
Abstract
Background
MicroRNAs (miRNAs) play crucial roles in the development of hepatocellular carcinoma (HCC). Hsa-microRNA-27b-3p (hsa-miR-27b) is involved in the formation and progression of various cancers, but its role and clinical value in HCC remain unclear.
Methods
The expression of hsa-miR-27b in HCC was examined by quantitative real-time PCR (qRT-PCR) and in situ hybridization (ISH) assays of clinical samples. Cell Counting Kit-8 assays (CCK-8), 5-ethynyl-2′-deoxyuridine (EdU) incorporation assays, Transwell assays, filamentous actin (F-actin) staining and western blot analyses were used to determine the effects of hsa-miR-27b on HCC cells in vitro. Subcutaneous xenograft and lung metastatic animal experiments were conducted to verify the role of hsa-miR-27b in HCC in vivo. In silico prediction, qRT-PCR, western blot, anti-Argonaute 2 (AGO2) RNA immunoprecipitation (RIP) and dual luciferase reporter assays were applied to identify the target genes of hsa-miR-27b. To detect the impacts of hsa-miR-27b on nuclear factor kappa B (NF-кB) signalling cascades mediated by transforming growth factor-activated kinase-binding protein 3 (TAB3), we performed qRT-PCR, western blot assays, immunofluorescence staining, immunohistochemistry (IHC) and dual-luciferase reporter assays. Recombinant oncolytic adenovirus (OncoAd) overexpressing hsa-miR-27b was constructed to detect their therapeutic value in HCC.
Results
The expression of hsa-miR-27b was lower in HCC than in adjacent non-tumourous tissues (ANTs), and the reduced expression of hsa-miR-27b was associated with worse outcomes in patients with HCC. Hsa-miR-27b significantly inhibited the proliferation, migration, invasion, subcutaneous tumour growth and lung metastasis of HCC cells. The suppression of hsa-miR-27b promoted the nuclear translocation of NF-κB by upregulating TAB3 expression. TAB3 was highly expressed in HCC compared with ANTs and was negatively correlated with the expression of hsa-miR-27b. The impaired cell proliferation, migration and invasion by hsa-miR-27b overexpression were recovered by ectopic expression of TAB3. Recombinant OncoAd with overexpression of hsa-miR-27b induced anti-tumour activity compared with that induced by negative control (NC) OncoAd in vivo and in vitro.
Conclusions
By targeting TAB3, hsa-miR-27b acted as a tumour suppressor by inactivating the NF-кB pathway in HCC in vitro and in vivo, indicating its therapeutic value against HCC.
Graphical Abstract

Similar content being viewed by others


Background
Hepatocellular carcinoma (HCC) is one of the most malignant tumours with a poor prognosis, and the incidence of HCC is still increasing globally [1]. It is critical to elucidate the underlying molecular mechanism of HCC formation and progression, to identify new potential diagnostic biomarkers and to develop effective therapeutic interventions for HCC [2]. miRNAs, small non-coding RNAs that consist of approximately 22 nucleotides, were found to have crucial roles in carcinogenesis [3]. miRNAs can regulate multiple genes by targeting messenger RNA (mRNA) transcripts, leading to mRNA translational repression or degradation [4]. The expression patterns of miRNAs are tissue specific; thus, miRNAs can provide a basis for an in-depth understanding of the mechanisms of tumour progression [5, 6].
Many studies have demonstrated that hsa-miR-27b acts as an oncogene or a tumour suppressor in different tumour types [7,8,9]. Studies have shown that the high expression of hsa-miR-27b promotes the proliferation and invasion of cervical cancer and breast cancer [10, 11]. However, hsa-miR-27b could increase the anti-tumour immune response by regulating proliferator-activated receptor γ expression in neuroblastoma cells [12]. In addition, hsa-miR-27b inhibited gastric cancer cell proliferation, migration and invasion in the liver [13]. With respect to HCC, hsa-miR-27b has been reported to be overexpressed in HCC and mediate the proliferation, cell-cycle progression and apoptosis of HCC cells [14]. He et al. showed that hsa-miR-27b was frequently upregulated in HCC and promoted the migration and invasion of cancer cells [15]. However, some studies showed the opposite results. Liang et al. explored the clinical importance of hsa-miR-27b in HCC and found that the expression of hsa-miR-27b in tumour tissues was lower than that in ANTs [16]. Hsa-miR-27b, which was found to be deleted in liver cancer, sensitized HCC cells to chemotherapy by activating p53-dependent apoptosis and reducing cytochrome P450 family 1 subfamily B member 1-mediated drug detoxification [17]. Interestingly, Fu et al. showed that premature hsa-miR-27b was frequently upregulated, while mature hsa-miR-27b was downregulated in HCC tissues compared with ANTs. Mechanistically, long non-coding RNA DNAJC3 anti-sense RNA 1 sponged premature hsa-miR-27b in the nucleus to suppress its maturation and promote cell proliferation [18]. To date, the expression pattern of hsa-miR-27b and its exact roles in HCC are unclear, and therapeutic applications of hsa-miR-27b in HCC remain to be elucidated.
The NF-кB pathway plays a critical role in regulating cellular processes such as immune responses [19], inflammatory reactions (20), apoptosis [21], proliferation and differentiation [22]. Dysfunction of this pathway is associated with the progression of numerous cancers and immune disorders [23]. Under normal conditions, NF-кB is inactivated by binding with the inhibitors of NF-кB (IκB) and is sequestered in the cytoplasm [24]. Upon certain cell stimulation, such as tumour necrosis factor alpha (TNF-α), interleukin-1 and lipopolysaccharide, the activated IκB kinase (IKK) complex induces the phosphorylation and degradation of IκB proteins, which subsequently liberates NF-κB to the nucleus and initiates transcriptional processes [19, 25]. TAB3 is dysregulated in a variety of cancers [26,27,28] and participates in the activation of the NF-кB pathway [29, 30]. Alfredo Criollo et al. reported that overexpression of TAB3 suppressed, while depletion of TAB3 triggered, autophagy in HeLa cells [31]. Overexpression of TAB3 induced constitutive cellular activation of NF-κB signalling [29]. Chen et al. demonstrated that TAB3 was upregulated in ovarian cancer cells and tissues and predicted poor prognosis of patients [32]. In addition, knockdown of TAB3 could retain RELA proto-oncogene (p65) in the cytoplasm, indicating inactivation of the NF-κB pathway [32]. Mechanistically, TAB3 phosphorylates the IKK complex by forming the transforming growth factor-activated kinase 1 (TAK1)/transforming growth factor-activated kinase-binding protein 2 (TAB2)/TAB3 complex, which leads to the degradation of IкB and the nuclear translocation of NF-кB [33]. Although the functions of TAB3 in NF-кB signalling have been well studied, the underlying role of TAB3 in the progression of HCC remains unclear.
In this research, the decreased expression of hsa-miR-27b in HCC tissues was validated by analysis of patient data from our own clinical samples and public database. We demonstrated that hsa-miR-27b exerted a tumour-suppressor effect in HCC by targeting TAB3. Moreover, an oncolytic virus carrying hsa-miR-27b succeeded in suppressing tumour growth in a xenograft animal model, indicating the therapeutic role of hsa-miR-27b in patients with HCC.
Materials and methods
Patients and tissue specimens
A total of 71 pairs of HCC and ANTs from January 2015 to December 2016 were collected from patients with HCC who underwent hepatectomy at the Hepatic Surgery Center, Tongji Hospital of Huazhong University of Science and Technology (HUST) (Wuhan, China). HCC diagnosis was confirmed by histopathology. The median follow-up period was 27 months (range, 1–45 months). Forty-three pairs of snap-frozen HCC specimens were used to obtain total RNA and protein. All procedures were approved by the Ethics Committee of Tongji Hospital, HUST, and conducted according to the principles of the Declaration of Helsinki. Prior written and informed consent was obtained from each patient.
