cultivated rice, and from 1,083 cultivated indica and japonica varieties, to construct a comprehensive map of rice genome variation. A total of 7,970,359 non-singleton SNPs were identified from the 1,529 available rice genome sequences, the ancestral alleles of 9.3% of which are identical to those of O. rufipogon. These genotype data and population genetics analyses provide insights into the relationships between rice diversity and domestication processes.
The NGS strategy also provides new opportunities for RNA sequence diversity analysis or epigenomic studies in rice. The functional complexity of the rice transcriptome and its contribution to phenotype remains to be fully elucidated. Gene expression microarrays have been used traditionally for high-throughput measurements of gene expression levels (Jiao et al. [2005]; Furutani et al. [2006]; Li et al. [2006b]; Satoh et al. [2007]). Recently, with the development of the next-generation high-throughput DNA sequencing technologies, RNA-seq has shown advantages over microarrays by allowing accurate, efficient and reproducible estimations of transcript abundance of either known or unknown transcripts with a larger dynamic range using less RNA sample (Wilhelm et al. [2008]; Fullwood et al. [2009]). Furthermore, RNA-seq can detect genes expressed at low levels and refine the structure of transcripts (Wang et al. [2009]; Wilhelm and Landry [2009]). Using high-throughput paired-end RNA-seq, a substantial number of novel transcripts and exons have been detected, and a far greater amount of alternative splicing has been identified that was shown previously (Zhang et al. [2010]). The updated research focused on comparative analyses of the epigenome and comprehensive analyses of the eQTL (He et al. [2010]; Lu et al. [2010]). Additionally, NGS strategy has been applied recently in massive program of sequencing of small RNA populations from different rice tissues. Jeong et al. ([2011]) also identified 76 new rice miRNAs that play critical roles in a variety of developmental processes. Wang et al. ([2012]) sequenced an accession of O. rufipogon to c. 55× coverage, identified miRNAs using small RNAs generated from three different tissues of O. rufipogon and identified miRNA targets in O. rufipogon by degradome sequencing. The authors found that rice miRNA genes have experienced a complex evolutionary process during domestication. This plethora of genetic diversity RNA data is also an important genetic resource for rice breeding.
Evolution analysis based on resequencing
Cultivated rice is thought to have been domesticated from wild rice thousands of years ago. However, the evolutionary origins and domestication processes of cultivated rice have long been debated. A wide range of genetic and archeological studies have been carried out to investigate rice phylogenetics and the demographic history of rice domestication. Some population genetics studies have indicated that indica and japonica originated independently, and some demographic analyses have suggested that domesticated rice has a single origin (Kovach et al. [2007]; Molina et al. [2011]). With the development of NGS technologies, analysis of the global diversity of rice germplasm and sequencing-based genome variation analysis can provide in-depth insights into rice domestication.
Recently, Huang et al. ([2012a]) systematically constructed a comprehensive map of rice genome variation with 6,119,311 SNPs using the 1,529 rice genome sequences. Phylogenetic tree analysis indicated that O. sativa and japonica are descended from O. rufipog on-I and Or-III (the O. rufipogon species were classified into three types, i.e., Or-1, Or-II and Or-III), respectively. A total of 213,188 indica-japonica-differentiated SNPs were found, and only 9,595 SNPs were fixed between O. rufipogon and O. sativa. The differentiation was enhanced during domestication, with the divergence index Fst expanding from 0.18 in O. rufipogon to 0.55 in O. sativa. The level of genetic differentiation between indica and Or-I was modest (Fst = 0.17), and that is 0.36 in japonica. The authors found that only about 33% of the genetic diversity of Or-III persisted in japonica. Moreover, indica contains approximately 75% of the genetic diversity observed in Or-I. In a search for signatures of selection, the authors identified 32 selective sweeps in the rice genome and 55 domestication loci by searching for signatures of domestication using an integrated genomics approach. The authors detected a series of gene introgression events. The most well-characterized domestication genes, such as Bh4, PROG1, sh4, qSW5 and OsCl, were among the 55 loci detected in the total population (Zhu et al. [2011]; Shomura et al. [2008]; Saitoh et al. [2004]). However, an additional three genes, qSH1, Waxy and Rc, were detected only in the japonica panel. SNP-based phylogenetic tree analysis showed that the middle region of the Pearl River district in Guangxi Province, southern China, is probably the origin of development of cultivated rice. It can be speculated that japonica was possibly first domesticated and then crossed with local wild rice in Southeast Asia to generate indica.
Also Xu et al. ([2012]), used a haplotype map of 6.5 million SNPs from 50 rice genome sequences to identify thousands of genes with significantly lower diversity in cultivated rice than that in wild rice; these genes represent candidate regions selected during domestication. A total of 73 candidate genes that underwent sweeps in both japonica and indica were identified by comparing polymorphism levels in cultivated and wild species. These polymorphism levels were calculated by the reduction of diversity values (ROD = 1 − π cul /π wild ), based on the ratio of the diversity in cultivated rice to the diversity in wild rice. Two well-known rice domestication genes, prog1 (Jin et al. [2008]; Tan et al. [2008]) and sh4 (Li et al. [2006a]), were successfully identified in the putative artificial selection gene set. Gene families related to morphology, growth and transcriptional regulation were enriched among many of the candidate genes. All of these functionally uncharacterized or unknown candidate genes related to artificial selection provide useful guidance for rapidly identifying genes of agronomic significance in rice. Population structure and phylogenetic analyses not only support the hypothesis that japonica and indica were domesticated independently, but they also suggest that japonica was domesticated from the Chinese strain of Oryza rufipogon. The data generated in this study provide a valuable resource for rice improvement.
