
Abstract
Osteoporosis, or bone loss, is a progressive, systemic skeletal disease that affects millions of people worldwide. Osteoporosis is generally age related, and it is underdiagnosed because it remains asymptomatic for several years until the development of fractures that confine daily life activities, particularly in elderly people. Most patients with osteoporotic fractures become bedridden and are in a life-threatening state. The consequences of fracture can be devastating, leading to substantial morbidity and mortality of the patients. The normal physiologic process of bone remodeling involves a balance between bone resorption and bone formation during early adulthood. In osteoporosis, this process becomes imbalanced, resulting in gradual losses of bone mass and density due to enhanced bone resorption and/or inadequate bone formation. Several growth factors underlying age-related osteoporosis and their signaling pathways have been identified, such as osteoprotegerin (OPG)/receptor activator of nuclear factor B (RANK)/RANK ligand (RANKL), bone morphogenetic protein (BMP), wingless-type MMTV integration site family (Wnt) proteins and signaling through parathyroid hormone receptors. In addition, the pathogenesis of osteoporosis has been connected to genetics. The current treatment of osteoporosis predominantly consists of antiresorptive and anabolic agents; however, the serious adverse effects of using these drugs are of concern. Cell-based replacement therapy via the use of mesenchymal stem cells (MSCs) may become one of the strategies for osteoporosis treatment in the future.
Similar content being viewed by others


Background
Osteoporosis, a bone disease involving the appearance of porous bone, is characterized by low bone mass and microarchitectural deterioration of bone tissues, leading to reduced bone strength and a consequent increase in fracture risk [1, 2]. Osteoporosis is increasingly recognized as a major public health concern that affects more than 200 million people worldwide and causes more than 8.9 million fractures, and mainly hip fractures, per year [3]. The incidence of osteoporosis has dramatically risen because the life expectancy of the population has been increasing in every geographic region [4]. The consequences of osteoporosis are significant, as is the financial burden, estimated at approximately US $17 billion for more than 2 million fractures in the US [5]. The prevention of this disease and its associated fractures is considered essential to health maintenance, quality of life, and independence in the elderly population.
According to the World Health Organization (WHO) criteria, osteoporosis is defined as having a bone mineral density (BMD) value that is 2.5 standard deviations or more (T-score ≤ -2.5) below the average value for young healthy women, as measured by dual-energy x-ray absorptiometry (DXA), which is the most validated technique (i.e., the gold standard) [1, 2]. A low BMD not only is a major risk factor for fractures but also is an independent risk factor for death [6]. BMD testing of the hip and spine is required for a densitometric diagnosis of osteoporosis. Measurements of bone strength other than bone density at these sites may predict fracture risk but cannot be used to diagnose osteoporosis [7]. BMD remains the best tool to assess fracture risk, but it cannot predict the fracture risk in certain cases, particularly in type 2 diabetes patients, who usually have a higher BMD and an increased fracture risk [8]. The majority of osteoporotic fractures occur in individuals with a BMD level within the osteopenic range (-2.5 < T-score < -1) or even with normal BMD levels [9]. The Fracture Risk Assessment (FRAX) tool integrates BMD and clinical risk factors such as age, gender, the history of fracture, the parental history of hip fracture, current smoking, excessive alcohol intake, rheumatoid arthritis, glucocorticoid use, and other causes of secondary osteoporosis. FRAX has been shown to be a more applicable prediction tool to estimate the risk and probability of fracture in an individual over the next 10 years. FRAX can also provide general clinical guidance for treatment decisions [10].
Over the past few decades, there have been great advances in understanding of the physiologic process of bone remodeling, together with associated pathologic conditions. The underlying pathogenesis of osteoporosis involves an imbalance of bone homeostasis that results from many causes, such as hormone deficiency, genetic disorders, use of certain medications, and medical conditions. Osteoporosis is characterized by low bone mass and density, which lead to an increased fracture risk. The aims of treatment for osteoporosis are to reduce bone loss and maintain bone density, especially in patients who have fractures or a high risk of fractures. Many therapeutic drugs for treating osteoporosis are available in the market, most of which have relied on bone resorption inhibition, such as bisphosphonate, and several drugs are being developed for treatment in the future. However, controversies have confounded the treatment of osteoporosis. Thus, new treatments based on the promotion of bone regeneration or alternative cell-based therapy for osteoporosis patients are expected to be investigated. Stem cells are expected to have great therapeutic potential, particularly in regenerative medicine. Specifically, stem cells could be a promising cell source for cell-based therapy for osteoporosis. In the present review, the current understanding of mesenchymal stem cells (MSCs) and their roles in osteoporosis, the genetics and transcriptional regulation involved in the pathogenesis of osteoporosis, the signaling pathways associated with osteoporosis and trends in using stem cells as cell-based therapy for osteoporosis is summarized.
Pathogenesis
Imbalance of bone homeostasis
Bone formation generally comprises three basic steps: synthesis of extracellular protein matrix (osteoid) by osteoblasts; matrix mineralization by coating the protein matrix with a layer of mineral, and predominantly calcium phosphate in the form of crystals of hydroxyapatite; and bone remodeling, which is a process that occurs throughout human life. Bone remodeling is essential to maintain the integrity of the skeleton and serves as storage for mineral homeostasis [11]. Via an interactive process called coupling, this process is balanced by the functions of bone-resorbing osteoclasts and bone-forming osteoblasts in early adulthood. When the bone loses its mineral content and density and develops osteopenia, this culminates in osteoporosis, which is associated with a risk of bone fractures [11, 12].
Osteoporosis is normally related to increasing age, consistent with the fact that most of the older population is affected by this condition. It has been shown that genetics could be another explanation for the pathogenesis of osteoporosis. The results of laboratory studies have indicated that osteoporosis is caused by an imbalance of the coupling interactive process, with increased bone resorption relative to bone formation. In this regard, the imbalance is a consequence of changes at the cellular level, by which osteoclast development is enhanced but osteoblast differentiation is insufficient because of impaired activity and enhanced apoptosis [13, 14].
Underlying transcriptional regulation and genetics
Although MSCs have the ability to undergo multipotent differentiation, cell fate determination and differentiation toward either osteoblasts or adipocytes are well regulated by lineage-specific transcription factors such as runt-related transcription factor 2 (Runx2) and osterix (Osx) for osteoblasts and peroxisome proliferator-activated receptor gamma (PPARγ) for adipocytes, suggesting an inverse correlation between osteogenesis and adipogenesis [15–19]. During these processes, intrinsic (genetic) and/or environmental (local and/or systemic) conditions interplay to specify cell fate toward one of the possible lineages. Several lines of evidence have demonstrated that osteoporotic MSCs have defects in intrinsic signals that cause functional alterations, leading to poor osteogenic differentiation capacity and favoring increased adipogenesis [13, 20]. A recent study of microarray analyses demonstrated that MSCs from elderly patients with primary osteoporosis have a distinct transcriptome compared with control MSCs and elderly donor non-osteoporotic MSCs, as shown by enhanced mRNA expression of osteoporosis-associated genes (RUNX2, lipoprotein receptor-related protein 5; LRP5, collagen type 1 alpha1; COL1A1), genes involved in osteoclastogenesis (CSF1, PTH1R), and genes coding for inhibitors of wingless-type MMTV integration site family (Wnt) and bone morphogenetic protein (BMP) signaling, indicating intrinsic deficiencies in self-renewal and differentiation potential in osteoporotic MSCs [21]. Interestingly, transcriptional alterations may reflect epigenetic changes as part of the process of age-related osteoporosis [21].
Nevertheless, the regulatory mechanisms underlying the pathogenesis of osteoporosis have been linked to genetics. Several approaches, including linkage analysis in families, animal studies, candidate gene association studies, and genome-wide association studies (GWAS), have been used to identify the genes responsible for osteoporosis [22]. Linkage analysis is the classical approach that is used to study BMD variation [23, 24]. Linkage studies in animals such as mice, rats and primates provide another way to identify genes that regulate bone density and other phenotypes relevant to osteoporosis. The Alox15 gene has been found to regulate bone density in mice, and this finding was confirmed in Alox15-knockout mice showing increased BMD [25]. Candidate gene association studies have been widely used in the field of osteoporosis, analyzing polymorphic variants in candidate genes and relating them to the carriage of a specific allele or haplotype. Candidate genes such as sclerostin (SOST), COL1A1, ESR1, LRP5, TGF-β1 and VDR have been extensively investigated on a large scale [26–40]. Due to advances in genotyping technologies, GWAS have been applied to study osteoporosis, and large numbers of single-nucleotide polymorphisms (SNPs) have been identified. A GWAS by Richards et al. reported the identification of SNPs that are significantly associated with decreased BMD and increased risks of osteoporotic fractures and osteoporosis when they are located near the TNFRSF11B (osteoprotegerin or OPG) and LRP5 genes [41]. Another study, by Styrkarsdottir et al., used an extended GWAS to identify four new genome-wide significant loci; this loci were near the SOST gene at 17q21, the MARK3 gene at 14q32, the SP7 gene at 12q13 and the TNFRSF11A (receptor activator of nuclear factor kB or RANK) gene at 18q21 and were associated with the heritability of BMD [42]. However, genetic studies of osteoporosis-susceptibility genes need to be further explored.