Cell lines and culture
The HCC cell lines HLF, MHCC97H and HCCLM3 were obtained from the Liver Cancer Institute, Zhongshan Hospital, Fudan University, as previously described [34]. The human normal liver cell line WRL68 (PWE-HU129) was purchased from Meilune Biological Technology Co. (Dalian, China). The HCC cell lines Hep3B (GDC0070) and Huh7 (GDC0134) were purchased from the China Center for Type Culture Collection (CCTCC, Wuhan, China). The HCC cell line PLC/PRF/5 (TCHu119) was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). For all cell lines, short tandem repeat (STR) analysis was performed to confirm cell identity. All cell lines were maintained in Dulbecco’s modified Eagle medium (DMEM) (HyClone, UT, USA) with 10% foetal bovine serum (FBS) (Gibco) at 37 °C in a 5% CO2 humid atmosphere.
ISH and IHC analysis
ISH was performed using the ISH Kit (Boster, Bioengineering Company, Wuhan, China). All procedures were performed following the manufacturer’s instructions. Samples were stained with haematoxylin, dehydrated with alcohol, washed with xylene and sealed with flavour sealing tablets. Oligo (5′ digoxin-ACAAAGTTCTGTAGTGC-ACTGA) was used as an ISH probe for hsa-miR-27b. IHC was performed as described previously [35]. The dilutions of antibodies for the IHC procedure are listed in Additional file 1: Table S1. Representative images of ISH and IHC were captured and processed using a DM2300 microscope and ScopeImage 9.0 software (Nanjing Jiangnan Novel Optics Co., Ltd., China). ISH and IHC staining scores were independently determined by three pathologists without prior knowledge of patient information. The overall scores were defined by multiplying the percentage of positive cells by the staining intensity score as described previously [35].
RNA isolation and qRT-PCR
Total RNA from human specimens and cancer cells was extracted by TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. We used a reverse-transcription system (Vazyme, Nanjing, China) and miRcute Plus miRNA First-Strand cDNA Synthesis (Tiangen, Beijing, China) kits to generate cDNA of mRNA and miRNA, respectively. Relative quantification of hsa-miR-27b and the mRNA expression levels were determined by a miRcute Plus miRNA qPCR Detection Kit (Tiangen, Beijing, China) and SYBR Green PCR kit (Vazyme, Nanjing, China) according to the manufacturers’ protocol. GAPDH and U6 small nuclear RNA were used as internal controls for mRNA and miRNA, respectively. We used the comparative CT (2−∆∆CT) method to perform relative quantification analysis. The primers used in this study are listed in Additional file 1: Table S2.
Cell transfection and infection
HCC cells were seeded in six-well plates at 70% density and cultured overnight. Hsa-miR-27b mimics and inhibitors and their respective NC (RiboBio, Guangzhou, China) were transfected with Lipofectamine 3000 (Invitrogen, USA). Small interference RNA (siRNA) targeting TAK1 was used to transiently knock down its expression in HCC cells. si-NC: 5′-UUGUACUACACAAAAGUACUG-3′; si1-TAK1: 5′-GGUAGUAAUUACAGUGAAA-3′; si2-TAK1: 5′-CCCGTGTGAACCATCCTAATA-3′.
Lentiviruses for stably overexpressing or knocking down TAB3 were generated by Shanghai GeneChem Co., Ltd. (Shanghai, China) and used to infect the indicated cells for 48 h. Transfected cells were then selected by culturing in medium containing 2.5 μg/ml puromycin for 2 weeks. The small hairpin RNA (shRNA) target sequences for knocking down the indicated genes were as follows: scramble: 5′-GCCTAAGGTTAAGTCGCCCTCG-3′; sh-TAB3: 5′-CCTCCTTCATACATGCACATA-3′.
CCK-8 assays
Cells were seeded into 96-well plates at 1500 cells per well and incubated at 37 °C. The culture medium was changed to 100 μl of 10% CCK-8 (Dojindo, Kumamoto, Japan) solution at the indicated time and incubated in a cell incubator for 2 h. Optical density (OD) was measured by a Universal Microplate Reader ELx 800 (Bio-Tek, USA) at a wavelength of 450 nm. For each group, the absorbance values were determined by five replicates. After extraction of the blank value, the average gross OD values were used for data analysis.
Transwell assays
Transwell chambers (8 μm pore size, Corning, NY, USA) were used for cell migration assays. In Transwell migration assays, HCC cells suspended in 200 μl of serum-free DMEM (HyClone, UT, USA) were added to the upper chambers, and the lower chambers were filled with 500 μl of DMEM with 10% FBS. In the Transwell invasion assay, before cells were seeded in the upper chamber, chambers were coated with Matrigel (BD Biosciences, NJ, USA) for 3 h. Following 24 h (migration assays) or 48 h (invasion assays) of incubation at 37 °C, the migrated or invaded cells were fixed with 4% paraformaldehyde and stained with 1% crystal violet solution. Stained cells were counted in three random microscopic fields per well by a Nikon Digital ECLIPSE C1 system (Nikon Corporation).
F-actin assay
The indicated cells were seeded in 96-well plates at a density of 1000 cells per well and cultured overnight. Cells were then fixed with 4% paraformaldehyde for 15 min at room temperature. After two washes with phosphate-buffered saline (PBS), 100 µl of Alexa Fluor 555-conjugated phalloidin (Life Technologies, USA) was added into each well for 1 h at room temperature. PBS washes were performed twice, and the nuclei were counterstained with DAPI. The cells were then observed using the EVOS FL auto-imaging system (Life Technologies, USA).
Dual-luciferase reporter assay
The DNA sequence containing the predicted binding site with hsa-miR-27b in the 3′-untranslated region (3′UTR) of TAB3 or the 3′UTR-TAB3 mutant was cloned into the psiCHECK-2-vector (Promega, Madison, WI, USA). Approximately 1 × 105 cells per well were seeded in 24-well plates. After 24 h, the recombinant plasmid psiCHECK-2-3′UTR-TAB3 or psiCHECK-2-3′UTR-TAB3 mutant was co-transfected into cells with hsa-miR-27b mimics, hsa-miR-27b inhibitors or their respective NC using Lipofectamine 3000 (Invitrogen, USA). Total cell protein was extracted with Passive Lysis Buffer (Promega, Madison, WI, USA), and luciferase activity was determined using the Dual-Luciferase Reporter 1000 Assay System (Promega, Madison, WI, USA) with a GloMax 20/20 luminometer (Promega, Madison, WI, USA). Renilla luciferase values were normalized against firefly luciferase activity, and the ratio of Renilla-to-firefly luciferase activity is presented. For NF-кB signalling activation analysis, NF-кB luciferase reporter plasmid containing the minimal promoter with multiple tandem NF-кB binding sites (pNF-кB-Luc, Clontech, Palo Alto, CA, USA) and its control vector (pTAL-Luc, Clontech) were transfected into HCC cells by Lipofectamine 3000 (Invitrogen, USA). The indicated cell lines were treated with TNF-α (10 ng/ml) (MCE, USA).
Immunofluorescence staining
HCC cells were cultured on sterile coverslips in 24-well plates and treated with 20 ng/ml TNF-α (MCE, USA) for 10 min. After treatment, the cells were fixed in 4% paraformaldehyde for 15 min at room temperature, permeabilized with 0.5% Triton X-100 solution for 10 min and incubated with primary antibody at 4 °C overnight. Alexa Flour 488-conjugated anti-rabbit IgG and Alexa Flour 555-conjugated anti-mouse IgG (Beyotime Institute of Biotechnology, Shanghai, China) were used to incubate cells for 1 h at 37 °C. Nuclei were stained with DAPI (Wuhan Goodbio Biotechnology Co., Ltd., Wuhan, China). Images were captured by an EVOS FL auto imaging system (Life Technologies, USA).
Animal experiments
All BALB/c nude mice were purchased from Beijing HFK Bioscience Co., Ltd., and maintained under specific-pathogen-free conditions. For the xenograft tumour model, 1 × 106 indicated tumour cells were suspended in 100 μl of serum-free DMEM and inoculated subcutaneously into the flanks of 4-week-old nude mice. All experimental mice were monitored for 30 days and sacrificed to compare the volume and weight of subcutaneous tumours. For the in vivo metastasis assays, 1 × 106 cells in 100 μl of serum-free DMEM were injected into the tail vein of nude mice. After 2 months, the nude mice were sacrificed, and the lungs were dissected. All of the metastatic foci in the lung were calculated to evaluate the development of pulmonary metastasis.