QTL/gene identification by sequencing-based genotyping in rice
The majority of important agronomic traits in rice are controlled by multiple genes (namely, QTLs). QTL mapping is important for understanding the mechanisms underlying complex agronomic traits via genotyping and phenotyping of a classical population (RIL, DH or BCF2) derived from a cross between two cultivars. Conventional QTL mapping is a powerful method for QTL identification and cloning; however, compared with resequencing-based linkage maps or bin maps, it is generally regarded as a time-consuming and laborious process because lower resolution linkage maps are constructed with low-throughput molecular markers (usually simple sequence repeats, SSR) (Huang et al. [2012b]). Therefore, sequencing-based genotyping provides a more powerful tool for large-scale QTL/gene discovery.
Gao et al. ([2013]) resequenced and genotyped 132 core RILs derived from a cross between the two rice varieties PA64s and 93–11 to construct a SNP-based ultra-high-density linkage map using the NGS method. A total of 43 yield-associated QTLs, including 20 newly identified QTLs, were mapped using a 3,524-SNPs linkage map. Ten QTLs were further mapped using a larger RIL population and two QTLs, qSN8 and qSPB1, were delimited to regions each covering one candidate yield-related gene, DTH8 and LAX1 (Table 2). This precise QTL mapping from core to larger RIL populations using a sequencing-based approach will greatly facilitate QTL cloning and molecular breeding.
Wang et al. ([2011]) resequenced 150 RILs of 93-11/Nipponbare to construct SNP-based ultra-high-density linkage maps and identified 49 QTLs for 14 agronomic traits. Xu et al. ([2010]) resequenced 128 CSSLs of 93-11/Nipponbare to construct a bin map. Nine QTLs for culm length were fine-mapped and one QTL was located in a 791,655-bp region containing the rice “green revolution” gene sd1. Xie et al. ([2010]) sequenced 238 RILs of Zhenshan 97/Minghui 63 and constructed a 209,240-SNPs genetic map. Using the SNP bin map, Yu et al. ([2011]) identified 22 QTLs for four yield traits. The mapping interval of GS3 for grain length was narrowed down from a 6.0-Mb region in the RFLP/SSR genetic map to a 197-kb region. This indicated that the high-density SNP bin map could improve the power of QTL detection. Compared with the PCR-based marker method, the sequencing-based method was faster and more precise for the determination of the recombination breakpoint, achieving a resolution of 40 kb recombinant block on average (Wang et al. [2011]).
Sequencing-based GWAS has recently been applied as a high-throughput QTL mapping method to dissect agronomic traits in a variety of rice germplasm resources. Han and Huang et al. ([2013]) published a brief scheme from a typical sequence-based GWAS involving the identification of high-quality haplotypes to associate molecular markers with phenotypes accurately. QTL mapping by GWAS in rice germplasm can be used as a complementary strategy for classical biparental cross-mapping of dissecting complex traits, but it seems to be effective only for genes with large effects.
Using the GWAS method, Huang et al. ([2010]) identified 49 QTLs for 14 agronomic traits via resequencing 517 rice landraces. Six loci were confirmed to be close to previously identified genes. They later used 950 worldwide rice varieties and identified 32 new loci associated with flowering time and 10 with grain-related traits, indicating that the use of a larger sample size increases the power to detect trait-associated QTLs using GWAS (Huang et al. [2012b]), Zhao et al. ([2011]) identified dozens of common variants influencing 34 complex traits via resequencing 413 diverse accessions of O. sativa. These GWAS research platforms in rice directly link molecular variation in genes and metabolic pathways with the germplasm resources, accelerating varietal development and crop improvement.
Large-scale genome sequencing can also be used for rapid mapping of rare and spontaneous mutations using resequencing technologies. The SHOREmap pipeline developed for this purpose has been incorporated into several modules and performs various functions, from mapping to de novo marker identification through deep sequencing, as well as annotation of candidate mutations in Arabidopsis (Schneeberger and Weigel [2011]). MutMap (mutation map), or Next-Generation Mapping (NGM), is a method for rapid isolation of mutant genes based on whole genome resequencing of small pooled DNA from an F2 or F3 segregating population (Austin et al. [2011]; Abe et al. [2012a]). Yang ([2013]) used bulked segregant analysis (BSA), combined with NGS (referred to as NGS-BSA) to identified six QTLs for cold tolerance at the seedling stage via resequencing DNA pools from 385 extremely tolerant F3 individuals of Nipponbare/LPBG. QTL-seq is another method for rapid identification of QTLs by resequencing RILs or F2 genetic populations composed of 20–50 individuals per population showing extreme opposites in trait values for a given phenotype in segregating progeny. Takagi et al. ([2013]) identified two QTLs for seedling vigor using 531 F2 individuals of Dunghan Shali/Hitomebore. Two loci were confirmed to be close to the previously reported QTLs qPHS3-2S and qPHS-1 (Yano et al. [2012]; Abe et al. [2012b]). QTL-seq can generally be applied in population genomics studies to rapidly identify genomic regions that have undergone artificial or natural selective sweeps.
Improvement of biparental genome sequences of a hybrid rice
Hybrid rice breeding has great potential for improving rice yield. Our research group has resequenced 132 RILs derived from a cross between PA64s and 93–11, the parents of the pioneer super hybrid rice LYP9, together with their parents using Solexa sequencing technologies. We generated 244 GB of raw data, with approximately four-fold depth per RIL, 48-fold depth for PA64s and 36-fold depth for 93–11. A linkage map was constructed with an average interval of approximately 0.392 cM between recombinant blocks (Gao et al. [2013]).
Based on the constructed map of the graphic genotypes, we distinguished the reads from 132 LYP9 RILs to fill in the remaining gaps in the PA64s genome sequence. A total of 29.3 Mb new sequences were used to fill in the gaps. In the non-repeated regions, 36,033 loci with homozygous genotypes were identified as single base errors. Using the Sanger strategy, the genome sequence of PA64s now comprises 382 Mb.