Signaling pathways associated with osteoporosis
Over several decades, signaling pathways in bone homeostasis have been extensively studied. Dysregulation of these signaling pathways is associated with bone diseases, including osteoporosis. Major signaling pathways that govern the bone regenerative process are OPG/RANK/RANK ligand (RANKL), Wnt, and BMP signaling.
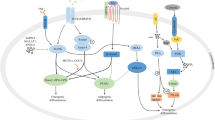
Bone homeostasis is maintained by the balanced function of osteoblasts and osteoclasts. The key regulators involved in this balancing process, equilibrating between bone formation and bone resorption, have been extensively explored. The OPG/RANK/RANKL system is one of the most important signaling pathways in bone metabolism (Fig. 1). Dysregulation of the OPG/RANK/RANKL system has been reported in osteoporosis. OPG, recently designated as TNFRSF11B and serving as a member of the tumor necrosis factor (TNF) receptor family, was first identified as a crucial component that is secreted by osteoblasts; bone marrow stromal cells [43]; and other cells, such as regulatory T (T reg) cells [44]. OPG protects the skeleton from excessive bone resorption by acting as a soluble decoy receptor that can bind to RANKL [45]. The binding of OPG and RANKL subsequently prevents RANKL from binding to its receptor, RANK [43]. The overexpression of the gene encoding OPG results in the development of high bone mass and reduced osteoclast numbers and activity [46]. OPG-deficient mice demonstrate osteoporosis, with an excessive number of osteoclasts [47, 48]. RANKL functions as an osteoclast-activating factor secreted by activated T cells and represents a potent molecule that binds to RANK, which is expressed on osteoclast precursors, known as preosteoclasts [49]. RANKL-RANK binding drives osteoclast differentiation and maturation. The activation of RANK through the binding of RANKL induces the activation of transcription factors such as c-fos, NFAT, and nuclear factor kappa B (NF-kB) in preosteoclasts and initiates several downstream signaling pathways, and especially the NF-kB pathway [50, 51]. RANKL-deficient mice exhibit osteopetrotic bones, or thickened bones, due to a defect in osteoclast development [52]. Moreover, RANKL relies on the presence of macrophage colony-stimulating factor (M-CSF), which is a cofactor for RANKL/RANK-mediated osteoclastogenesis [53]. However, experimental data revealed that RANKL alone could stimulate bone resorption in mice lacking M-CSF [54]. In contrast, M-CSF alone is insufficient to activate osteoclasts [55]. Therefore, RANKL plays a crucial role in osteoclastogenesis, and this phenomenon is required for bone resorption. Under physiologic conditions, OPG/RANKL is in equilibrium and preserves bone homeostasis. The OPG/RANKL ratio is an important factor to use to determine bone mass and skeletal integrity [56, 57]. Under osteoporotic conditions, RANKL is upregulated, which is associated with downregulation of OPG [58]. Moreover, several cytokines are elevated, and particularly TNF-α, IL-1, IL-4 and IL-6, in osteoporosis [59]. These proinflammatory cytokines modulate the RANKL/RANK ratio by stimulating and upregulating RANKL expression on T cells. Interestingly, this emerging role of the OPG/RANK/RANKL system not only is relevant to bone biology but also has been discovered beyond the immunological system. The cross-regulation between bone and immune cells is considered as a bone immunological niche [60]. Considering bone resorption, data in the literature have revealed that impairment of T cell subpopulations and their proinflammatory cytokine patterns are implicated in the pathogenesis of osteoporosis. At the bone tissue level, Th1 and Th2 cells play a role through their secreted cytokines, including RANKL, mediating osteoclast formation and activity, which are linked to bone resorption [61]. Furthermore, Th17 cells, a distinct lineage of proinflammatory T helper cells, were more recently identified as a potent T cell subpopulation that has a role in bone destruction [62]. Th17 cells have been found to increase in number in many bone diseases, and particularly osteoporosis [63]. Th17 cells produce IL-17, which functions in mediating osteoclast differentiation [63, 64]. It has been shown that Th17 cells also produce RANKL, directly contributing to bone loss [62]. Additionally, the Th17 population in the bone marrow and peripheral blood is large in estrogen-deficient osteoporosis [65]. Collectively, Th1/Th2/Th17 cells and their cytokines might play a key role as potent pro-osteoclastogenic mediators underlying the pathogenesis of osteoporotic development.

Bone homeostasis regulation by OPG/RANK/RANKL system. RANKL which secreted by activated T cells functions as an osteoclast-activating factor by binding to its receptor, RANK, which is expressed on preosteoclasts. RANKL-RANK binding induces the activation of several transcription factors in preosteoclasts and initiates several downstream signaling pathways that drive osteoclast differentiation and maturation. OPG which secreted by osteoblasts, bone marrow stromal cells, and T reg cells acts as a soluble receptor that can bind to RANKL and subsequently prevents RANKL-RANK binding. Under physiologic conditions, OPG/RANKL is in equilibrium and preserves bone homeostasis. Under osteoporotic conditions, RANKL is upregulated, which is associated with downregulation of OPG. Several proinflammatory cytokines are secreted from T helper cells (Th1/Th2/Th17) stimulating and upregulating RANKL expression and mediating osteoclast formation and activity, which are linked to increased bone resorption
Wnts are secreted glycoproteins that, when bound to their cognate receptors, can stimulate intracellular signaling cascades that play important roles in cell developmental processes, including osteogenesis [66, 67]. The binding of Wnt ligand to Frizzled receptor and the LRP6 coreceptors to form a complex stimulates the canonical Wnt/β-catenin pathway, whereas binding to the ROR2/RYK coreceptors stimulates non-canonical Wnt signaling [68]. In fact, signaling induced by Wnt/β-catenin is well established and generally plays a role in osteoblastogenesis by promoting the commitment and differentiation of MSCs toward the osteoblast lineage, which in turn suppresses adipogenesis through the inhibition of PPARγ-induced genes [66, 69]. Wnt/β-catenin signaling plays a role in osteoblast maturation and indirectly reduces osteoclastogenesis by stimulating the secretion of OPG, a natural inhibitor of RANKL [70, 71]. Considering the components of Wnt signaling, humans and mice with altered expression of LRP5 and Wnt10b have alterations in bone mass [72, 73]. Loss-of-function LRP5 mutation causes abnormality in bone formation [72]. A genetic study found that WNT10B polymorphisms have an impact on low bone mass and osteoporosis risk [74]. As previously stated, Wnt10b seems to be the most positive modulator of bone regeneration and homeostasis. Supporting these findings, a decreased number and decreased function of osteoblasts have been found in Wnt10b -/- knockout mice, coupled with a 30 % reduction in bone volume and BMD [75, 76]. Stevens et al. have found that heterozygous Wnt10b +/- mice showed a significant reduction in trabecular bone at 6 months of age, and both the number of bone marrow-derived MSCs and osteoblast differentiation were affected [76]. In another study, signaling through Wnt2, Wnt3 or Wnt3a induced proliferation and maintained the self-renewal of MSCs, whereas Wnt5a, Wnt5b or Wnt11 supported osteogenesis [77, 78]. β-catenin deficiency arrests osteoblast development at an early stage in mesenchymal osteoblastic precursors and impairs the maturation and mineralization of committed osteoblasts [67, 79]. Rodríguez et al. reported that osteoporotic MSCs had a diminished proliferation rate as well as decreased mRNA expression of Wnt signaling and the downstream components GSK-3β, LRP6 and OSX [28, 80]. In addition, dickkopf-1 (DKK-1) and SOST are endogenous inhibitors of the canonical Wnt/β-catenin pathway that is specific to bone [70, 81]. Genes coding for these inhibitors show enhanced expression in osteoporotic MSCs in humans [35]. Clinically, the serum DKK-1 level has been found to be significantly higher in patients with low BMD and postmenopausal osteoporosis [82]. Several findings have revealed crosstalk between Wnt signaling and other signaling factors, such as BMPs. In particular, BMP-2 has a synergistic effect with Wnt ligands and β-catenin, inducing bone formation through Wnt/β-catenin signaling and downstream T cell factor/lymphoid enhancer factor (TCF/LEF) transcriptional activity [83, 84].