For the OncoAd-mediated treatment of liver cancer, Hep3B tumour xenografts were established by subcutaneous inoculation of 5 × 105 cells into the right flank of 4-week-old nude mice. Twenty-one days after inoculation, the animals were randomly divided into three groups (n = 7 mice per group) and intra-tumourally injected with either PBS solution, OncoAd NC or OncoAd hsa-miR-27b (diluted in PBS) at multiplicities of infection (MOIs) of 5 × 108. The injections were performed once every other day for a total of four injections. Tumours were measured every 3 days, and volume was calculated by the following formula: volume = (length × width2)/2.
Recombinant adenovirus production
OncoAd hsa-miR-27b and OncoAd NC were constructed by Shanghai GeneChem Co., Ltd. (Shanghai, China). The u6-hsa-miR-27b cassette was cloned into the oncolytic adenoviral vector with the E1B 55-KD gene deletion. The recombinant adenovirus was transfected into 293T cells for amplification. The viral particles were collected, and the titre was detected according to the manufacturer’s operating instructions after purification.
Cytotoxicity assay
Hep3B and WRL68 cells were used for cytotoxicity assays in 96-well or 24-well plates by CCK-8 assays or crystal violet staining. Briefly, cells were pre-seeded in 96-well plates and cultured overnight. OncoAd at the indicated MOIs was used to treat cells for 24 h. Cell viability was measured by CCK-8 assays. Additionally, cells were prepared in the same manner in 24-well plates and cultured for 48 h. Then, the cells were fixed with 4% paraformaldehyde and stained with crystal violet staining solution.
Nuclear and cytoplasmic extraction assay
A total of 1 × 106 HCC cells were collected in a 1.5 ml microcentrifuge tube and then centrifuged at 500g for 5 min. The cells were washed by suspending the cell pellet with PBS. The supernatant was removed, leaving the cell pellet as dry as possible. Cytoplasmic and nuclear fraction protein extraction was performed using an NE-PER nuclear and cytoplasmic extraction reagent kit (Thermo Fisher Scientific, USA) according to the instructions.
In silico analysis
The miRNA expression profiles were retrieved from The Cancer Genome Atlas (TCGA) database (https://tcga-data.nci.nih.gov/tcga/) and the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/gds). Normalization and batch effect elimination were carried out for the two gene datasets using the Limma package in R. Statistical significance was calculated using the Kruskal–Wallis test. The correlation between the survival time and TAB3 expression in patients with HCC was assessed using the GEPIA website (http://gepia.cancer-pku.cn/). The miRDB (http://mirdb.org/miRDB), miRWalk (http://www.umm.uniheidelberg.de/apps-/zmf/mirwalk/) and TargetScan (http://www.targetscan.org/vert_71/) databases were used for miRNA target‐gene prediction.
EdU incorporation assays
HCC cells were seeded in 96-well plates at a density of 1000 cells per well and incubated overnight. Then, the cells were transfected with the indicated mimics or inhibitors for 72 h and subjected to an EdU incorporation assay using a Cell-Light EdU Apollo 567 In Vitro Imaging Kit (RiboBio, Guangzhou, China) according to the manufacturer’s instructions.
RIP assay
We performed RIP using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Darmstadt, Germany) following the manufacturer’s instructions. A detailed description was published previously [35].
Co-immunoprecipitation (co-IP)
The experiments were performed as described previously [36]. Briefly, a total of 1 × 107 cells were lysed with IP lysis buffer for 20 min at 4 °C (IP-lysis buffer: 50 mM Tris–HCl, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 10% glycerol and protease inhibitor cocktail, pH 7.4) and then centrifuged at 13,000 rpm for 10 min. The supernatant was incubated with 30 μl of pre-cleared protein G-conjugated agarose (GE Healthcare Life Sciences) for 4 h at 4 °C. The indicated antibodies were added into cleared supernatants overnight at 4 °C. One per cent of the lysate fraction was reserved as an input. The next day, 25 μl of protein G-conjugated agarose was used to precipitate the antibody–protein mixture at 4 °C for 4 h. The beads were then washed five times in IP wash buffer (50 mM Tris–HCl, 300 mM NaCl, 1% Triton X-100 and 1 mM EDTA, pH 7.4) to remove unbound antibodies. The eluate was separated from the beads by heating at 95 °C for 10 min.
Statistical analyses
We used SPSS 19.0 (IBM, Chicago, IL, USA) or Prism 6.0 (GraphPad Software, La Jolla, CA, USA) software to analyse the data. Student’s t-test (two-tailed) or analysis of variance (ANOVA) test was used to compare tests under the assumptions of normality and the equality of variance. Otherwise, non-parametric tests (Mann–Whitney U-test or Kruskal–Wallis) were used. Categorical data were analysed by χ2 test or Fisher’s exact test. Kaplan–Meier analyses were used to assess the survival between subgroups (log-rank test). A Cox proportional hazards model was used to determine the independent factors of survival based on the variables selected in the univariate and multivariate analyses. The results are presented as mean ± standard deviation (SD). p-Values less than 0.05 were considered statistically significant.
Results
Hsa-miR-27b is downregulated in HCC tissues
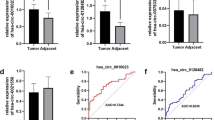
To investigate the role of hsa-miR-27b in HCC, we first analysed its expression level in cohorts of patients with HCC from the TCGA and the GEO (GSE115106) databases. The results showed that hsa-miR-27b expression was significantly decreased in HCC tissues compared with normal liver tissues (Fig. 1A). qRT-PCR analysis of 43 pairs of HCC samples verified the decreased expression of hsa-miR-27b in HCC compared with ANTs (Fig. 1B). ISH was then performed on 71 pairs of HCC specimens (Additional file 1: Table S3) to explore the relationship between hsa-miR-27b and the clinical features of patients with HCC. The ISH scores of hsa-miR-27b were significantly higher in ANTs than in counterpart HCC tissues (Fig. 1C, D and Additional file 1: Fig. S1A). Chi-squared analysis indicated that lower expression of hsa-miR-27b was significantly associated with higher serum α-fetoprotein (AFP) levels and advanced Child–Pugh grade (Additional file 1: Table S4). Kaplan–Meier analysis showed that patients with lower hsa-miR-27b levels exhibited shorter overall survival (OS) (Fig. 1E). In addition, univariate and multivariate Cox regression analyses demonstrated that lower expression of hsa-miR-27b was an independent risk factor for poor prognosis (Fig. 1F and Additional file 1: Table S5). Taken together, these results suggested that hsa-miR-27b was downregulated in HCC and that high expression of hsa-miR-27b predicted better prognosis.

Hsa-miR-27b is downregulated in HCC specimens, and low expression of hsa-miR-27b predicts poor prognosis. A Hsa-miR-27b expression in HCC samples and normal liver tissues was analysed using TCGA (HCC, n = 370; normal liver, n = 50; unpaired t-test) and GSE115016 (HCC, n = 12; normal liver, n = 12; unpaired t-test) datasets. B qRT-PCR analysis of the expression of hsa-miR-27b in HCC and adjacent non-tumourous tissues (ANTs) (Mann–Whitney test). Data were normalized to U6 and are shown as the fold change compared with patient 7. C ISH scoring of hsa-miR-27b in 71 pairs of HCC tissues (Mann–Whitney test). D Representative ISH images of hsa-miR-27b in paired HCC samples. Scale bar: overview images, 100 μm; magnified images, 200 μm. E Kaplan–Meier analysis of the correlation between hsa-miR-27b expression and overall survival in the cohort of patients with HCC (log-rank test). F Forest plot of the multivariate Cox proportional hazards model for overall survival. The bars correspond to 95% confidence intervals. *p < 0.05; **p < 0.01. Data are shown as mean ± SD. T, tumour; N, normal liver; ANTs, adjacent non-tumourous tissues; HCC, hepatocellular carcinoma; HR, hazard risk; CI, confidence interval
Hsa-miR-27b suppresses the proliferation, migration and invasion of HCC cells in vitro
We first determined hsa-miR-27b expression levels in the normal liver cell line WRL68 and six HCC cell lines by qRT-PCR (Fig. 2A). The results showed that the expression level of hsa-miR-27b was lower in the HCC cell lines than in WRL68. On the basis of the endogenous expression of hsa-miR-27b in HCC cell lines, we overexpressed hsa-miR-27b in MHCC97H and Huh7 cells with hsa-miR-27b mimics and knocked down hsa-miR-27b in Hep3B and HCCLM3 cells with hsa-miR-27b inhibitors (Fig. 2B). CCK-8 and EdU incorporation assays showed that hsa-miR-27b overexpression markedly inhibited and downregulation of hsa-miR-27b promoted proliferation of HCC cells (Fig. 2C, D and Additional file 1: Fig. S1B, C). To determine the effect of hsa-miR-27b on the migration and invasion of HCC cells, we carried out Transwell assays. Enhanced expression of hsa-miR-27b significantly impaired the migration and invasion capacities in MHCC97H and Huh7 cells, and vice versa in Hep3B and HCCLM3 cells (Fig. 2E and Additional file 1: Fig. S1D). Since epithelial–mesenchymal transition (EMT) is a key process in HCC metastasis [37], we first carried out fluorescence staining of F-actin in HCC cells with hsa-miR-27b manipulation. Overexpression of hsa-miR-27b inhibited actin cytoskeleton organization, while downregulation stimulated it (Fig. 2F and Additional file 1: Fig. S1E). Additionally, hsa-miR-27b increased the expression of epithelial markers (ZO-1) and decreased the expression of mesenchymal markers (N-cadherin and ZEB1), as evidenced by western blot analysis (Fig. 2G).