Similarly, we also updated the published 93–11 genome sequences, which comprise 423 Mb, including 369.8 Mb quality sequences located on chromosomes, filling in approximately 3.8 Mb of new sequences to cover 1,493 gaps. Using the linkage map obtained from the RILs, we realigned the PDRs (parental derived reads) to the 93–11 genome sequence and corrected 62,650 SNP errors (Figure 1). The high-quality genome sequences of PA64s and 93–11 were improved by RIL linkage mapping, providing the basis for detailed genetic analysis. Therefore, resequencing a segregating population can be used to improve multiple reference genome sequences. The improved sequence information, which was uploaded to the rice genome website (http://rice.genomics.org.cn/rice/), may be beneficial for improving super hybrid rice (Gao et al. [2013]).

An example of falsely anchored scaffolds revealed by linkage map and syntenic analysis. The region of falsely anchored scaffolds disturbed the normal linkage relationships in the graphic map of the genotypes and was not supported by normal syntenic analysis with the reference.
Future perspectives
The birth of NGS technologies is a landmark event in functional genomics, creating a new era of rice resequencing in a highly accelerated manner based on the high-quality rice reference genomes. It provides the large amount of genome sequencing data available in rice, including more than 1,500 one-fold rice genome sequences from natural populations and thousands of low-fold genome sequences from various rice lines of crossed-based genetic populations of DH, RILs F2 or ILs (introgression lines) (Tables 1 and 2), even the increasing reference genome sequences such as Nipponbare, 9311, and Kasalath (Kanamori et al. [2013]). The genome sequences of PA64s and GLA4 are further improving. Furthermore, increasing numbers of rice germplasm resources and genetic materials are being resequenced; for example, the International Rice Research Institute (IRRI), Chinese Academy of Agricultural Sciences (CAAS) and Beijing Genomics Institute (BGI) –Shenzhen, have collaboratively been resequencing 3,000 accessions of global rice germplasm from the IRRI genebank since 2010. The increasing amount of sequenced genome information has been applied broadly in analyses of genetic diversity, genome variation, RNA sequences and domestication processes, even in cultivars improvement. The resequencing-based method shows a series of obvious advantages; it is approximately 20-times faster in terms of data collection and 35-times more precise in detecting recombination breakpoints, compared to genetic mapping with PCR-based markers (Huang et al. [2009]). However, one of the major drawbacks that limit the use of NGS technology, especially in de novo sequencing, is the short sequencing read lengths. Owing to significant inter- and intra-species chromosomal structural changes induced in rice by InDels, duplications, inversions, translocations and transpositions (Wu et al. [2009]; Hurwitz et al. [2010]; Lin et al. [2012]), assembly and mapping of NGS short-read sequences is complex and relatively difficult. High-quality sequencing approaches have been suggested in conjunction with high-throughput sequencing for comparative genomics analyses and genome evolution studies (Alkan et al. [2011]). A technology which combines the massive throughput of the NGS with the long read lengths achieved by electrophoresis-based Sanger sequencing, would enable rapid, high-quality production of de novo genome sequences (Hert et al. [2008]). Future directions in the field of DNA sequencing are the ability to use individual molecules without any library preparation or amplification, the identification of specific nucleotide modifications, and generation of longer sequence reads (Kircher and Kelso [2010]). Therefore, it is necessary to develop multiple sequencing approaches and platforms, including the third-generation long read technologies, high-quality long-insert clones and new assembly algorithms.
Despite the advantages of efficiency in resequencing, the availability of a series of reasonable and adequate genetic resources is a prerequisite for the implementation of this technology. More than 100,000 accessions of diverse rice germplasm in global genebanks are available, but in fact, genetic diversity in modern rice cultivars has become increasingly narrow. In order to enhance the identification and use of the genetic diversity in rice, Frankel ([1984]) proposed the development of “core collections” for effective use of these germplasm. Subsequently, a series of “core collections” has been developed based on agro-morphological or biochemical traits and ecogeographical information as well as molecular markers and genome information (Kojima et al. [2005]; Huang et al. [2009]). Kojima et al. ([2005]) developed a rice diversity research set of germplasm (RDRS) from the NIAS (National Institute of Agrobiological Sciences, Japan) Genebank for in-depth genetic diversity analysis via development of genetic materials, such as substitute backcrossed lines or chromosome introgression lines. Yamamoto et al. ([2010]) clarified the dynamics of the pedigree haplotypes using 151 representative Japanese cultivars and 1,917 polymorphism SNPs between Nipponbare and Koshihikari. This information can be used to predict particular haplotypes associated with desirable phenotypes during the Japanese rice breeding process, and help in using such haplotype blocks for rice improvement. Sampling and population sizes directly affect genome variation analysis; therefore, Huang et al. ([2012a]) used more germplasm resources and a larger natural population to detect genome variation. Using this approach, at least 7,970,359 SNPs were identified from 1,529 genome sequences that were successfully used in the search for signatures of selection. We sequenced LYP9 RILs, and established an ideal platform for molecular breeding. Accumulated data on the various traits and on genome polymorphism in the collection will be beneficial for the construction of a genotype-phenotype database and will enhance the efficient use of rice genetic resources for the development of new cultivars.