In addition to Wnt signaling, the BMPs, belonging to the transforming growth factor beta (TGF-β) superfamily, are responsible for numerous cell regulatory processes, including osteogenic differentiation and regulation of bone formation [85]. Upon binding of BMP ligand, signal transduction is initiated through the interaction between two serine-threonine kinase cell surface receptors (BMP receptors (BMPRs)). In particular, BMPR-IA and BMPR-IB are involved in MSC differentiation [86]. BMP-2, BMP-4, BMP-7, BMP-9, and BMP-13 are commonly studied in the context of MSC differentiation-related osteoblastogenesis and bone formation [87, 88]. Notably, BMP-2 promotes Runx2 expression in mesenchymal osteoprogenitors and also promotes Osx and distal-less homeobox 5 (Dlx5) expression in osteoblasts [89–93]. BMP-3 is an exception because it inhibits osteogenesis [94]. BMPs function as both autocrine and paracrine factors, and their synthesis is induced by BMPs themselves via local feedback mechanisms. Evidence has shown that MSCs from osteoporosis patients are impaired in function and this alteration is associated with BMP signaling [95, 96]. However, BMP antagonists have been described, including noggin (NOG) and gremlin (GREM). Overexpression of NOG, as shown in transgenic mouse studies, results in decreased BMD because of increased inhibition of bone formation [97, 98]. SNPs in the NOG gene are associated with osteoporosis-related phenotypes in humans [99]. GREM is detectable in the skeleton, and its overexpression causes osteopenia and fractures [100]. Genetic variants of GREM2 are associated with BMD, and GREM2 is considered a susceptibility gene for osteoporosis [101].
Osteoporosis treatments
Current options for the treatment of osteoporosis are predominantly drug-based agents that either inhibit bone resorption or directly stimulate bone generation to increase bone mass. Non-pharmacological treatments via calcium and vitamin D consumption have been given to patients who have a high risk of osteoporosis related to insufficient calcium and vitamin D intake and postmenopause [102, 103]. Pharmacological treatments are given to patients who are diagnosed with osteoporosis who have already had a fracture or who have a high risk of osteoporotic fracture or re-fracture. Bisphosphonates, which are synthetic compounds that decrease bone resorption by promoting osteoclast apoptosis [104, 105], are the most common medications prescribed as first-line drugs for osteoporosis treatment. Several bisphosphonates have been approved as drugs for the treatment of osteoporosis, including alendronate [106], ibandronate [107], risedronate [108], and zoledronate [109]. However, serious side effects, such as osteonecrosis of the jaw and atypical femoral fractures, have been described in patients under long-term bisphosphonate treatment [110, 111]. Although serious adverse events are rare for current antiresorptive compounds and do not represent a major concern, the development of drugs with higher efficacy in improvement of bone quality and prevention of fractures is still necessary. Other antiresorptive drugs that can serve as alternatives for osteoporosis treatment include denosumab, a RANKL inhibitor that blocks the main pathway involved in osteoclast formation and activation [112], and calcitonin, a naturally occurring peptide [113]. Hormone therapy, such as therapy with estrogen [114] and with selective estrogen receptor modulators (SERMs) acting as estrogen agonists, such as raloxifene [115], has been used in postmenopausal women to slow the bone breakdown process, maintain bone density, and reduce fracture risk. However, long-term side effects, and particularly the development of breast cancer, and risks of cardiovascular events and thromboembolism limit the use of estrogen and SERMs as treatment strategies for osteoporosis [116, 117].
In contrast with antiresorptive drugs, anabolic drugs that can increase bone formation, rather than preventing bone loss, are of interest to rebuild bone, increase bone strength, and reduce the risk of fractures in osteoporosis patients. To date, approved anabolic drugs have been limited to parathyroid hormone (PTH) and its analog, teriparatide (recombinant human PTH(1–34)), which are considered as treatments for patients with severe osteoporosis. Nevertheless, it was reported that administering a high dose of teriparatide for a long period increased the incidence of osteosarcoma in an animal study [118]. Although evidence of osteosarcoma has not been reported in patients taking teriparatide, treatment with teriparatide is not allowed beyond 2 years according to the FDA. One therapeutic drug, strontium ranelate, which is thought to have dual actions on bone metabolism, both increasing bone formation and decreasing bone resorption, represents as a potential agent for the treatment of postmenopausal osteoporosis to reduce the risk of vertebral and hip fractures [119]. Considering the costs and disadvantages of prolonged treatment with drugs and hormones in osteoporosis patients, cell therapy may be a good alternative candidate therapeutic strategy to treat osteoporosis in the future.
Stem cell-based therapy for osteoporosis
Cell therapy has attracted considerable clinical attention for the treatment of various diseases for many decades. Stem cells are believed to be an ideal source of cells for cell replacement therapy for bone diseases due to their properties of self-renewal and plasticity, which can repair or regenerate damaged tissues. Candidate stem cell types include embryonic stem (ES) cells, induced pluripotent stem (iPS) cells and somatic stem cells such as MSCs. The use of ES and iPS cells is limited due to ethical issues and virus-based derivation methods [120]. It seems likely that the use of MSCs overcomes such limitations and is more practical in other disease models. In recent years, MSCs have become dramatically interesting for the treatment of osteoporosis. MSCs have the ability to self-renew and to grow into specific tissues, such as cartilage, bone, and adipose tissue. Human MSCs are defined by their phenotypic expression of CD105, CD73 and CD90; their absence of expression of hematopoietic markers such as CD45, CD34, and CD14; and their ability to differentiate into osteogenic, adipogenic and chondrogenic lineages under permissive conditions [121]. It has been reported that MSCs can avoid allogeneic rejection by being hypoimmunogenic, modulating the T cell phenotype, and creating an immunosuppressive locus [122]. Moreover, MSC-derived osteogenic cells show immunoprivileged and immunomodulatory properties similar to those of their parental MSCs [123]. With regard to the pathogenesis of osteoporosis, resulting in bone mass reduction, transplantation of MSCs might promote new bone formation and strengthen the bone, contributing to improvement of bone quality and prevention of fractures. After transplantation, MSCs contribute to bone formation by two possible mechanisms of action: (1) MSCs’ homing to a damaged site or pathologic area and then differentiating into bone-forming cells to repair the degenerated tissue and (2) MSCs’ acting in a paracrine manner by secreting certain growth factors that modify the environment and recruit resident cells to repair the degenerated tissue [124, 125].
Sources of mesenchymal stem cells: advantages and disadvantages
Bone marrow-derived MSCs
Bone marrow is the most commonly used tissue source of adult MSCs. Bone marrow-derived MSCs (BM-MSCs) have been extensively studied in bone regeneration and repair due to their high efficiency in osteogenic differentiation. Studies in animal models have revealed that both allogeneic and autologous BM-MSC transplantation is applicable in the treatment of osteoporosis. Ichioka et al. demonstrated that normal allogeneic BM-MSCs could increase trabecular bone and attenuate the loss of BMD after being directly injected into the bone marrow cavity of an irradiated P6 substrain of senescence-accelerated mice (SAMP6), an osteoporotic mouse model that exhibits age-dependent inhibition of osteoblastogenesis and osteoclastogenesis along with enhanced adipogenesis [126]. A similar result was also observed in an ovariectomy (OVX)-induced rat model of osteoporosis after receipt of allogeneic BM-MSCs isolated from healthy rats [127]. Autologous BM-MSC transplantation was reported to improve bone formation and to strengthen osteoporotic bone in an OVX-induced rabbit model of osteoporosis [128] as well as in goats with long-term estrogen deficiency, mimicking the postmenopausal osteoporosis that occurs in humans [129]. However, use of autologous BM-MSCs for osteoporosis treatment in elderly patients is limited due to the age-related decline in the overall BM-MSC number [130]. Recently, use of autologous BM-MSCs for the treatment of osteoporosis has been performed in clinical trial study. Autologous BM-MSCs were collected 30 days before infusion, and the cells were cultured under GMP conditions to establish the dose range. In this study, the cells were subjected to the process of fucosylation before intravenous infusion into osteoporosis patients. However, this study is still in the process of recruiting participants and is thus not yet completed (ClinicalTrials.gov Identifier: NCT02566655).