Hsa-miR-27b inhibits the proliferation and migration of HCC cells in vitro. A Endogenous expression of hsa-miR-27b in WRL68 and HCC cell lines. Data were normalized to U6 and are shown as fold change compared with HLF cells (n = 3). B Overexpression or knockdown of hsa-miR-27b in HCC cells using hsa-miR-27b mimics or inhibitors, respectively, as measured by qRT-PCR. Data were normalized to U6 and are shown as fold change relative to the control cells (n = 3, Mann–Whitney test). C The proliferation of the indicated cells with overexpression or knockdown of hsa-miR-27b as measured by CCK-8 assays (n = 5, Kruskal–Wallis test). D Representative images of the EdU incorporation assay (left). Scale bar: 100 μm; quantification of EdU-positive cells (right) (n = 3, Mann–Whitney test). E Representative images (top) and quantification (bottom) of cells that migrated or invaded in the indicated groups (n = 3, Mann–Whitney test). Scale bar, 200 μm. F Representative pictures of phalloidin staining for F-actin (red) in the indicated cells. DAPI was used to show the nucleus (blue). Scale bar, 20 μm. G Western blot analysis for EMT markers as indicated. GAPDH was used as a loading control. All experiments were repeated three times. *p < 0.05. **p < 0.01. Data are shown as mean ± SD. NC, negative control
Hsa-miR-27b inhibits tumour growth and lung metastasis in vivo
We stably overexpressed hsa-miR-27b in MHCC97H and Huh7 cells (MHCC97H/hsa-miR-27b, Huh7/hsa-miR-27b) and knocked down hsa-miR-27b in Hep3B and HCCLM3 cells (Hep3B/KD hsa-miR-27b, HCCLM3/KD hsa-miR-27b) by lentivirus (Fig. 3A). HCC cells were subcutaneously injected into nude mice to investigate the effects of hsa-miR-27b on tumourigenicity in vivo. Thirty days after inoculation, the mice were sacrificed to assess tumour growth. Tumour volumes and weights were significantly lower in the mice injected with the hsa-miR-27b-overexpressing MHCC97H cells than in the mice injected with the control cells. The opposite results were observed in the hsa-miR-27b-knockdown Hep3B cells (Fig. 3B). IHC assays of Ki-67 showed that tumour cells with higher hsa-miR-27b levels exhibited weaker proliferation than their corresponding controls (Fig. 3C). In vivo tumour lung metastasis assays showed that the mice bearing MHCC97H cells with hsa-miR-27b overexpression developed fewer metastatic nodules than the mice in the control group. Consistently, knocking down hsa-miR-27b in Hep3B enhanced its metastatic ability, as evidenced by a higher incidence of lung metastasis and more metastatic nodules than in the control group (Fig. 3D).

Hsa-miR-27b inhibits the growth and lung metastasis of HCC cells in vivo. A qRT-PCR analysis of hsa-miR-27b stable overexpression or knockdown efficacy in the indicated cells. Data were normalized to U6 and are shown as fold change relative to the control cells (n = 3, Mann–Whitney test). B Macroscopic images of subcutaneous tumours from mice (n = 7) bearing hsa-miR-27b-overexpressing MHCC97H cells or hsa-miR-27b-knockdown Hep3B cells (left); quantification of the volumes and weights of subcutaneous tumours in the indicated groups (right); unpaired t-test. C Representative haematoxylin and eosin (H&E) staining and IHC of Ki67 in subcutaneous xenografts. Quantification of staining scores was calculated (n = 7, unpaired t-test); Scale bar, 200 μm. D Lung metastasis experiments were performed in nude mice with the indicated cells. D1 and D2 Representative macroscopic and H&E staining images of lung metastases. D3 Quantification of visible tumour nodules. MHCC97H: control group (n = 7); hsa-miR-27b group (n = 7); unpaired t-test. Hep3B: control group (n = 7); KD hsa-miR-27b group (n = 7); Mann–Whitney test. D4 Lung metastatic incidence in different groups; *p < 0.05. **p < 0.01. Data are shown as mean ± SD. ctrl, control; KD, knockdown
TAB3 is the downstream target of hsa-miR-27b
To explore the downstream effectors of hsa-miR-27b, we generated a Venn diagram to overlap the predicted hsa-miR-27b targets from three independent websites: miRDB, miRWalk and TargetScan (Additional file 1: Fig. S2A). The top ten predicted targets were selected for qRT-PCR verification (Fig. 4A1 and Additional file 1: Fig. S2B). The mRNA and protein levels of TAB3, which is involved in the activation of NF-κB signalling, were downregulated by hsa-miR-27b overexpression and upregulated by hsa-miR-27b knockdown (Fig. 4A). Moreover, the mRNA and protein levels of TAB3 decreased coordinately with increasing doses of the hsa-miR-27b mimics in MHCC97H cells (Fig. 4B). The expression of TAB3 in tumours was significantly higher than that in non-tumourous liver tissue, as shown by western blot (Fig. 4C and Additional file 1: Fig. S2C). Additionally, the hsa-miR-27b level was negatively correlated with the mRNA (r = −0.2395, p < 0.05) and protein levels of TAB3 (r = −0.4124, p < 0.05) (Fig. 4D). IHC analysis of subcutaneous tumour sections from nude mice showed that the expression level of TAB3 increased in the hsa-miR-27b knockdown group and decreased in the hsa-miR-27b overexpression group (Additional file 1: Fig. S2D). AGO2 was initially reported as the catalytic centre of the RNA-induced silencing complex [38]. Thus, we conducted an anti-AGO2 RIP assay. The results showed that hsa-miR-27b and TAB3 mRNA were enriched in anti-AGO2 precipitate. Moreover, the abundance of TAB3 mRNA in anti-AGO2 precipitate increased in the hsa-miR-27b-overexpressing MHCC97H cells and decreased in the hsa-miR-27b-knockdown Hep3B cells (Fig. 4E). The potential binding site of hsa-miR-27b in the TAB3 3′UTR was predicted by miRBase (Fig. 4F), and a dual luciferase reporter assay showed that the relative luciferase activity of TAB3-3′UTR was decreased in hsa-miR-27b-overexpressing MHCC97H and Huh7 cells, but increased in hsa-miR-27b-knockdown Hep3B and HCCLM3 cells. However, co-transfection of TAB3-MUT and the hsa-miR-27b mimics or inhibitors had no such effect (Fig. 4F and Additional file 1: Fig. S2E). These results demonstrated that hsa-miR-27b bound to the 3′UTR of TAB3 mRNA and decreased its expression in an AGO2-dependent manner.