Broadening genetic diversity in rice is one of the most important breeding measures required for overcoming the bottleneck in increasing yield. Discovering valuable genes and alleles from the global rice germplasm demands a long-term collective effort to increasing genetic diversity. The currently available genome information indicates that much more natural allelic variations are present in wild rice than in domesticated rice. Breeders have mainly utilized intersubspecies heterosis and elite genes of wild rice germplasm to broaden genetic diversity, such as hybrid rice sterility lines Zhenshan97A with the wild rice CMS-DA (cytoplasmic male sterility) gene and the indica sterile line PA64s on a japonica background. The functional allelic loci obtained from sequencing-based GWAS mapping have greatly accelerated diverse germplasm mining and molecular breeding. More than 700 cloned rice genes and the sequencing-based GWAS loci will help fulfill the ultimate goal of determining the function of every gene in the rice genome in future through a highly coordinated effort facilitated by the International Rice Functional Genomics Project (IRFGP) (Zhang et al. [2008]; Ikeda et al. [2013]). The identification of the functional genes in cultivated rice and the high degree of natural allelic variation in wild rice will be beneficial for developing elite super rice varieties such as new idiotype super rice with indica-japonica heterosis (Liu et al. [2012]; Guo and Ye [2014]) or green super rice (Zhang [2007]) through genome selection or molecular breeding via gene design.
Conclusion
In this review, we summarize the current status of whole genome resequencing of diverse germplasm resources in rice using NGS technology. More than 1,500 accessions of rice germplasm resources from natural populations and thousands of various rice lines of biparental populations have been resequenced, and effectively utilized in evolutionary analysis, rice genomics and functional genomics studies. We discuss the limitations, application and future prospects of rice resequencing.
Abbreviations
- NGS:
-
Next-generation sequencing
- GWAS:
-
Genome-wide association studies
- SNP:
-
Single ncleotide polymorphisms
- BAC:
-
Bacterial artificial chromosome
- SV:
-
Structure variation
- InDels:
-
Insertions/deletions
- CNV:
-
Copy number variations
References
Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R: Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotech 2012, 30: 174–178. 10.1038/nbt.2095
Abe A, Takagi H, Fujibe T, Aya K, Kojima M, Sakakibara H, Uemura A, Matsuoka M, Terauchi R: OsGA20ox1 , a candidate gene for a major QTL controlling seeding vigor in rice. Theor Appl Genet 2012, 125: 647–657. 10.1007/s00122-012-1857-z
Alkan C, Sajjadian S, Eichler EE: Limitations of next-generation genome sequence assembly. Nat Methods 2011, 8: 61–65. 10.1038/nmeth.1527
Ansorge WJ: Next-generation DNA sequencing techniques. New Biotech 2009, 25(4):195–203. 10.1016/j.nbt.2008.12.009
Ashikari M, Sakakibara H, Lin SY, Yamamoto T, TTakashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M: Cytokinin oxidase regulates rice grain production. Science 2005, 309: 741–745. 10.1126/science.1113373
Austin RS, Vidaurre D, Stamatiou G, Breit R, Provart NJ, Bonetta D, Zhang JF, Fung P, Gong Y, Wang P, McCourt P, Guttman DS: Next-generation mapping of Arabidopsis genes. Plant J 2011, 67: 715–725. 10.1111/j.1365-313X.2011.04619.x
Doi K, Izawa T, Fuse T, Yamanouchi U, Kubo T, Shimatani Z, Yano M, Yoshimura A: Ehd1 , a B-type response regulator in rice, confers short-day promotion of flowering and controls FT-like gene expression independently of Hd1 . Genes Dev 2004, 18: 926–936. 10.1101/gad.1189604
Duan MJ, Sun ZZ, Shu LP, Tan YN, Yu D, Sun XW, Liu RF, Li YJ, Gong SY, Yuan DY: Genetic analysis of an elite super-hybrid rice parent using high-density SNP markers. Rice 2013, 6: 21. 10.1186/1939-8433-6-21
Fan CC, Xing YZ, Mao HL, Lu TT, Han B, Xu CG, Li XH, Zhang QF: GS3 , a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet 2006, 112(6):1164–1171. 10.1007/s00122-006-0218-1
Feuillet C, Leach JE, Rogers J, Schnable PS, Eversole K: Crop genome sequenceing: lessons and rationals. Trends Plant Sci 2011, 16: 77–88. 10.1016/j.tplants.2010.10.005
Frankel OH: Genetic perspectives of germplasm conservation. In “Genetic manipulation: impact on man and society”. Edited by: Arber WK, Llinensee K, Peacock WJ, Stralinger P. Cambridge University Press, Cambridge; 1984:161–170.
Fullwood MJ, Wei CL, Liu ED, Ruan YJ: Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res 2009, 19: 521–532. 10.1101/gr.074906.107
Furutani I, Sukegawa S, Kyozuka J: Genome-wide analysis of spatial and temporal gene expression in rice panicle development. Plant J 2006, 46: 503–511. 10.1111/j.1365-313X.2006.02703.x
Gao ZY, Zeng DL, Cui X, Zhou YH, Yan MX, Huang DN, Li JY, Qian Q: Map-based cloning of the ALK gene, which controls the gelatinization temperature of rice. Sci. China C Life Sci 2003, 46: 661–668. 10.1360/03yc0099
Gao Q, Yue GD, Li WQ, Wang JY, Xu JH, Yin Y: Recent progress using high-throughput sequencing technologies in plant molecular breeding. J Integr Plant Biol 2012, 54(4):215–227. 10.1111/j.1744-7909.2012.01115.x
Gao ZY, Zhao SC, He WM, Guo LB, Peng YL, Wang JJ, Guo XS, Zhang XM, Rao YC, Zhang C, Dong GJ, Zheng FY, Lu CX, Hu J, Zhou Q, Liu HJ, Wu HY, Xu J, Ni PX, Zeng DL, Liu DH, Tian P, Gong LH, Ye C, Zhang GH, Wang J, Tian FK, Xue DW, Liu Y, Zhu L, et al.: Dissecting yield-associated loci in super hybrid rice by resequencing recombinant inbred lines and improving parental genome sequences. Proc Natl Acad Sci U S A 2013, 110(35):14492–14497. 10.1073/pnas.1306579110
Garries AJ, Tai TH, Coburn J, Kresovich S, McCouch S: Genetic structure and diversity in Oryza sativa L. Genetics 2005, 169: 1631–1638. 10.1534/genetics.104.035642
Goff SA, Ricke D, Lan TH, Presting G, Wang RL, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchison D, Martin C, Katagiri F, Lange BM, Moughamer T, Xia Y, Budworth P, Zhong JP, Miguel T, Paszkowski U, Zhang SP, Colbert M, Sun WL, Chen LL, Cooper B, Park S, Wood TC, Mao L, Quail P, et al.: A draft sequence of the rice genome ( Oryza sativa L. ssp. japonica ). Science 2002, 296: 92–100. 10.1126/science.1068275
Guo LB, Ye GY (2014) Use of major quantitative trait loci to improve grain yield of rice. Rice Sci 21(2). doi:10.1016/S1672–6308(13)60174–2 Guo LB, Ye GY (2014) Use of major quantitative trait loci to improve grain yield of rice. Rice Sci 21(2). doi:10.1016/S1672-6308(13)60174-2
Guo LB, Cheng SH, Qian Q: Highlights in sequencing and analysis of rice genome. Chinese J Rice Sci 2004, 18(6):557–562.