Adipose tissue-derived MSCs
Adipose tissue provides an attractive source of MSCs that has become increasingly popular in many stem cell applications. Adipose tissue-derived MSCs (AD-MSCs) are isolated from white adipose tissues via a minimally invasive approach and can be expanded and differentiated into classical mesenchymal lineages involved in adipogenesis, osteogenesis, and chondrogenesis [131, 132]. AD-MSCs are more easily isolated and more abundant and produce higher yields in terms of cell number compared with BM-MSCs [133]. However, the yield of AD-MSCs and their proliferative and differentiation capacities vary depending on the tissue harvesting site [134] and the age of the donor [135]. For application in cell therapy for osteoporosis, AD-MSCs were reported to function as an effective autologous cell-based approach for the treatment of osteoporosis. SAMP6 osteoporosis mice showed significant improvement in several trabecular bone parameters after a single intratibial transplantation of isogenic AD-MSCs [136]. A preclinical study of the in vivo function of human AD-MSCs by Cho et al. revealed that human AD-MSCs could prevent OVX-induced bone loss in nude mice over 8 weeks, even though there was no evidence of long-term engraftment of infused human AD-MSCs in the bone of recipient mice [137]. The effect of human AD-MSC therapy likely occurs in a paracrine manner by the secretion of various bone-related growth factors, e.g., hepatocyte growth factor, BMP-2, and RANKL, and extracellular matrix (ECM) proteins, e.g., fibronectin, which might promote osteogenic differentiation, bone remodeling and repair in the recipients [124]. Moreover, Xinhai et al. demonstrated that autologous AD-MSCs enhanced bone regeneration in OVX-induced rabbit models of osteoporosis due to not only their own osteogenic differentiation but also their promotion of osteogenesis and inhibition of adipogenesis by osteoporotic BM-MSCs through activation of BMP-2 and the BMPR-IB signaling pathway [125]. Recently, a clinical trial has studied the use of human AD-MSCs for the treatment of proximal humeral fractures in individuals over 50 years old, representing a model for fractures of osteoporotic bone. In this study, AD-MSCs were wrapped around hydroxyapatite microgranules embedded in a fibrin gel to allow cellularized composite graft augmentation. Clinical/radiological follow-up was performed after 6, 9 and 12 months, and functional assessment was performed after 6 weeks and 6 and 12 months using the Quick DASH score and the Constant score. Unfortunately, the study was terminated, and no results are available (ClinicalTrials.gov Identifier: NCT01532076).
Perinatal-derived MSCs
Although BM- and AD-MSCs are effective sources, the therapeutic potential of these adult MSCs can be affected by the donor’s lifestyle and age. Perinatal tissues are alternative sources of MSCs that have attracted growing interest in bone regenerative medicine [138]. Not only are these cells younger than adult MSCs, but perinatal-derived MSCs also have the major advantage of an easy and noninvasive harvesting procedure without any risk to the donor. A comparative study of MSCs isolated from different perinatal tissue sites, including the umbilical cord, umbilical cord blood (UCB), amnion, and chorion, revealed that these tissues exhibit similar characteristics to BM-MSCs, including similar phenotypic features, growth properties, differentiation capacities, secretory protein profiles, and low immunogenic properties [138]. However, these stem cell sources are still limited by their low capacity to differentiate compared with BM- and AD-MSCs, and they have not been clearly examined in preclinical studies.
Placenta-derived MSCs
The placenta is an easily accessible source of perinatal MSCs that provides a high yield of MSCs. Placenta-derived MSCs (PL-MSCs) express common markers of MSCs and exhibit adipogenic, osteogenic, and neurogenic differentiation capacities [139]. Sanvoranart et al. demonstrated that PL-MSCs responded to bortezomib, a chemotherapeutic agent that improves osteolytic lesions in multiple myeloma, via enhancement of osteogenic differentiation, similarly to BM-MSCs [140]. This finding suggests the potential therapeutic application of PL-MSCs in osteopenia and osteoporosis patients.
Umbilical cord-derived MSCs
The umbilical cord contains various cell types, including vessels, connective tissues, and Wharton’s jelly. After isolation, these heterogeneous cells are observed to possess differential vimentin and cytokeratin expression in culture, but not variable capacities to differentiate into chondrogenic, adipogenic, and osteogenic lineages [141]. In vivo bone formation by umbilical cord-derived MSCs (UC-MSCs) was demonstrated by Diao et al., who loaded human UC-MSCs into scaffolds, implanted the scaffolds into BALB/c nude mice subcutaneously and found that the human UC-MSCs could efficiently form bone after implantation for 12 weeks [142].
Wharton’s jelly-derived MSCs
Wharton’s jelly is the mucoid connective tissue that surrounds the umbilical cord vein and that functions in the protection of the vasculature from pressure. Fibroblast-like cells were first isolated from Wharton’s jelly by McElreavey et al. in 1991 [143]. These fibroblast-like cells were characterized as MSCs due to their expression of MSC phenotypic markers and their capacities to differentiate into osteogenic, adipogenic, and chondrogenic lineages [144]. A comparative study of human derived-MSCs demonstrated that Wharton’s jelly-derived MSCs (WJ-MSCs) exhibited the strongest inhibitory effects on T cell proliferation and the weakest expression of immune-related genes, such as genes encoding major histocompatibility complex (MHC) II and human leukocyte antigen (HLA), compared with BM-, AD-, and PL-MSCs [145]. These immunomodulatory and immunosuppressive properties of WJ-MSCs make them more applicable for clinical use as cell therapy. A study in canines by Kang et al. revealed that canine WJ-MSCs were capable of forming new bone in recipients with bone defects after orthotopic implantation with beta tricalcium phosphate (β-TCP) for 20 weeks. The capacity of WJ-MSCs to undergo osteogenic differentiation in vitro and new bone formation in vivo was similar to that of other MSCs isolated from canine bone marrow, adipose tissue, and UCB [146]. Hence, WJ-MSCs can potentially be used in clinical bone engineering for further treatment of bone defect diseases.
Trends in stem cell therapy for osteoporosis
The main hurdles for stem cell-based therapy for osteoporosis are long-term engraftment and the uncertainty of stem cell fate after transplantation. Certain reports have revealed that long-term engraftment of MSCs appears to be low and that the function of MSCs might be mediated through a paracrine mechanism, rather than through sustained engraftment in injured tissues [137, 147, 148]. Senescence of MSCs has been investigated as one of the key factors affecting the growth of MSCs in vitro, possibly hampering the cells’ long-term survival after transplantation [149]. Many ongoing studies are aiming to develop high-quality in vitro MSC cultures to increase the survival and engraftment rates. These developing methodologies include modification of MSCs by certain factors and improvement of in vitro MSC culture systems and differentiation procedures. The adjustment of culture conditions before transplantation, such as hypoxic preconditioning of MSC cultures in vitro, has been performed to increase the proliferation rate and to enhance the differentiation potential as well as to induce mobilization and homing of MSCs following transplantation [150, 151]. Genetically modified MSCs have been developed to ensure their homing, differentiation capacity, survival, and long-term engraftment at the injury sites of recipients. Immortalization of MSCs by knockdown of p53, a cell cycle regulator, in combination with overexpression of human telomerase reverse transcriptase (hTERT), the catalytic component of telomerase that leads to telomere elongation, could promote proliferation and increase the lifespan of MSCs while retaining the cells’ differentiation properties [152]. A combination of cell and gene therapy by overexpression of certain growth factors in MSCs has been promoted as being advantageous for MSC-based therapy [153, 154]. For example, the ectopic expression of basic fibroblast growth factor (bFGF) and platelet-derived growth factor B (PDGF-B) enhanced the in vitro proliferation and osteogenesis of BM-MSCs while inhibiting their adipogenesis [154]. MSCs are also an attractive cellular vehicle for the in vivo delivery of therapeutic genes, such as the genes encoding BMP-2 and RANK-Fc (a soluble inhibitor of RANKL), which could increase bone formation in osteoporosis animal models [155, 156]. Upon transplantation in vivo, the expressed transgene exerted its effect on both the host mesenchymal tissue (paracrine effect) and the transplanted MSCs (autocrine effect), contributing to the induction of bone formation in the recipients. These strategies of MSC modification are advantageous for the treatment of osteoporosis, which is characterized by increased bone resorption, and the therapies aim to maintain bone density and reduce the risk of fractures. To achieve effective MSC-based therapy for osteoporosis, the poor bone marrow homing and engraftment of MSCs after their systemic transplantation have to be improved. One emerging approach to overcome these limitations involves the overexpression of molecules involved in the bone homing of transplanted MSCs. Ectopic expression of α4 integrin on MSCs greatly increased bone marrow homing after systemic injection through the tail vein in immunocompetent mice. α4 integrin forms a heterodimer with endogenous β1 integrin and functions as a cell adhesion molecule, interacting with ECM proteins such as fibronectin and vascular cell adhesion protein 1 (VCAM-1) and thereby mediates the bone marrow homing and engraftment of MSCs [157]. Another study demonstrated that genetic modification of MSCs with CXCR4, the receptor for stromal-derived factor 1 (SDF-1), which mediates the bone marrow homing and engraftment of hematopoietic stem cells (HSCs), could also increase the bone marrow homing of MSCs and restore bone formation in mice with glucocorticoid-induced osteoporosis [158]. The development of in vitro differentiation procedures is quite important for MSCs used as cell therapy, especially for the treatment of localized osteoporosis and healing fractures resulting from osteoporosis. Technology consisting of three-dimensional (3D) in vitro culture models using biomaterial scaffolds has been developed, with the aim of mimicking the in vivo microenvironment to induce efficient tissue formation in vitro [159]. The biomaterial scaffolds must be slowly biodegradable and can act as a biocompatible matrix to support cell growth. In a recent preclinical study, Müller et al. demonstrated the combination of osteoconductive biomaterials with genetically modified human BM-MSCs in a bone defect rat model. The BM-MSCs were transduced with BMP-2 and loaded into β-TCP scaffolds before implantation into recipient rats. The researchers showed that when combined with BMP-2-transduced BM-MSCs, the scaffolds provided better results than scaffolds with recombinant BMP-2-treated BM-MSCs did [160]. This combination may represent a promising strategy for healing large-area bone defects in osteoporosis.