TAB3 is the downstream target of hsa-miR-27b. A1, A2 qRT-PCR (A1) and western blot (A2) analysis of TAB3 mRNA and protein levels after overexpression or knockdown of hsa-miR-27b in HCC cells. Data were normalized with GAPDH and are shown as the fold change relative to the control cells. GAPDH was used as a loading control for western blot analysis (n = 3 for qRT-PCR, Mann–Whitney test). B MHCC97H cells were transfected with hsa-miR-27b mimics at the indicated concentrations. qRT-PCR analysis of TAB3 mRNA (top) and western blot analysis of TAB3 protein levels (bottom) were performed. Data were normalized to GAPDH and are shown as fold change compared with the cells treated with an NC for qRT-PCR (n = 3, one-way ANOVA). GAPDH was used as a loading control for western blot analysis. C Relative TAB3 expression in HCC tissues (n = 43) compared with ANTs (n = 43) analysed by western blots. D Linear regression analysis between relative TAB3 mRNA (left) or protein levels (right) and hsa-miR-27b expression in HCC specimens (n = 43). E RNA immunoprecipitation assays of 3′UTR-TAB3 enrichments by anti-IgG or anti-AGO2 antibodies in the hsa-miR-27b-overexpressing MHCC97H cells (top) or the hsa-miR-27b-knockdown Hep3B cells (bottom). Data are shown as percentage of 10% input (n = 3, Kruskal–Wallis test). F Schematic diagram of hsa-miR-27b binding sequences in the 3′UTR of TAB3 and mutants in the 3′UTR of TAB3 (TAB3-MUT) (top). Dual luciferase reporter assay of MHCC97H and Hep3B cells co-transfected with wild-type (WT) or mutated TAB3 3′UTR luciferase reporter plasmids with hsa-miR-27b mimics or inhibitors (n = 3, Mann–Whitney test). A, B, E and F were repeated three times. *p < 0.05. **p < 0.01. Data are shown as mean ± SD. NC, negative control; ANT: adjacent non-tumourous tissues; HCC, hepatocellular carcinoma; 3′UTR, 3′ untranslated region; ctrl, control; KD, knockdown
TAB3 mediates the impacts of hsa-miR-27b in HCC
IHC analysis of TAB3 in paired clinical HCC specimens was performed to analyse the clinical relevance of TAB3. The average scoring of TAB3 was significantly higher in HCC than in its corresponding ANTs (Fig. 5A and Additional file 1: Fig. S3A). Kaplan–Meier survival analysis from the GEPIA database showed that patients with HCC with higher expression of TAB3 protein had significantly shorter OS times than patients with HCC with lower TAB3 expression (Fig. 5B). To investigate the role of TAB3 in HCC, we first analysed its expression level in the HCC cell line (Fig. 5C). We then used lentivirus to stably knock down TAB3 in MHCC97H and Huh7 cells with relatively low endogenous hsa-miR-27b expression, and overexpressed TAB3 in HCCLM3 and Hep3B cells with relatively high endogenous hsa-miR-27b expression (Fig. 5D and Additional file 1: Fig. S3B). The CCK-8 assay showed that downregulation of TAB3 inhibited, and overexpression of TAB3 promoted, cell proliferation in HCC cells (Fig. 5E and Additional file 1: Fig. S3C). Transwell assays showed that cells with higher TAB3 expression levels displayed stronger migration and invasion than those with lower TAB3 expression levels (Fig. 5F and Additional file 1: Fig. S3D).

The impacts of hsa-miR-27b on HCC cells were mediated by TAB3. A Representative images (left) and quantification (unpaired t-test) (right) of TAB3 IHC staining in 71 pairs of HCC tissues and ANTs. Scale bar: left, 100 μm; right, 200 μm. B Kaplan–Meier analysis of the relationship between TAB3 expression and overall survival of patients with HCC in the GEPIA database (log-rank test). C TAB3 mRNA and protein expression in HCC cell lines as determined by qRT-PCR (top) and western blots (bottom). Data were normalized to GAPDH and are shown as fold change compared with Hep3B cells for qRT-PCR (n = 3). GAPDH was used as a loading control for western blot analysis. D qRT-PCR (top) and western blot (bottom) analysis of TAB3 overexpression or knockdown efficacy in the indicated cells. Data were normalized to GAPDH and are shown as fold change relative to their respective control cells for qRT-PCR (n = 3, Mann–Whitney test). GAPDH was used as a loading control for western blot analysis. E CCK-8 assay of the indicated cells (n = 5, Kruskal–Wallis test). F Representative images of cell migration and invasion in the indicated groups (top). Scale bar, 200 μm. Quantification of cell migration and invasion (n = 3, Mann–Whitney test) (bottom). G CCK-8 assay for the indicated cell groups (n = 5, Kruskal–Wallis test). H Representative images and quantification of migrated and invaded cells in the indicated cell groups (n = 3, Mann–Whitney test). Scale bar, 200 μm. I Western blotting for EMT markers as indicated. GAPDH was used as a loading control. C–I were repeated three times. *p < 0.05. **p < 0.01. Data are shown as mean ± SD. ANTs, adjacent non-tumourous tissues; HCC, hepatocellular carcinoma; vec, vector; OE, overexpression; scr, scramble; sh, small hairpin RNA; ctrl, control; vec, vector; pc3.1-, pcDNA3.1-; KD, knockdown; sh, small hairpin RNA
To confirm whether TAB3 is critically involved in hsa-miR-27b-mediated cell growth, migration and invasion, we enhanced TAB3 expression in the hsa-miR-27b-overexpressing MHCC97H cells, or knocked down TAB3 in the hsa-miR-27b-knockdown Hep3B cells. The impaired proliferation, migration and invasion induced by hsa-miR-27b overexpression were recovered by overexpressing TAB3 in MHCC97H cells, and vice versa in the hsa-miR-27b-knockdown Hep3B cells with TAB3 downregulation (Fig. 5G–I).
Hsa-miR-27b inhibits NF-кB activity by targeting TAB3 in HCC cells
Gene set enrichment analysis (GSEA) for the patient cohort from TCGA showed that the level of hsa-miR-27b was inversely correlated with the NF-кB-activated gene signatures, suggesting that hsa-miR-27b might be involved in the de-activation of NF-кB signalling, which was consistent with its negative regulation of TAB3 expression (Fig. 6A). Furthermore, cyclin D1 (CCND1), matrix metallopeptidase 9 (MMP9), vascular endothelial growth factor C (VEGFC), Myc proto-oncogene (c-myc) and signal transducer and activator of transcription 3 (STAT3), the downstream effectors of NF-кB signalling [39], were found to be negatively regulated by hsa-miR-27b, implying that hsa-miR-27b downregulation contributed to NF-кB activation in HCC cells (Fig. 6B). A dual-luciferase reporter assay showed that overexpression of hsa-miR-27b reduced NF-кB-induced luciferase activity in MHCC97H and Huh7 cells (Fig. 6C). Immunofluorescence staining showed that overexpressing hsa-miR-27b led to a significant cytoplasmic location of p65 and weaker fluorescence intensity of p-p65, whereas silencing hsa-miR-27b resulted in p65 accumulation in the nucleus and stronger signals for p-p65 (Fig. 6D). IHC analysis of subcutaneous tumour sections from nude mice showed that the expression level of p-p65 increased in the low hsa-miR-27b expression group and decreased in the high hsa-miR-27b expression group (Fig. 6E). Collectively, these results suggested that hsa-miR-27b inhibited NF-кB signalling in HCC.