Han B, Huang XH: Sequencing-based genome-wide association study in rice. Curr Opin Plant Biol 2013, 16: 133–138. 10.1016/j.pbi.2013.03.006
He GM, Zhu XP, Elling A, Chen LB, Wang XF, Guo L, Liang M, He H, Zhang HR, Chen F, Qi Y, Chen R, Deng X: Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell 2010, 22: 17–33. 10.1105/tpc.109.072041
He Z, Zhai W, Wen H, Tang T, Wang Y, Lu X, Greenberg A, Hudson R, Wu C, Shi S: Two evolutionary histories in the genome of rice: the roles of domestication genes. PLoS Genet 2011, 7: 9.
Hert DG, Fredlake CP, Barron AE: Advantages and limitations of next-generation sequencing technologies: a comparison of electrophoresis and non-electrophoresis methods. Electrophoresis 2008, 29: 4618–4626. 10.1002/elps.200800456
Hu J, Zhu L, Zeng D, Gao Z, Guo L, Fang Y, Zhang G, Dong G, Yan M, Liu J, Qian Q: Identification and characterization of NARROW AND ROLLED LEAF 1 , a novel gene regulating leaf morphology and plant architecture in rice. Plant Mol Biol 2010, 73(3):283–292. 10.1007/s11103-010-9614-7
Huang XH, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, Guan JP, Fan DL, Weng QJ, Huang T, Dong GJ, Sang T, Han B: High-throughput genotyping by whole-genome resequencing. Genome Res 2009, 19: 1068–1076. 10.1101/gr.089516.108
Huang XH, Wei XH, Sang T, Zhao Q, Feng Q, Zhao Y, Li CY, Zhu CR, Lu TT, Zhang ZW, Li M, Fan DL, Guo YL, Wang A, Wang L, Deng LW, Li WJ, Lu YQ, Weng QJ, Liu KY, Huang T, Zhou TY, Jing YF, Li W, Lin Z: Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet 2010, 42(11):961–967. 10.1038/ng.695
Huang XH, Kurata N, Wei XH, Wang ZX, Wang A, Zhao Q, Zhao Y, Liu KY, Lu HY, Li WJ, Guo YL, Lu YQ, Zhou CC, Fan DL, Weng QJ, Zhu CR, Huang T, Zhang L, Wang YC, Feng L, Furuumi HY, Kubo T, Miyabayashi T, Yuan XP, Xu Q: A map of rice genome variation reveals the origin of cultivated rice. Nature 2012, 490: 497–503. 10.1038/nature11532
Huang XH, Zhao Y, Wei XH, Li CY, Wang A, Zhao Q, Li WJ, Guo YL, Deng LW, Zhu CR, Fan DL, Lu YQ, Weng QJ, Liu KY, Zhou TY, JingYF SLZ, Dong GJ, Huang T, Lu TT, Feng Q, Qian Q, Li JY, Han B: Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat Genet 2012, 44(1):32–41. 10.1038/ng.1018
Huang XH, Lu TT, Han B: Resequencing rice genomes: an emerging new era of rice genomics. Trends Genet 2013, 29(4):225–230. 10.1016/j.tig.2012.12.001
Hurwitz BL, Kudrna D, Yu Y, Sebastian A, Zuccolo A, Jackson S, Ware D, Wing R, Stein L: Rice structural variation: a comparative analysis of structural variation between rice and three of its closest relatives in the genus Oryza . Plant J 2010, 63: 990–1003. 10.1111/j.1365-313X.2010.04293.x
Ikeda M, Miura K, Aya K, Kitano H, Matsuoka M: Genes offering the potential for designing yield-related traits in rice. Curr Opin Plant Biol 2013, 16: 213–220. 10.1016/j.pbi.2013.02.002
The map-based sequence of the rice genome Nature 2005, 436: 793–800. 10.1038/nature03895
Jeong DH, Park S, Zhai J, Gurazada SG, De Paoli E, Meyers BC, Green PJ: Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 2011, 23: 4185–4207. 10.1105/tpc.111.089045
Jeong IS, Yoon UH, Lee GS, Ji HS, Lee HJ, Han CD, Hahn JH, An GH, Kim YH: SNP-based analysis of genetic diversity in anther-derived rice by whole genome sequencing. Rice 2013, 6(1):6. 10.1186/1939-8433-6-6
Jiao YL, Jia PX, Wang XF, Sua N, Yu L, Zhang DF, Ma LG, Feng Q, Jin ZQ, Li L, Xue YB, Cheng ZK, Zhao HY, Han B, Deng XW: A tiling microarray expression analysis of rice chromosome 4 suggests a chromosome-level regulation of transcription. Plant Cell 2005, 17: 1641–1657. 10.1105/tpc.105.031575
Jiao Y, Wang Y, Xue D, Wang J, Yan M, Liu G, Dong G, Zeng D, Lu Z, Zhu X, Qian Q, Li J: Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat Genet 2010, 42(6):541–544. 10.1038/ng.591
Jin J, Huang W, Gao JP, Yang J, Shi M, Zhu MZ, Luo D, Lin HX: Genetic control of rice plant architecture under domestication. Nat Genet 2008, 40: 1365–1369. 10.1038/ng.247
Kanamori H, Fujisawa M, Katagiri S, Oono Y, Fujisawa H, Karasawa W, Kurita K, Sasaki H, Mori S, Hamada M, Mukai Y, Yazawa T, Mizuno H, Namiki N, Sasaki T, Katayose Y, Matsumoto T, Wu J: A BAC physical map of aus rice cultivar 'Kasalath', and the map-based genomic sequence of 'Kasalath' chromosome 1. Plant J 2013, 76: 699–708. 10.1111/tpj.12317
Kircher M, Kelso J: High-throughput DNA sequencing–concepts and limitations. Bioessays 2010, 32(6):524–536. 10.1002/bies.200900181