Alternative approaches involving improvement of native BM-MSCs or the local biologic environment at defect sites are of interest and are under investigation. Using a biomaterial scaffold combined with gene delivery for BMP-7 and PDGF-B expression has been shown to enhance the recruitment of BM-MSCs to defect sites and to promote their differentiation into osteoblasts, resulting in increased new bone formation in segmental femoral defects in ovariectomized rats [161]. α5β1 integrin, which mediates osteoblast differentiation in adult human MSCs through ECM-integrin interaction, is considered to be a target for promoting the osteogenic differentiation of BM-MSCs. The use of agonists that target α5β1 integrin can promote MSC recruitment and differentiation into osteoblasts and can also increase the survival of mature osteoblasts, leading to increased bone formation and repair in vivo [162]. Small molecules and microRNAs (miRNAs) are topics of interest in this area and may be applicable for osteoporosis treatment. Many miRNAs have been found to regulate the osteogenic differentiation of MSCs by various mechanisms [163]. Several miRNAs, e.g., miR-27a, miR-346, and miR-1423p, have been demonstrated to directly target inhibitors of the Wnt/β-catenin pathway, such as glycogen synthase kinase 3 beta (GSK3-β), SFRP1, and APC [164–166], resulting in modulation of the Wnt/β-catenin pathway and promotion of osteogenic differentiation of MSCs. Certain miRNAs, e.g., miR-20a, promote osteogenic differentiation by downregulating genes involved in adipogenic lineages, such as the gene encoding PPARγ [167]. By contrast, certain miRNAs negatively regulate osteogenic differentiation by targeting osteogenic genes, e.g., RUNX2, OSX, and SATB2. Inhibition strategy using an antagomir sequence against these miRNAs might attenuate the expression of the osteogenic genes and subsequently induce osteogenic differentiation [168–170]. The discovery of small molecules that target MSCs for fate determination by using high-throughput screening (HTS) techniques provides an advantage in drug development for osteoporosis treatment [171]. Small molecules may directly stimulate signaling pathways or target genes involved in osteogenic differentiation of MSCs [172–174]. For example, simvastatin, a 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, could promote osteogenic differentiation by activating the BMP-2 pathway in an ovariectomized rat model, leading to increasing BMD and bone volume [173, 175]. Given this finding together with their other advantages, i.e., their small size, high stability, non-immunogenicity, and, most of all, cell permeability, small molecules have undoubted potential for treating osteoporosis.
Conclusion
Osteoporosis is a systemic bone disorder defined by low BMD occurring due to an imbalance of osteoclastic and/or osteoblastic activities. The current therapeutics for osteoporosis are based on medicine for the prevention of further bone loss. The serious side effects caused by prolonged treatment have led to a need for an alternative approach for the life-long treatment of osteoporosis. Cell therapy appears to fulfill this demand, and MSCs provide a promising source of cells for clinical application in the treatment of osteoporosis. MSCs have been widely used for osteoporosis research as well as in other bone diseases due to not only their intrinsic ability to differentiate into osteoblasts but also their availability and ease of isolation, with high cell yields, from various tissues. Moreover, the immunoprivileged and immunosuppressive properties of MSCs make them more applicable in allogeneic cell replacement therapy. To date, over 400 clinical trials of MSC therapies have been registered with ClinicalTrials.gov (http://www.clinicaltrials.gov/); these trials have involved many diseases and conditions, such as bone disorders (osteoarthritis, osteogenesis imperfecta, osteoporosis, and rheumatoid arthritis), diabetes mellitus, graft-versus-host disease, and spinal cord injury. However, many questions remain unanswered, and many features have to be validated, such as the long-term engraftment and senescence of MSCs and suitable sources of MSCs for transplantation. In conclusion, much more work is needed to clarify the clinical applications of MSCs; however, the evidence certainly indicates that MSCs will play an important role in cell therapy for osteoporosis in the near future.
Abbreviations
3D, three-dimensional; AD, adipose tissue-derived; Alox15, arachidonate 15-lipoxygenase; bFGF, basic fibroblast growth factor; BM, bone marrow-derived; BMD, bone mineral density; BMP, bone morphogenetic protein; BMPR, bone morphogenetic protein receptor; CD, cluster of differentiation; COL1A1, collagen type 1 alpha1; CSF1, colony-stimulating factor 1; CXCR4, chemokine (c-x-c motif) receptor type 4; DKK-1, dickkopf-1; Dlx5, distal-less homeobox 5; DXA, dual-energy x-ray absorptiometry; ES, embryonic stem; ESR1, estrogen receptor 1; GREM, gremlin; GSK3β, glycogen synthase kinase 3 beta; GWAS, genome-wide association studies; HLA, human leukocyte antigen; HSC, hematopoietic stem cell; iPS, induced pluripotent stem; LRP5, lipoprotein receptor-related protein 5; MARK3, microtubule affinity regulation kinase 3; MHC, major histocompatibility complex; MSC, mesenchymal stem cell; NOG, noggin; OPG, osteoprotegerin; OSX, osterix; OVX, ovariectomy; PDGF-B, platelet-derived growth factor B; PL, placenta-derived; PPARγ, peroxisome proliferator-activated receptor gamma; PTH1R, parathyroid hormone 1 receptor; RANKL, receptor activator of nuclear factor kappa B ligand; RUNX2, runt-related transcription factor 2; SDF-1, stromal-derived factor-1; SERM, selective estrogen receptor modulator; SNPs, single nucleotide polymorphisms; SOST, sclerostin; SP7, Sp7 transcription factor 7; TCF/LEF, T cell factor/lymphoid enhancer factor; TGF-β, transforming growth factor beta; TNFRSF, tumor necrosis factor receptor superfamily; UC, umbilical cord-derived; UCB, umbilical cord blood; VDR, vitamin D receptor; WHO, World Health Organization; WJ, Wharton’s jelly-derived; Wnt, wingless-type MMTV integration site family; β-TCP, beta tricalcium phosphate.
References
Kanis JA. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis: synopsis of a WHO report. WHO Study Group. Osteoporos Int. 1994;4:368–81.
Kanis JA, Melton 3rd LJ, Christiansen C, Johnston CC, Khaltaev N. The diagnosis of osteoporosis. J Bone Miner Res. 1994;9:1137–41.
Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int. 2006;17:1726–33.
Dhanwal DK, Dennison EM, Harvey NC, Cooper C. Epidemiology of hip fracture: Worldwide geographic variation. Indian J Orthop. 2011;45:15–22.
Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res. 2007;22:465–75.
David C, Confavreux CB, Mehsen N, Paccou J, Leboime A, Legrand E. Severity of osteoporosis: what is the impact of co-morbidities? Joint Bone Spine. 2010;77 Suppl 2:S103–6.
Bonnick SL, Harris ST, Kendler DL, McClung MR, Silverman SL. Management of osteoporosis in postmenopausal women: 2010 position statement of The North American Menopause Society. Menopause. 2010;17:25–54.
Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes--a meta-analysis. Osteoporos Int. 2007;18:427–44.
Eriksen EF. Treatment of osteopenia. Rev Endocr Metab Disord. 2012;13:209–23.
Dawson-Hughes B, Tosteson AN, Melton 3rd LJ, Baim S, Favus MJ, Khosla S, Lindsay RL. Implications of absolute fracture risk assessment for osteoporosis practice guidelines in the USA. Osteoporos Int. 2008;19:449–58.
Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–37.
Zebaze RM, Ghasem-Zadeh A, Bohte A, Iuliano-Burns S, Mirams M, Price RI, Mackie EJ, Seeman E. Intracortical remodelling and porosity in the distal radius and post-mortem femurs of women: a cross-sectional study. Lancet. 2010;375:1729–36.
Marie PJ, Kassem M. Osteoblasts in osteoporosis: past, emerging, and future anabolic targets. Eur J Endocrinol. 2011;165:1–10.
Pino AM, Rosen CJ, Rodriguez JP. In osteoporosis, differentiation of mesenchymal stem cells (MSCs) improves bone marrow adipogenesis. Biol Res. 2012;45:279–87.
Marie PJ. Transcription factors controlling osteoblastogenesis. Arch Biochem Biophys. 2008;473:98–105.
James AW. Review of Signaling Pathways Governing MSC Osteogenic and Adipogenic Differentiation. Scientifica (Cairo). 2013;2013:684736.