Hsa-miR-27b inhibits NF-кB signalling activity by targeting TAB3 in HCC cells. A GSEA of the correlation between hsa-miR-27b and NF-кB target gene signatures using the cohort of patients with HCC in the TCGA database. B NF-кB target gene mRNA (left) and protein (right) levels in the indicated cells. Data were normalized to GAPDH and are shown as fold change relative to the control cells for qRT-PCR (n = 3, Mann–Whitney test). GAPDH was used as a loading control for western blotting. C Dual luciferase reporter analysis for NF-кB transcriptional activity in the indicated cells. Data were normalized to the NC cells (n = 3, Mann–Whitney test). D Confocal images of immunofluorescence staining of p65 and p-p65 in the indicated cells. Scale bar, 200 μm (n = 3, Mann–Whitney test). E Representative IHC images and quantification of IHC scoring for p65 and phospho-p65 in subcutaneous tumours (n = 7, unpaired t-test). Scale bar, 200 μm. F Western blot analysis of protein expression in the indicated cells treated with TNF-α (10 ng/ml). GAPDH was used as a loading control. G Western blot analysis of p65 expression in the nucleic/cytoplasmic fractions after treatment with TNF-α (10 ng/ml) in the indicated cells. GAPDH or lamin A/C was used as a positive control for the cytoplasmic or nucleic fraction. B–G were repeated three times. *p < 0.05. **p < 0.01. Data are shown as mean ± SD. ES, enrichment score; NES, normalized enrichment score; NC, negative control; KD, knockdown; ctrl, control. CE, cytoplasmic extracts; NE, nuclear extracts
Co-IP assay demonstrated that TAB3 bound with TAK1 in HCC cells (Additional file 1: Fig. S3E), which was consistent with previous reports [40]. Additionally, impairing the expression of TAK1 in TAB3-overexpressed HCC cells impeded the enhanced NF-кB activity evidenced by dual luciferase reporter assays (Additional file 1: Fig. S3F, G). These results indicated that the impact of TAB3 on NF-кB signalling was mediated by TAK1. To determine whether TAB3 mediated the modulation of NF-кB signalling by hsa-miR-27b, we overexpressed TAB3 in the hsa-miR-27b-overexpressing MHCC97H cells and knocked down TAB3 in the hsa-miR-27b-knockdown Hep3B cells. The impaired p-IKKβ, p-IκBα and p-p65 levels induced by hsa-miR-27b overexpression were recovered by ectopic expression of TAB3 in MHCC97H cells, while the enhanced IκBα expression was also restored. Consistent results were obtained in the hsa-miR-27b knockdown cells with TAB3 downregulation (Fig. 6F). Additionally, the decreased/elevated nuclear location of p65 in the hsa-miR-27b-overexpressing/knockdown HCC cells was rescued by overexpressing/knocking down TAB3 (Fig. 6G). Taken together, these results demonstrated that decreased expression of TAB3 mediated the de-activation of NF-κB signalling by hsa-miR-27b, and the TAB3/IKKβ/IκBα signalling axis was modulated by hsa-miR-27b.
OncoAd hsa-miR-27b inhibits HCC growth
OncoAd represents a promising therapeutic approach that exhibits anti-tumour effects through selective tumour cell killing [41]. Herein, we constructed an OncoAd overexpressing hsa-miR-27b with the U6 promoter (Fig. 7A). The replication of OncoAd measured by qRT-PCR demonstrated that they selectively replicated in tumour cells (Additional file 1: Fig. S4A). OncoAd hsa-miR-27b efficiently overexpressed hsa-miR-27b in WRL68 and Hep3B cells (Additional file 1: Fig. S4B). To evaluate the HCC-specific cytotoxicity of OncoAd, we infected hepatoma Hep3B cells and liver WRL68 cells with OncoAd hsa-miR-27b or the control virus OncoAd NC at the indicated MOIs. A cytopathic effect was observed in the Hep3B cells infected with OncoAd hsa-miR-27b, and the cytopathic effect of OncoAd hsa-miR-27b was superior to that of OncoAd NC, while no obvious cytotoxicity was observed in WRL68 cells at the same MOIs (Fig. 7B, C). In addition, the CCK-8 assay indicated that OncoAd hsa-miR-27b showed higher cytotoxicity than OncoAd NC at the same MOI in a time-dependent manner (Additional file 1: Fig. S4C). Moreover, the proportion of cracked nuclei exhibited by Hoechst 33342 staining was significantly increased in the OncoAd hsa-miR-27b-treated Hep3B cells compared with the OncoAd NC-treated cells, while no such effect was observed in the OncoAd hsa-miR-27b- or OncoAd NC-treated WRL68 cells (Additional file 1: Fig. S4D).

OncoAd hsa-miR-27b suppressed HCC growth in vitro and in vivo. A Schematic diagram of the structures of the OncoAd. B Hep3B cells were treated with OncoAd hsa-miR-27b and OncoAd NC at the indicated MOI. After 48 h, cell viabilities were measured by CCK-8 assays (n = 5, unpaired t-test). C Hep3B and WRL68 cells were infected with OncoAd at the indicated MOIs for 2 days. Images of cells stained with crystal violet indicated the cytopathic effect of OncoAd hsa-miR-27b and OncoAd NC. D Growth curve of subcutaneous tumours with intra-tumoural injection of PBS (n = 7), OncoAd NC (n = 7) or OncoAd hsa-miR-27b (n = 7). OncoAd were intra-tumourally injected every 2 days after inoculating subcutaneous tumours for 21 days (Day 0); PBS was injected as a control. The arrow represents the first action of intra-tumoural injection, two-way ANOVA. E and F Macroscopic images and quantification of tumour weights of the Hep3B xenografts treated with PBS (n = 7), OncoAd NC (n = 7) or OncoAd hsa-miR-27b (n = 7) at 21 days post-intra-tumoural injection, one-way ANOVA. G Representative images of H&E staining and IHC staining of the indicated protein in Hep3B xenografts; scale bar, 200 μm. Quantification of Ki-67 and p-p65-positive cells, as well as IHC scoring of TAB3 and p65 (n = 7, one-way ANOVA). B and C were repeated three times. *p < 0.05, **p < 0.01. Data are shown as mean ± SD. NC, negative control; OncoAd, oncolytic adenovirus. MOI, multiplicities of infection; ITR, inverted terminal repeat
The in vivo anti-tumour efficacy of OncoAd hsa-miR-27b was evaluated in a Hep3B xenograft animal model. The average volumes and weights of subcutaneous tumours were significantly lower in the mice with administration of OncoAd hsa-miR-27b than in the mice administered PBS or OncoAd NC (Fig. 7D–F). In addition, OncoAd hsa-miR-27b succeeded in downregulating TAB3 and p-p65 expression levels in Hep3B xenografts. Moreover, the OncoAd hsa-miR-27b-treated tumours showed weaker proliferation measured by Ki67 staining than that in the other groups (Fig. 7G). These results demonstrated the anti-tumoural potential of OncoAd hsa-miR-27b in HCC.
Discussion
Recent studies have reported that hsa-miR-27b is involved in tumour progression through multiple pathways. The functions of hsa-miR-27b in cancer development are highly contextual [42]. Jin et al. showed that hsa-miR-27b was highly upregulated in human breast cancer, and knockdown of hsa-miR-27b repressed breast cancer growth [10]. Inconsistently, Tao et al. reported that hsa-miR-27b suppressed gastric cancer cell proliferation and colony formation [43]. In HCC, the current views of the role of hsa-miR-27b are contradictory [14,15,16]. A copy number variation analysis of TCGA database showed that the hsa-miR-27b gene locus was lost in 12 of 49 liver cancer samples, and the expression of hsa-miR-27b was found to be downregulated compared with that in normal liver tissues in TCGA and ArrayExpress databases [17]. The decreased expression of hsa-miR-27b in HCC was further confirmed using qRT-PCR by two independent research groups [16, 17]. By analysing the TCGA database (372 HCC tissues versus 50 normal adjacent liver tissues) and using qRT-PCR assay of 68 pairs of HCC specimens (patients with hepatitis B virus and without metastases), Liang et al. also determined that the level of hsa-miR-27b were decreased in HCC. Besides, Fu et al. showed that hsa-miR-27b was downregulated at mature miRNA level but upregulated at premature level (66 pairs of tissue samples) [18]. However, opposite results were obtained by Sun et al. [14] and He et al. [15]. qRT-PCR analysis of 90 and 35 pairs of HCC patient specimens demonstrated that the expression of hsa-miR-27b was higher in HCC tissues than in their corresponding normal liver tissues [14, 15]. In our study, we comprehensively analysed the expression pattern of hsa-miR-27b using bioinformatic analysis (two datasets), qRT-PCR (n = 43) and ISH examinations (n = 71). We concluded that the expression of hsa-miR-27b was downregulated in tumour tissues and that lower expression of hsa-miR-27b in HCC was an independent risk factor for shorter OS.