Kojima Y, Ebana K, Fukuoka S, Nagamine T, Kawase M: Development of an RFLP-based rice diversity research set of germplasm. Breed Sci 2005, 55: 431–440. 10.1270/jsbbs.55.431
Komatsu M, Maekawa M, Shimamoto K, Kyozuka J: The LAX1 and FRIZZY PANICLE 2 genes determine the inflorescence architecture of rice by controlling rachis-branch and spikelet development. Dev Biol 2001, 231(2):364–373. 10.1006/dbio.2000.9988
Konishi S, Izawa T, Lin SY, Ebana K, Fukuta Y, Sasaki T, Yano M: An SNP caused loss of seed shattering during rice domestication. Science 2006, 312: 1392–1396. 10.1126/science.1126410
Kovach MJ, Sweeney MT, McCouch SR: New insights into the history of rice domestication. Trends Genet 2007, 23: 578–587. 10.1016/j.tig.2007.08.012
Li C, Zhou A, Sang T: Rice domestication by reducing shattering. Science 2006, 311: 1936–1939. 10.1126/science.1123604
Li L, Wang XF, Stolc V, Li XY, Zhang DF, Su N, Tongprasit W, Li S, Cheng Z, Wang J, Deng XW: Genome-wide transcription analyses in rice using tiling microarrays. Nat Genet 2006, 38: 124–129. 10.1038/ng1704
Li SC, Xie KL, LiWB ZT, RenY WSQ, Deng QM, Zheng AP, Zhu J, Liu HN, Wang LX, Ai P, Gao FY, Huang B, Cao XM, Li P: Re-sequencing and genetic variation identification of a rice line with ideal plant architecture. Rice 2012, 5(1):18. 10.1186/1939-8433-5-18
Lin H, Xia P, Wing RA, Zhang Q, Luo M: Dynamic intra-Japonica subspecies variation and resource application. Mol Plant 2012, 5: 218–230. 10.1093/mp/ssr085
Liu J, Tao HJ, Shi S, Ye WJ, Qian Q, Guo LB: Genetics and breeding improvement for panicle type in rice. Chin J Rice Sci 2012, 26(2):227–234.
Lu T, Lu G, Fan D, Zhu C, Li W, Zhao Q, Feng Q, Zhao Y, Guo Y, Li W, Huang X, Han B: Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res 2010, 20: 1238–1249. 10.1101/gr.106120.110
Luo AD, Qian Q, Yin HF, Liu XQ, Yin CX, Lan Y, Tang JY, Tang ZS, Cao SY, Wang XJ, Xia K, Fu XD, Luo D, Chu CC: EUI1 , encoding a putative cytochrome P450 monooxygenase, regulates internode elongation by modulating gibberellin responses in rice. Plant Cell Physiol 2006, 47(2):181–191. 10.1093/pcp/pci233
McNally KL, Childs KL, Bohnert R, Davidson RM, Zhao KY, Ulat V, Zeller G, Clark RM, Hoen DR, Bureau TE, Stokowski R, Ballinger DG, Frazer KA, Cox DR, Padhukasahasram B, Bustamante CD, Weigel D, Mackill DJ, Bruskiewich RM, Rätsch G, Buell CR, Leung H, Leach JE: Genome wide SNP variation reveals relationships among landraces and modern varieties of rice. Proc Natl Acad Sci U S A 2009, 106(30):12273–12278. 10.1073/pnas.0900992106
Miura K, Ashikari M, Matsuoka M: The role of QTLs in breeding of high-yielding rice. Trends Plant Sci 2011, 16(6):319–326. 10.1016/j.tplants.2011.02.009
Molina J, Sikora M, Garud N, Flowers JM, Rubinstein S, Reynolds A, Huang P, Jackson S, Schaal BA, Bustamante CD, Boyko AR, Purugganan MD: Molecular evidence for a single evolutionary origin of domesticated rice. Proc Natl Acad Sci U S A 2011, 108: 8351–8356. 10.1073/pnas.1104686108
Qi J, Qian Q, Bu QY, Li SY, Chen Q, Sun JQ, Liang WX, Zhou YH, Chu CC, Li XG, Ren FG, Palme K, Zhao BR, Chen JF, Chen M, Li CY: Mutation of the rice Narrow leaf1 gene, which encodes a novel protein, affects vein patterning and polar auxin transport. Plant Physiol 2008, 147(4):1947–1959. 10.1104/pp.108.118778
Saitoh K, Onishi K, Mikami I, Thidar K, Sano Y: Allelic diversification at the C ( OsC1 ) locus of wild and cultivated rice: nucleotide changes associated with phenotypes. Genetics 2004, 168: 997–1007. 10.1534/genetics.103.018390
Sasaki A, Ashikari M, Ueguchi-Tanaka M, Itoh H, Nishimura A, Swapan D, Ishiyama K, Saito T, Kobayashi M, Khush G: A mutant gibberelin-synthesis gene in rice. Nature 2002, 416: 701–702. 10.1038/416701a
Satoh K, Doi K, Nagata T, Kishimoto N, Suzuki K, Otomo Y, Kawai J, Nakamura M, Kishikawa TH, Kanagawa S, Arakawa T, Takahashi-Iida J, Murata M, Ninomiya N, Sasaki D, Fukuda S, Tagami M, Yamagata H, Kurita K, Kamiya K, Yamamoto M, Kikuta A, Bito T, Fujitsuka N, Ito K, Kanamori H, Choi R, Nagamura Y, Matsumoto T, Murakami K, et al.: Gene organization in rice revealed by full-length cDNA mapping and gene expression analysis through microarray. PLoS One 2007, 2: e1235. 10.1371/journal.pone.0001235
Schneeberger K, Weigel D: Fast forward genetics enabled by new sequencing technologies. Trends Plant Sci 2011, 16: 282–288. 10.1016/j.tplants.2011.02.006
Shomura A, Izawa T, Ebana K, Ebitani T, Kanegae H, Konishi S, Yano M: Deletion in a gene associated with grain size increased yields during rice domestication. Nat Genet 2008, 40: 1023–1028. 10.1038/ng.169