Valenti MT, Garbin U, Pasini A, Zanatta M, Stranieri C, Manfro S, Zucal C, Dalle Carbonare L. Role of ox-PAPCs in the differentiation of mesenchymal stem cells (MSCs) and Runx2 and PPARgamma2 expression in MSCs-like of osteoporotic patients. PLoS One. 2011;6:e20363.
Zhang X, Yang M, Lin L, Chen P, Ma KT, Zhou CY, Ao YF. Runx2 overexpression enhances osteoblastic differentiation and mineralization in adipose--derived stem cells in vitro and in vivo. Calcif Tissue Int. 2006;79:169–78.
Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–50.
Cao JJ. Effects of obesity on bone metabolism. J Orthop Surg Res. 2011;6:30.
Benisch P, Schilling T, Klein-Hitpass L, Frey SP, Seefried L, Raaijmakers N, Krug M, Regensburger M, Zeck S, Schinke T, Amling M, Ebert R, Jakob F. The transcriptional profile of mesenchymal stem cell populations in primary osteoporosis is distinct and shows overexpression of osteogenic inhibitors. PLoS One. 2012;7:e45142.
Ralston SH. Genetics of osteoporosis. Ann N Y Acad Sci. 2010;1192:181–9.
Peacock M, Koller DL, Lai D, Hui S, Foroud T, Econs MJ. Sex-specific quantitative trait loci contribute to normal variation in bone structure at the proximal femur in men. Bone. 2005;37:467–73.
Wilson SG, Reed PW, Andrew T, Barber MJ, Lindersson M, Langdown M, Thompson D, Thompson E, Bailey M, Chiano M, Kleyn PW, Spector TD. A genome-screen of a large twin cohort reveals linkage for quantitative ultrasound of the calcaneus to 2q33-37 and 4q12-21. J Bone Miner Res. 2004;19:270–7.
Klein RF, Allard J, Avnur Z, Nikolcheva T, Rotstein D, Carlos AS, Shea M, Waters RV, Belknap JK, Peltz G, Orwoll ES. Regulation of bone mass in mice by the lipoxygenase gene Alox15. Science. 2004;303:229–32.
Grant SF, Reid DM, Blake G, Herd R, Fogelman I, Ralston SH. Reduced bone density and osteoporosis associated with a polymorphic Sp1 binding site in the collagen type I alpha 1 gene. Nat Genet. 1996;14:203–5.
Mann V, Ralston SH. Meta-analysis of COL1A1 Sp1 polymorphism in relation to bone mineral density and osteoporotic fracture. Bone. 2003;32:711–7.
Ralston SH, Uitterlinden AG, Brandi ML, Balcells S, Langdahl BL, Lips P, Lorenc R, Obermayer-Pietsch B, Scollen S, Bustamante M, Husted LB, Carey AH, Diez-Perez A, Dunning AM, Falchetti A, Karczmarewicz E, Kruk M, van Leeuwen JP, van Meurs JB, Mangion J, McGuigan FE, Mellibovsky L, del Monte F, Pols HA, Reeve J, Reid DM, Renner W, Rivadeneira F, van Schoor NM, Sherlock RE, Ioannidis JP, Investigators, G. Large-scale evidence for the effect of the COLIA1 Sp1 polymorphism on osteoporosis outcomes: the GENOMOS study. PLoS Med. 2006;3:e90.
Mann V, Hobson EE, Li B, Stewart TL, Grant SF, Robins SP, Aspden RM, Ralston SH. A COL1A1 Sp1 binding site polymorphism predisposes to osteoporotic fracture by affecting bone density and quality. J Clin Invest. 2001;107:899–907.
Stewart TL, Roschger P, Misof BM, Mann V, Fratzl P, Klaushofer K, Aspden R, Ralston SH. Association of COLIA1 Sp1 alleles with defective bone nodule formation in vitro and abnormal bone mineralization in vivo. Calcif Tissue Int. 2005;77:113–8.
Sano M, Inoue S, Hosoi T, Ouchi Y, Emi M, Shiraki M, Orimo H. Association of estrogen receptor dinucleotide repeat polymorphism with osteoporosis. Biochem Biophys Res Commun. 1995;217:378–83.
Albagha OM, Pettersson U, Stewart A, McGuigan FE, MacDonald HM, Reid DM, Ralston SH. Association of oestrogen receptor alpha gene polymorphisms with postmenopausal bone loss, bone mass, and quantitative ultrasound properties of bone. J Med Genet. 2005;42:240–6.
Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346:1513–21.
van Meurs JB, Rivadeneira F, Jhamai M, Hugens W, Hofman A, van Leeuwen JP, Pols HA, Uitterlinden AG. Common genetic variation of the low-density lipoprotein receptor-related protein 5 and 6 genes determines fracture risk in elderly white men. J Bone Miner Res. 2006;21:141–50.
Balemans W, Foernzler D, Parsons C, Ebeling M, Thompson A, Reid DM, Lindpaintner K, Ralston SH, Van Hul W. Lack of association between the SOST gene and bone mineral density in perimenopausal women: analysis of five polymorphisms. Bone. 2002;31:515–9.
Uitterlinden AG, Arp PP, Paeper BW, Charmley P, Proll S, Rivadeneira F, Fang Y, van Meurs JB, Britschgi TB, Latham JA, Schatzman RC, Pols HA, Brunkow ME. Polymorphisms in the sclerosteosis/van Buchem disease gene (SOST) region are associated with bone-mineral density in elderly whites. Am J Hum Genet. 2004;75:1032–45.
Langdahl BL, Knudsen JY, Jensen HK, Gregersen N, Eriksen EF. A sequence variation: 713-8delC in the transforming growth factor-beta 1 gene has higher prevalence in osteoporotic women than in normal women and is associated with very low bone mass in osteoporotic women and increased bone turnover in both osteoporotic and normal women. Bone. 1997;20:289–94.
Yamada Y, Miyauchi A, Goto J, Takagi Y, Okuizumi H, Kanematsu M, Hase M, Takai H, Harada A, Ikeda K. Association of a polymorphism of the transforming growth factor-beta1 gene with genetic susceptibility to osteoporosis in postmenopausal Japanese women. J Bone Miner Res. 1998;13:1569–76.
Langdahl BL, Uitterlinden AG, Ralston SH, Trikalinos TA, Balcells S, Brandi ML, Scollen S, Lips P, Lorenc R, Obermayer-Pietsch B, Reid DM, Armas JB, Arp PP, Bassiti A, Bustamante M, Husted LB, Carey AH, Perez Cano R, Dobnig H, Dunning AM, Fahrleitner-Pammer A, Falchetti A, Karczmarewicz E, Kruk M, van Leeuwen JP, Masi L, van Meurs JB, Mangion J, McGuigan FE, Mellibovsky L, Mosekilde L, Nogues X, Pols HA, Reeve J, Renner W, Rivadeneira F, van Schoor NM, Ioannidis JP, investigators, A., investigators, D., investigators, E., investigators, E., investigators, F., investigators, L., investigators, E. and Study, G. Large-scale analysis of association between polymorphisms in the transforming growth factor beta 1 gene (TGFB1) and osteoporosis: the GENOMOS study. Bone. 2008;42:969–81.
Morrison NA, Qi JC, Tokita A, Kelly PJ, Crofts L, Nguyen TV, Sambrook PN, Eisman JA. Prediction of bone density from vitamin D receptor alleles. Nature. 1994;367:284–7.
Richards JB, Rivadeneira F, Inouye M, Pastinen TM, Soranzo N, Wilson SG, Andrew T, Falchi M, Gwilliam R, Ahmadi KR, Valdes AM, Arp P, Whittaker P, Verlaan DJ, Jhamai M, Kumanduri V, Moorhouse M, van Meurs JB, Hofman A, Pols HA, Hart D, Zhai G, Kato BS, Mullin BH, Zhang F, Deloukas P, Uitterlinden AG, Spector TD. Bone mineral density, osteoporosis, and osteoporotic fractures: a genome-wide association study. Lancet. 2008;371:1505–12.
Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T, Jonsdottir T, Saemundsdottir J, Snorradottir S, Center JR, Nguyen TV, Alexandersen P, Gulcher JR, Eisman JA, Christiansen C, Sigurdsson G, Kong A, Thorsteinsdottir U, Stefansson K. New sequence variants associated with bone mineral density. Nat Genet. 2009;41:15–7.
Boyce BF, Xing L. The RANKL/RANK/OPG pathway. Curr Osteoporos Rep. 2007;5:98–104.
Zaiss MM, Sarter K, Hess A, Engelke K, Bohm C, Nimmerjahn F, Voll R, Schett G, David JP. Increased bone density and resistance to ovariectomy-induced bone loss in FoxP3-transgenic mice based on impaired osteoclast differentiation. Arthritis Rheum. 2010;62:2328–38.
Min H, Morony S, Sarosi I, Dunstan CR, Capparelli C, Scully S, Van G, Kaufman S, Kostenuik PJ, Lacey DL, Boyle WJ, Simonet WS. Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis. J Exp Med. 2000;192:463–74.