Multiple researchers have documented that the NF-кB signalling pathway is constitutively activated and may act as a promising target for the treatment of HCC [23, 44]. TAB3 is involved in regulating differentiation and progression in several cancer types by modulating NF-кB signalling [27, 29, 30, 45]. In HCC, hsa-miR-195 suppressed cell proliferation and migration in vitro and in vivo by decreasing the expression of multiple NF-κB downstream effectors via direct targeting of IKKα and TAB3 [46]. Zhao et al. showed that hsa-miR-26b suppresses NF-κB signalling and thereby sensitizes HCC cells to doxorubicin-induced apoptosis by binding the 3′UTR of TAK1 and TAB3 [25]. In addition, hsa-miR-195, hsa-miR-342-3p and hsa-miR-26b target the NF-κB pathway by binding to the 3′UTR of TAB3 [25, 46, 47].
Although miRNA-mediated NF-кB signalling inhibition is a promising cancer treatment method, miRNAs as cancer therapeutics need to overcome the problems of instability. As new technologies emerging in the construction of miRNAs, their mimetics, and delivery vehicles, miRNAs are becoming more viable for many cancer treatments, especially in chemo- and radiotherapy combinations [48]. Prior studies have noted that OncoAd may provide a promising platform for miRNA-based cancer therapy [41]. OncoAd co-expressing hsa-miR-34a and interleukin-24 (IL-24) induced increased anti-tumour activity compared with OncoAd expressing hsa-miR-34a or IL-24 alone [49]. Callegari et al. developed a novel OncoAd, Ad-199T, which could replicate more efficiently in tumour tissues in an hsa-miR-199-dependent manner in HCC HepG2 cells [50]. In this study, we constructed OncoAd hsa-miR-27b to deliver hsa-miR-27b for the treatment of HCC. A cytopathic effect was observed in the Hep3B cells infected with OncoAd hsa-miR-27b, and little cytotoxicity was observed in normal liver cells.
Conclusions
In this study, we demonstrated that hsa-miR-27b was a tumour suppressor in HCC. Hsa-miR-27b suppressed the proliferation, migration and invasion of cancer cells to inhibit HCC growth and metastasis. Besides, TAB3 was identified as the target downstream effector of hsa-miR-27b. Moreover, we demonstrated that the inactivation of NF-кB signalling by hsa-miR-27b was mediated by TAB3 through the IKKβ/IκBα axis. Compared with OncoAd NC, OncoAd hsa-miR-27b induced stronger anti-tumour activity in vivo and in vitro. Therefore, our results implied that hsa-miR-27b could serve as a potential diagnostic and therapeutic target for HCC.
Availability of data and materials
Datasets used and/or analysed in this study can be obtained from the corresponding author on reasonable request.
Abbreviations
- AFP :
-
α-Fetoprotein
- ANTs:
-
Adjacent normal tissues
- AGO2 :
-
Argonaute 2
- CCK-8:
-
Cell Counting Kit-8
- Bcl-2 :
-
BCL2 apoptosis regulator
- CCND1 :
-
Cyclin D1
- c-myc :
-
Myc proto-oncogene
- co-IP:
-
Co-immunoprecipitation
- Edu:
-
5-Ethynyl-2′-deoxyuridine
- EMT:
-
Epithelial–mesenchymal transition
- F-actin :
-
Filamentous actin
- GEO:
-
Gene Expression Omnibus
- GSEA:
-
Gene set enrichment analysis
- HCC:
-
Hepatocellular carcinoma
- IHC:
-
Immunohistochemistry
- IKB-α :
-
I-kappa-B-alpha
- IKK :
-
Inhibitor of NF-кB kinase
- IL-24 :
-
Interleukin-24
- ISH:
-
In situ hybridization
- ITR:
-
Inverted terminal repeat
- LIHC:
-
Liver hepatocellular carcinoma
- miRNA:
-
MicroRNA
- Hsa-miR-27b :
-
Hsa-microRNA-27b-3p
- MMP9 :
-
Matrix metallopeptidase 9
- mRNA:
-
Messenger RNA
- MOI:
-
Multiplicities of infection
- NC:
-
Negative control
- NF-кB :
-
Nuclear factor kappa-B
- O.D.:
-
Optical density
- OncoAd hsa-miR-27b :
-
Oncolytic adenovirus with overexpression of hsa-miR-27b
- OncoAd NC:
-
Oncolytic adenovirus with negative control
- OS:
-
Overall survival
- p65 :
-
RELA proto-oncogene
- PBS:
-
Phosphate-buffered saline
- p-IKKβ :
-
Phospho-inhibitor kappa B kinase-β
- qRT-PCR:
-
Quantitative real-time PCR
- RIP:
-
RNA immunoprecipitation
- STAT3 :
-
Signal transducer and activator of transcription 3
- TAK1 :
-
Transforming growth factor β-activated protein kinase 1
- TAB2 :
-
Transforming growth factor-activated kinase-binding protein 2
- TAB3 :
-
Transforming growth factor-activated kinase (TAK1)-binding protein 3
- TCGA:
-
The Cancer Genome Atlas
- TNF-α :
-
Tumour necrosis factor α
- TPM:
-
Transcripts per million
- VEGFC :
-
Vascular endothelial growth factor C
- ZEB1 :
-
Zinc finger E-box binding homeobox 1
- ZO-1 :
-
Tight junction protein 1
- 3ʹUTR:
-
3′-Untranslated region
References
Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5–16.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–85.
Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71.
Hammond SM. An overview of microRNAs. Adv Drug Deliv Rev. 2015;87:3–14.
Yang X, Chen J, Liao Y, Huang L, Wen C, Lin M, Li W, et al. miR-27b-3p promotes migration and invasion in colorectal cancer cells by targeting HOXA10. Biosci Rep. 2019;39:BSR20191087.
Chen D, Si W, Shen J, Du C, Lou W, Bao C, Zheng H, et al. miR-27b-3p inhibits proliferation and potentially reverses multi-chemoresistance by targeting CBLB/GRB2 in breast cancer cells. Cell Death Dis. 2018;9:188.
Sun Y, Xu T, Cao YW, Ding XQ. Antitumor effect of miR-27b-3p on lung cancer cells via targeting Fzd7. Eur Rev Med Pharmacol Sci. 2017;21:4113–23.
Jin L, Wessely O, Marcusson EG, Ivan C, Calin GA, Alahari SK. Prooncogenic factors miR-23b and miR-27b are regulated by Her2/Neu, EGF, and TNF-α in breast cancer. Cancer Res. 2013;73:2884–96.
Zhang S, Liu F, Mao X, Huang J, Yang J, Yin X, Wu L, et al. Elevation of miR-27b by HPV16 E7 inhibits PPARγ expression and promotes proliferation and invasion in cervical carcinoma cells. Int J Oncol. 2015;47:1759–66.
Lee JJ, Drakaki A, Iliopoulos D, Struhl K. miR-27b targets PPARγ to inhibit growth, tumor progression and the inflammatory response in neuroblastoma cells. Oncogene. 2012;31:3818–25.
Feng Q, Wu X, Li F, Ning B, Lu X, Zhang Y, Pan Y, et al. miR-27b inhibits gastric cancer metastasis by targeting NR2F2. Protein Cell. 2017;8:114–22.
Sun XF, Sun JP, Hou HT, Li K, Liu X, Ge QX. MicroRNA-27b exerts an oncogenic function by targeting Fbxw7 in human hepatocellular carcinoma. Tumour Biol. 2016;37:15325–32.
He S, Zhang J, Lin J, Zhang C, Sun S. Expression and function of microRNA-27b in hepatocellular carcinoma. Mol Med Rep. 2016;13:2801–8.
Liang H, Ai-Jun J, Ji-Zong Z, Jian-Bo H, Liang Z, Yong-Xiang Y, Chen Y. Clinicopathological significance of miR-27b targeting Golgi protein 73 in patients with hepatocellular carcinoma. Anticancer Drugs. 2019;30:186–94.
Mu W, Hu C, Zhang H, Qu Z, Cen J, Qiu Z, Li C, et al. miR-27b synergizes with anticancer drugs via p53 activation and CYP1B1 suppression. Cell Res. 2015;25:477–95.