Subbaiyan GK, Waters DLE, Katiyar SK, Sadananda AR, Vaddadi S, Henry RJ: Genome-wide DNA polymorphisms in elite indica rice inbreds discovered by whole-genome sequencing. Plant Biotechnol J 2012, 10: 623–634. 10.1111/j.1467-7652.2011.00676.x
Sweeney MT, Thomson MJ, Pfeil BE, McCouch S: Caught red-handed: Rc encodes a basic helix-loop-helix protein conditioning red pericarp in rice. Plant Cell 2006, 18: 283–294. 10.1105/tpc.105.038430
Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C, Uemura A, Utsushi H, Tamiru M, Takuno S, Innan H, Cano LM, Kamoun S, Terauchi R: QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 2013, 74: 174–183. 10.1111/tpj.12105
Tan LB, Li XR, Liu FX, Sun XY, Li CG, Zhu ZF, Fu YC, Cai HW, Wang XK, Xie DX, Sun CQ: Control of a key transition from prostrate to erect growth in rice domestication. Nat Genet 2008, 40: 1360–1364. 10.1038/ng.197
Wang ZY, Zheng FQ, Shen GZ, Gao JP, Snustad DP, Li MG, Zhang JL, Hong MM: The amylase content in rice endosperm is related to the posttranscriptional regulation of the waxy gene. Plant J 1995, 7: 613–622. 10.1046/j.1365-313X.1995.7040613.x
Wang Z, Gerstein M, Snyder M: RNA-Seq: A revolutionary tool for transcriptomics. Nat Rev Genet 2009, 10: 57–63. 10.1038/nrg2484
Wang L, Wang A, Huang XH, Zhao Q, Dong GJ, Qian Q, Sang T, Han B: Mapping 49 quantitative trait loci at high resolution through sequencing-based genotyping of rice recombinant inbred lines. Theor Appl Genet 2011, 122: 327–340. 10.1007/s00122-010-1449-8
Wang Y, Bai XF, Yan CH, Gui YJ, Wei XH, Zhu QH, Guo LB, Fan LJ: Genomic dissection of small RNAs in wild rice ( Oryza rufipogon ): lessons for rice domestication. New Phytol 2012, 196(3):914–925. 10.1111/j.1469-8137.2012.04304.x
Wei XJ, Xu JF, Guo HN, Jiang L, Chen SH, Yu CY, Zhou ZL, Hu PS, Zhai HQ, Wan JM: DTH8 suppresses flowering in rice, influencing plant height and yield potential simultaneously. Plant Physiol 2010, 153(4):1747–1758. 10.1104/pp.110.156943
Wilhelm BT, Landry JR: RNA-Seq—quantitative measurement of expression through massively parallel RNA-sequencing. Methods 2009, 48: 249–257. 10.1016/j.ymeth.2009.03.016
Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, Penkett CJ, Rogers J, Bahler J: Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 2008, 453: 1239–1243. 10.1038/nature07002
Wing RA, Ammiraju JSS, Luo MZ, Kim HR, Yu Y, Kudrna D, Goicoechea JL, Wang WM, Nelson W, Rao K, Brar D, Mackill DJ, Han B, Soderlund C, Stein L, SanMiguel P, Jackson S: The Oryza map alignment project: the golden path to unlocking the genetic potential of wild rice species. Plant Mol Biol 2005, 59: 53–62. 10.1007/s11103-004-6237-x
Wu JZ, Fujisawa M, Tian ZX, Yamagata H, Kamiya K, Shibata M, Hosokawa S, Ito Y, Hamada M, Katagiri S, Kurita K, Yamamoto M, Kikuta A, Machita K, Karasawa W, Kanamori H, Namiki N, Mizuno H, Ma JX, Sasaki T, Matsumoto T: Comparative analysis of complete orthologous centromeres from two subspecies of rice reveals rapid variation of centromere organization and structure. Plant J 2009, 60: 806–819. 10.1111/j.1365-313X.2009.04002.x
Xie WB, Feng Q, Yu HH, Huang XH, Zhao Q, Xing YZ, Yu SB, Han B, Zhang QF: Parent-independent genotyping for constructing an ultrahigh-density linkage map based on population sequencing. Proc Natl Acad Sci U S A 2010, 107(23):10578–10583. 10.1073/pnas.1005931107
Xu JJ, Zhao Q, Du PN, Xu CW, Wang BH, Feng Q, Liu QQ, Tang SZ, Gu MH, Han B, Liang GH: Developing high throughput genotyped chromosome segment substitution lines based on population whole-genome re-sequencing in rice. BMC Genomics 2010, 11: 656–669. 10.1186/1471-2164-11-656
Xu X, Liu X, Ge S, Jensen JD, Hu FY, Li X, Dong Y, Gutenkunst RN, Fang L, Huang L, Li JX, He WM, Zhang GJ, Zheng XM, Zhang FM, Li YR, Yu C, Kristiansen K, Zhang XQ, Wang J, Wright M, McCouch S, Nielsen R, Wang J, Wang W: Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotech 2012, 30(1):105–111. 10.1038/nbt.2050
Yamamoto T, Nagasaki H, Yonemaru J, Ebana K, Nakajima M, Shibaya T, Yano M: Fine definition of the pedigree haplotypes of closely related rice cultivars by means of genome-wide discovery of single-nucleotide polymorphisms. BMC Genomics 2010, 11: 267. 10.1186/1471-2164-11-267
Yang ZM: Mapping of QTLs for cold tolerance and a dominant genic male sterility gene SMS in rice. A Ph.D Dissertation of Zhejiang University, Hangzhou, China; 2013.
Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L, Fuse T, Baba T, Yamamoto K, Umebara Y, Nagamura Y: Hd1 , a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 2000, 12: 2473–2483. 10.1105/tpc.12.12.2473
Yano K, Takashi T, Nagamatsu S, Kojima M, Sakakibara H, Kitano H, Matsuoka M, Aya K: Efficacy of microarray profiling data combined with QTL mapping for the identification of a QTL gene controlling the initial growth rate in rice. Plant Cell Physiol 2012, 53: 729–739. 10.1093/pcp/pcs027
Yu J, Hu SN, Wang J, Wong GK, Li SG, Liu B, Deng YJ, Dai L, Zhou Y, Zhang XQ, Cao ML, Liu J, Sun JD, Tang JB, Chen YJ, Huang XB, Lin W, Ye C, Tong W, Cong LJ, Geng JN, Han YJ, Li L, Li W, Hu GQ, Huang XG, Li WJ, Li J, Liu ZW, Li L, et al.: A draft sequence of the rice genome ( Oryza sativa L. ssp. indica ). Science 2002, 296: 79–92. 10.1126/science.1068037
Yu BS, Lin ZW, Li HX, Li XJ, Li JY, Wang YH, Zhang X, Zhu ZF, Zhai WX, Wang XK, Xie DY: TAC1 , a major quantitative trait locus controlling tiller angle in rice. The Plant Journal 2007, 52: 891–898. 10.1111/j.1365-313X.2007.03284.x
Yu HH, Xie WB, Wang J, Xing YZ, Xu CG, Li XH, Xiao JH, Zhang QF: Grains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS One 2011, 6(3):e17595. 10.1371/journal.pone.0017595
Zhang Q: Strategies for developing Green Super Rice. Proc Natl Acad Sci U S A 2007, 104: 16402–16409. 10.1073/pnas.0708013104
Zhang Q, Li J, Xue Y, Han B, Deng CW: Rice 2020: a call for an international coordinated effort in rice functional genomics. Mol Plant 2008, 1: 715–719. 10.1093/mp/ssn043
Zhang G, Guo G, Hu X, Zhang Y, Li Q, Li R, Zhuang R, Lu Z, He Z, Fang X, Chen L, Tian W, Tao Y, Kristiansen K, Zhang X, Li S, Yang H, Wang J, Wang J: Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res 2010, 20: 646–654. 10.1101/gr.100677.109
Zhao KY, Tung CW, Eizenga GC, Wright MH, Ali ML, Price AH, Norton GJ, Islam MR, Reynolds A, Mezey J, McClung AM, Bustamante CD, McCouch SR: Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa . Nat Commun 2011, 2: 467. 10.1038/ncomms1467
Zhou H, Liu QJ, Li J, Jiang DG, Zhou LY, Wu P, Lu S, Li F, Zhu LY, Liu ZL, Chen LT, LiuYG ZCX: Photoperiod-and thermo-sensitive genic male sterility in rice are caused by a point mutation in a novel noncoding RNA that produces a small RNA. Cell Res 2012, 22(4):649–660. 10.1038/cr.2012.28
Zhu BF, Si LZ, Wang ZX, ZhouY ZJJ, Shangguan YY, Lu DF, Fan DL, Li CY, Lin HX, Qian Q, Sang T, Zhou B, Minobe Y, Han B: Genetic control of a transition from black to straw-white seed hull in rice domestication. Plant Physiol 2011, 155: 1301–1311. 10.1104/pp.110.168500
Zou JH, Chen ZX, Zhang SY, Zhang WP, Jiang GH, Zhao XF, Zhai WX, PanXB ZLH: Characterizations and fine mapping of a mutant gene for high tillering and dwarf in rice. Planta 2005, 222(4):604–612. 10.1007/s00425-005-0007-0
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LG and ZG performed the analysis, prepared the tables and figures. LG and QQ wrote the paper. All authors read and approve the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Guo, L., Gao, Z. & Qian, Q. Application of resequencing to rice genomics, functional genomics and evolutionary analysis. Rice 7, 4 (2014). https://doi.org/10.1186/s12284-014-0004-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12284-014-0004-7