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–19.
Mizuno A, Amizuka N, Irie K, Murakami A, Fujise N, Kanno T, Sato Y, Nakagawa N, Yasuda H, Mochizuki S, Gomibuchi T, Yano K, Shima N, Washida N, Tsuda E, Morinaga T, Higashio K, Ozawa H. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun. 1998;247:610–5.
Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–8.
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–9.
Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901.
Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, Morita I, Wagner EF, Mak TW, Serfling E, Takayanagi H. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202:1261–9.
Jones DH, Kong YY, Penninger JM. Role of RANKL and RANK in bone loss and arthritis. Ann Rheum Dis. 2002;61 Suppl 2:ii32–9.
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–76.
Kearns AE, Khosla S, Kostenuik PJ. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr Rev. 2008;29:155–92.
Hodge JM, Collier FM, Pavlos NJ, Kirkland MA, Nicholson GC. M-CSF potently augments RANKL-induced resorption activation in mature human osteoclasts. PLoS One. 2011;6:e21462.
Hofbauer LC, Schoppet M. Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. JAMA. 2004;292:490–5.
Rogers A, Eastell R. Circulating osteoprotegerin and receptor activator for nuclear factor kappaB ligand: clinical utility in metabolic bone disease assessment. J Clin Endocrinol Metab. 2005;90:6323–31.
Mainini G, Incoronato M, Urso L, Scaffa C. Serum osteoprotegerin correlates with age and bone mass in postmenopausal, but not in fertile age women. Clin Exp Obstet Gynecol. 2011;38:355–9.
Rifas L. Bone and cytokines: beyond IL-1, IL-6 and TNF-alpha. Calcif Tissue Int. 1999;64:1–7.
Pagliari D, Ciro Tamburrelli F, Zirio G, Newton EE, Cianci R. The role of “bone immunological niche” for a new pathogenetic paradigm of osteoporosis. Anal Cell Pathol (Amst). 2015;2015:434389.
Yuan FL, Li X, Lu WG, Zhao YQ, Li CW, Li JP, Sun JM, Xu RS. Type 17 T-helper cells might be a promising therapeutic target for osteoporosis. Mol Biol Rep. 2012;39:771–4.
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–82.
Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: a new candidate in the pathogenesis of osteoporosis. PLoS One. 2012;7:e44552.
Korn T, Oukka M, Kuchroo V, Bettelli E. Th17 cells: effector T cells with inflammatory properties. Semin Immunol. 2007;19:362–71.
Zhao R. Immune regulation of bone loss by Th17 cells in oestrogen-deficient osteoporosis. Eur J Clin Invest. 2013;43:1195–202.
Manolagas SC. Wnt signaling and osteoporosis. Maturitas. 2014;78:233–7.
Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–50.
Hoeppner LH, Secreto FJ, Westendorf JJ. Wnt signaling as a therapeutic target for bone diseases. Expert Opin Ther Targets. 2009;13:485–96.
Kang S, Bennett CN, Gerin I, Rapp LA, Hankenson KD, Macdougald OA. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma. J Biol Chem. 2007;282:14515–24.
Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202–9.
Glass 2nd DA, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–64.
Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, De Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Juppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, Van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, Warman ML, Osteoporosis-Pseudoglioma Syndrome Collaborative, G. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–23.
Bennett CN, Ouyang H, Ma YL, Zeng Q, Gerin I, Sousa KM, Lane TF, Krishnan V, Hankenson KD, MacDougald OA. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J Bone Miner Res. 2007;22:1924–32.
Perez-Castrillon JL, Olmos JM, Nan DN, Castillo J, Arozamena J, Montero A, Perez-Nunez MI, Riancho JA. Polymorphisms of the WNT10B gene, bone mineral density, and fractures in postmenopausal women. Calcif Tissue Int. 2009;85:113–8.
Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD, MacDougald OA. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci U S A. 2005;102:3324–9.
Stevens JR, Miranda-Carboni GA, Singer MA, Brugger SM, Lyons KM, Lane TF. Wnt10b deficiency results in age-dependent loss of bone mass and progressive reduction of mesenchymal progenitor cells. J Bone Miner Res. 2010;25:2138–47.
Boland GM, Perkins G, Hall DJ, Tuan RS. Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J Cell Biochem. 2004;93:1210–30.
Ling L, Nurcombe V, Cool SM. Wnt signaling controls the fate of mesenchymal stem cells. Gene. 2009;433:1–7.
Chen J, Long F. beta-catenin promotes bone formation and suppresses bone resorption in postnatal growing mice. J Bone Miner Res. 2013;28:1160–9.
Rodriguez JP, Garat S, Gajardo H, Pino AM, Seitz G. Abnormal osteogenesis in osteoporotic patients is reflected by altered mesenchymal stem cells dynamics. J Cell Biochem. 1999;75:414–23.
Cruciat CM, Niehrs C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb Perspect Biol. 2013;5:a015081.
Ahmed SF, Fouda N, Abbas AA. Serum dickkopf-1 level in postmenopausal females: correlation with bone mineral density and serum biochemical markers. J Osteoporos. 2013;2013:460210.
Mbalaviele G, Sheikh S, Stains JP, Salazar VS, Cheng SL, Chen D, Civitelli R. Beta-catenin and BMP-2 synergize to promote osteoblast differentiation and new bone formation. J Cell Biochem. 2005;94:403–18.
Zhang M, Yan Y, Lim YB, Tang D, Xie R, Chen A, Tai P, Harris SE, Xing L, Qin YX, Chen D. BMP-2 modulates beta-catenin signaling through stimulation of Lrp5 expression and inhibition of beta-TrCP expression in osteoblasts. J Cell Biochem. 2009;108:896–905.
Cheng H, Jiang W, Phillips FM, Haydon RC, Peng Y, Zhou L, Luu HH, An N, Breyer B, Vanichakarn P, Szatkowski JP, Park JY, He TC. Osteogenic activity of the fourteen types of human bone morphogenetic proteins (BMPs). J Bone Joint Surg Am. 2003;85-A:1544–52.
Chen D, Ji X, Harris MA, Feng JQ, Karsenty G, Celeste AJ, Rosen V, Mundy GR, Harris SE. Differential roles for bone morphogenetic protein (BMP) receptor type IB and IA in differentiation and specification of mesenchymal precursor cells to osteoblast and adipocyte lineages. J Cell Biol. 1998;142:295–305.
Knight MN, Hankenson KD. Mesenchymal Stem Cells in Bone Regeneration. Adv Wound Care (New Rochelle). 2013;2:306–16.
Chen G, Deng C, Li YP. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci. 2012;8:272–88.
Zhang C. Transcriptional regulation of bone formation by the osteoblast-specific transcription factor Osx. J Orthop Surg Res. 2010;5:37.
Lee MH, Kwon TG, Park HS, Wozney JM, Ryoo HM. BMP-2-induced Osterix expression is mediated by Dlx5 but is independent of Runx2. Biochem Biophys Res Commun. 2003;309:689–94.
Holleville N, Mateos S, Bontoux M, Bollerot K, Monsoro-Burq AH. Dlx5 drives Runx2 expression and osteogenic differentiation in developing cranial suture mesenchyme. Dev Biol. 2007;304:860–74.
Jeong HM, Jin YH, Kim YJ, Yum J, Choi YH, Yeo CY, Lee KY. Akt phosphorylates and regulates the function of Dlx5. Biochem Biophys Res Commun. 2011;409:681–6.
Choi YH, Jeong HM, Jin YH, Li H, Yeo CY, Lee KY. Akt phosphorylates and regulates the osteogenic activity of Osterix. Biochem Biophys Res Commun. 2011;411:637–41.
Daluiski A, Engstrand T, Bahamonde ME, Gamer LW, Agius E, Stevenson SL, Cox K, Rosen V, Lyons KM. Bone morphogenetic protein-3 is a negative regulator of bone density. Nat Genet. 2001;27:84–8.
Prall WC, Haasters F, Heggebo J, Polzer H, Schwarz C, Gassner C, Grote S, Anz D, Jager M, Mutschler W, Schieker M. Mesenchymal stem cells from osteoporotic patients feature impaired signal transduction but sustained osteoinduction in response to BMP-2 stimulation. Biochem Biophys Res Commun. 2013;440:617–22.
Haasters F, Docheva D, Gassner C, Popov C, Bocker W, Mutschler W, Schieker M, Prall WC. Mesenchymal stem cells from osteoporotic patients reveal reduced migration and invasion upon stimulation with BMP-2 or BMP-7. Biochem Biophys Res Commun. 2014;452:118–23.
Wu XB, Li Y, Schneider A, Yu W, Rajendren G, Iqbal J, Yamamoto M, Alam M, Brunet LJ, Blair HC, Zaidi M, Abe E. Impaired osteoblastic differentiation, reduced bone formation, and severe osteoporosis in noggin-overexpressing mice. J Clin Invest. 2003;112:924–34.