Fu C, Li J, Li P, Cheng D. LncRNA DNAJC3-AS1 promotes hepatocellular carcinoma (HCC) progression via sponging premature miR-27b. Cancer Manag Res. 2021;13:8575–83.
Lawrence T. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651.
Gasparini C, Feldmann M. NF-κB as a target for modulating inflammatory responses. Curr Pharm Des. 2012;18:5735–45.
Yi W, Wen Y, Tan F, Liu X, Lan H, Ye H, Liu B. Impact of NF-κB pathway on the apoptosis–inflammation–autophagy crosstalk in human degenerative nucleus pulposus cells. Aging (Albany NY). 2019;11:7294–306.
Bottero V, Withoff S, Verma IM. NF-κB and the regulation of hematopoiesis. Cell Death Differ. 2006;13:785–97.
Xia Y, Shen S, Verma IM. NF-κB, an active player in human cancers. Cancer Immunol Res. 2014;2:823–30.
Perkins ND. The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer. 2012;12:121–32.
Zhao N, Wang R, Zhou L, Zhu Y, Gong J, Zhuang SM. MicroRNA-26b suppresses the NF-κB signaling and enhances the chemosensitivity of hepatocellular carcinoma cells by targeting TAK1 and TAB3. Mol Cancer. 2014;13:35.
Ding J, Huang S, Wang Y, Tian Q, Zha R, Shi H, Wang Q, et al. Genome-wide screening reveals that miR-195 targets the TNF-α/NF-κB pathway by down-regulating IκB kinase α and TAB3 in hepatocellular carcinoma. Hepatology. 2013;58:654–66.
Zhao J, Gai L, Gao Y, Xia W, Shen D, Lin Q, Mao W, et al. TAB3 promotes human esophageal squamous cell carcinoma proliferation and invasion via the NFκB pathway. Oncol Rep. 2018;40:2876–85.
Chen J, Gu J, Feng J, Liu Y, Xue Q, Ni T, Wang Z, et al. TAB3 overexpression promotes cell proliferation in non-small cell lung cancer and mediates chemoresistance to CDDP in A549 cells via the NF-κB pathway. Tumour Biol. 2016;37:3851–61.
Jin G, Klika A, Callahan M, Faga B, Danzig J, Jiang Z, Li X, et al. Identification of a human NF-κB-activating protein, TAB3. Proc Natl Acad Sci USA. 2004;101:2028–33.
Zhou Q, Cheng C, Wei Y, Yang J, Zhou W, Song Q, Ke M, et al. USP15 potentiates NF-κB activation by differentially stabilizing TAB2 and TAB3. FEBS J. 2020;287:3165–83.
Criollo A, Niso-Santano M, Malik SA, Michaud M, Morselli E, Marino G, Lachkar S, et al. Inhibition of autophagy by TAB2 and TAB3. EMBO J. 2011;30:4908–20.
Chen Y, Wang X, Duan C, Chen J, Su M, Jin Y, Deng Y, et al. Loss of TAB3 expression by shRNA exhibits suppressive bioactivity and increased chemical sensitivity of ovarian cancer cell lines via the NF-κB pathway. Cell Prolif. 2016;49:657–68.
Adhikari A, Xu M, Chen ZJ. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene. 2007;26:3214–26.
Ding ZY, Jin GN, Wang W, Chen WX, Wu YH, Ai X, Chen L, et al. Reduced expression of transcriptional intermediary factor 1 γ promotes metastasis and indicates poor prognosis of hepatocellular carcinoma. Hepatology. 2014;60:1620–36.
Huang Z, Wen J, Yu J, Liao J, Liu S, Cai N, Liang H, et al. MicroRNA-148a-3p inhibits progression of hepatocellular carcinoma by repressing SMAD2 expression in an Ago2 dependent manner. J Exp Clin Cancer Res. 2020;39:150.
Liang J, Li G, Liao J, Huang Z, Wen J, Wang Y, Chen Z, et al. Non-coding small nucleolar RNA SNORD17 promotes the progression of hepatocellular carcinoma through a positive feedback loop upon p53 inactivation. Cell Death Differ. 2022;29:988–1003.
Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. 2016;65:798–808.
Shen J, Xia W, Khotskaya YB, Huo L, Nakanishi K, Lim SO, Du Y, et al. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature. 2013;497:383–7.
Pahl HL. Activators and target genes of Rel/NF-κB transcription factors. Oncogene. 1999;18:6853–66.
Besse A, Lamothe B, Campos AD, Webster WK, Maddineni U, Lin SC, Wu H, et al. TAK1-dependent signaling requires functional interaction with TAB2/TAB3. J Biol Chem. 2007;282:3918–28.
Rosewell Shaw A, Suzuki M. Recent advances in oncolytic adenovirus therapies for cancer. Curr Opin Virol. 2016;21:9–15.
Ding L, Ni J, Yang F, Huang L, Deng H, Wu Y, Ding X, et al. Promising therapeutic role of miR-27b in tumor. Tumour Biol. 2017;39:1010428317691657.
Tao J, Zhi X, Zhang X, Fu M, Huang H, Fan Y, Guan W, et al. miR-27b-3p suppresses cell proliferation through targeting receptor tyrosine kinase like orphan receptor 1 in gastric cancer. J Exp Clin Cancer Res. 2015;34:139.
Feng W, Xue T, Huang S, Shi Q, Tang C, Cui G, Yang G, et al. HIF-1α promotes the migration and invasion of hepatocellular carcinoma cells via the IL-8–NF-κB axis. Cell Mol Biol Lett. 2018;23:26.
Gao L, Zhao H, Sun H, Huang Y. TAB3 overexpression promotes NF-κB activation and inflammation in acute pancreatitis. Am J Blood Res. 2020;10:118–23.
Shi G, Shi J, Liu K, Liu N, Wang Y, Fu Z, Ding J, et al. Increased miR-195 aggravates neuropathic pain by inhibiting autophagy following peripheral nerve injury. Glia. 2013;61:504–12.
Zhao L, Zhang Y. miR-342-3p affects hepatocellular carcinoma cell proliferation via regulating NF-κB pathway. Biochem Biophys Res Commun. 2015;457:370–7.
Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–22.
Lou W, Chen Q, Ma L, Liu J, Yang Z, Shen J, Cui Y, et al. Oncolytic adenovirus co-expressing miRNA-34a and IL-24 induces superior antitumor activity in experimental tumor model. J Mol Med (Berl). 2013;91:715–25.
Callegari E, Elamin BK, D’Abundo L, Falzoni S, Donvito G, Moshiri F, Milazzo M, et al. Anti-tumor activity of a miR-199-dependent oncolytic adenovirus. PLoS ONE. 2013;8:e73964.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Innovative Research Group Project of the National Natural Science Foundation of China (No. 82172971, 31671348 and 81874189), Chen Xiao-Ping Foundation for the Development of Science and Technology of Hubei Province (CXPJJH12000001-2020317), the National Science and Technology Major Project (2018ZX10723204-003), National Key Research and Development Project (2018YFA0208904) and the Fundamental Research Funds for the Central Universities, HUST (2020JYCXJJ019).
Author information
Authors and Affiliations
Contributions
J.W. and Z.H.: designed the experiments and drafted the manuscript; J.W., Z.H. and Y.W.: data acquisition and interpretation, statistical analysis; J.W., J.Y., L.X. and J.L.: data acquisition; Y.W., J.L. and Z.H.: technical or material support; X.C., B.Z. and L.C.: study supervision and funding acquisition. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal experimental procedures were performed in accordance with the Basel Declaration, and was also approved by the Committee on the Ethics of Animal Experiments of Tongji Hospital (TJH-202005005, 2 March 2020). All human tissue samples were obtained in accordance with the Declaration of Helsinki (1975), and were approved by the ethics committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China) (TJ-IRB20211163, 30 November 2021).
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Additional Figures S1–S4 and Tables S1–S5.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wen, J., Huang, Z., Wei, Y. et al. Hsa-microRNA-27b-3p inhibits hepatocellular carcinoma progression by inactivating transforming growth factor-activated kinase-binding protein 3/nuclear factor kappa B signalling. Cell Mol Biol Lett 27, 79 (2022). https://doi.org/10.1186/s11658-022-00370-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11658-022-00370-4