Devlin RD, Du Z, Pereira RC, Kimble RB, Economides AN, Jorgetti V, Canalis E. Skeletal overexpression of noggin results in osteopenia and reduced bone formation. Endocrinology. 2003;144:1972–8.
Moffett SP, Dillon KA, Yerges LM, Goodrich LJ, Nestlerode C, Bunker CH, Wheeler VW, Patrick AL, Zmuda JM. Identification and association analysis of single nucleotide polymorphisms in the human noggin (NOG) gene and osteoporosis phenotypes. Bone. 2009;44:999–1002.
Gazzerro E, Pereira RC, Jorgetti V, Olson S, Economides AN, Canalis E. Skeletal overexpression of gremlin impairs bone formation and causes osteopenia. Endocrinology. 2005;146:655–65.
Cheung CL, Lau KS, Sham PC, Tan KC, Kung AW. Genetic variants in GREM2 are associated with bone mineral density in a southern Chinese population. J Clin Endocrinol Metab. 2013;98:E1557–61.
Dawson-Hughes B, Harris SS, Krall EA, Dallal GE. Effect of calcium and vitamin D supplementation on bone density in men and women 65 years of age or older. N Engl J Med. 1997;337:670–6.
Baccaro LF, Conde DM, Costa-Paiva L, Pinto-Neto AM. The epidemiology and management of postmenopausal osteoporosis: a viewpoint from Brazil. Clin Interv Aging. 2015;10:583–91.
Van Beek ER, Lowik CW, Papapoulos SE. Bisphosphonates suppress bone resorption by a direct effect on early osteoclast precursors without affecting the osteoclastogenic capacity of osteogenic cells: the role of protein geranylgeranylation in the action of nitrogen-containing bisphosphonates on osteoclast precursors. Bone. 2002;30:64–70.
Rodan GA, Reszka AA. Bisphosphonate mechanism of action. Curr Mol Med. 2002;2:571–7.
Papapoulos SE, Quandt SA, Liberman UA, Hochberg MC, Thompson DE. Meta-analysis of the efficacy of alendronate for the prevention of hip fractures in postmenopausal women. Osteoporos Int. 2005;16:468–74.
Inderjeeth CA, Glendenning P, Ratnagobal S, Inderjeeth DC, Ondhia C. Long-term efficacy, safety, and patient acceptability of ibandronate in the treatment of postmenopausal osteoporosis. Int J Womens Health. 2015;7:7–17.
Harrington JT, Ste-Marie LG, Brandi ML, Civitelli R, Fardellone P, Grauer A, Barton I, Boonen S. Risedronate rapidly reduces the risk for nonvertebral fractures in women with postmenopausal osteoporosis. Calcif Tissue Int. 2004;74:129–35.
Black DM, Delmas PD, Eastell R, Reid IR, Boonen S, Cauley JA, Cosman F, Lakatos P, Leung PC, Man Z, Mautalen C, Mesenbrink P, Hu H, Caminis J, Tong K, Rosario-Jansen T, Krasnow J, Hue TF, Sellmeyer D, Eriksen EF, Cummings SR. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med. 2007;356:1809–22.
Yarom N, Yahalom R, Shoshani Y, Hamed W, Regev E, Elad S. Osteonecrosis of the jaw induced by orally administered bisphosphonates: incidence, clinical features, predisposing factors and treatment outcome. Osteoporos Int. 2007;18:1363–70.
Abrahamsen B, Eiken P, Eastell R. Subtrochanteric and diaphyseal femur fractures in patients treated with alendronate: a register-based national cohort study. J Bone Miner Res. 2009;24:1095–102.
Cummings SR, San Martin J, McClung MR, Siris ES, Eastell R, Reid IR, Delmas P, Zoog HB, Austin M, Wang A, Kutilek S, Adami S, Zanchetta J, Libanati C, Siddhanti S, Christiansen C. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756–65.
Chesnut 3rd CH, Silverman S, Andriano K, Genant H, Gimona A, Harris S, Kiel D, LeBoff M, Maricic M, Miller P, Moniz C, Peacock M, Richardson P, Watts N, Baylink D. A randomized trial of nasal spray salmon calcitonin in postmenopausal women with established osteoporosis: the prevent recurrence of osteoporotic fractures study. PROOF Study Group Am J Med. 2000;109:267–76.
Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. Jama. 2002;288:321–33.
Ettinger B, Black DM, Mitlak BH, Knickerbocker RK, Nickelsen T, Genant HK, Christiansen C, Delmas PD, Zanchetta JR, Stakkestad J, Gluer CC, Krueger K, Cohen FJ, Eckert S, Ensrud KE, Avioli LV, Lips P, Cummings SR. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA. 1999;282:637–45.
Barrett-Connor E, Mosca L, Collins P, Geiger MJ, Grady D, Kornitzer M, McNabb MA, Wenger NK. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med. 2006;355:125–37.
Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R, Caan B, Chlebowski R, Curb D, Gass M, Hays J, Heiss G, Hendrix S, Howard BV, Hsia J, Hubbell A, Jackson R, Johnson KC, Judd H, Kotchen JM, Kuller L, LaCroix AZ, Lane D, Langer RD, Lasser N, Lewis CE, Manson J, Margolis K, Ockene J, O'Sullivan MJ, Phillips L, Prentice RL, Ritenbaugh C, Robbins J, Rossouw JE, Sarto G, Stefanick ML, Van Horn L, Wactawski-Wende J, Wallace R, Wassertheil-Smoller S. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women's Health Initiative randomized controlled trial. JAMA. 2004;291:1701–12.
Vahle JL, Sato M, Long GG, Young JK, Francis PC, Engelhardt JA, Westmore MS, Linda Y, Nold JB. Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone (1-34) for 2 years and relevance to human safety. Toxicol Pathol. 2002;30:312–21.
Marie PJ, Felsenberg D, Brandi ML. How strontium ranelate, via opposite effects on bone resorption and formation, prevents osteoporosis. Osteoporos Int. 2011;22:1659–67.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement Cytotherapy. 2006;8:315–7.
Ryan JM, Barry FP, Murphy JM, Mahon BP. Mesenchymal stem cells avoid allogeneic rejection. J Inflamm (Lond). 2005;2:8.
Liu H, Kemeny DM, Heng BC, Ouyang HW, Melendez AJ, Cao T. The immunogenicity and immunomodulatory function of osteogenic cells differentiated from mesenchymal stem cells. J Immunol. 2006;176:2864–71.
Lee K, Kim H, Kim JM, Kim JR, Kim KJ, Kim YJ, Park SI, Jeong JH, Moon YM, Lim HS, Bae DW, Kwon J, Ko CY, Kim HS, Shin HI, Jeong D. Systemic transplantation of human adipose-derived stem cells stimulates bone repair by promoting osteoblast and osteoclast function. J Cell Mol Med. 2011;15:2082–94.
Ye X, Zhang P, Xue S, Xu Y, Tan J, Liu G. Adipose-derived stem cells alleviate osteoporosis by enhancing osteogenesis and inhibiting adipogenesis in a rabbit model. Cytotherapy. 2014;16:1643–55.
Ichioka N, Inaba M, Kushida T, Esumi T, Takahara K, Inaba K, Ogawa R, Iida H, Ikehara S. Prevention of senile osteoporosis in SAMP6 mice by intrabone marrow injection of allogeneic bone marrow cells. Stem Cells. 2002;20:542–51.
Ocarino Nde M, Boeloni JN, Jorgetti V, Gomes DA, Goes AM, Serakides R. Intra-bone marrow injection of mesenchymal stem cells improves the femur bone mass of osteoporotic female rats. Connect Tissue Res. 2010;51:426–33.
Wang Z, Goh J, De Das S, Ge Z, Ouyang H, Chong JS, Low SL, Lee EH. Efficacy of bone marrow-derived stem cells in strengthening osteoporotic bone in a rabbit model. Tissue Eng. 2006;12:1753–61.
Cao L, Liu G, Gan Y, Fan Q, Yang F, Zhang X, Tang T, Dai K. The use of autologous enriched bone marrow MSCs to enhance osteoporotic bone defect repair in long-term estrogen deficient goats. Biomaterials. 2012;33:5076–84.
Stolzing A, Jones E, McGonagle D, Scutt A. Age-related changes in human bone marrow-derived mesenchymal stem cells: consequences for cell therapies. Mech Ageing Dev. 2008;129:163–73.
Arana M, Mazo M, Aranda P, Pelacho B, Prosper F. Adipose tissue-derived mesenchymal stem cells: isolation, expansion, and characterization. Methods Mol Biol. 2013;1036:47–61.
Strem BM, Hicok KC, Zhu M, Wulur I, Alfonso Z, Schreiber RE, Fraser JK, Hedrick MH. Multipotential differentiation of adipose tissue-derived stem cells. Keio J Med. 2005;54:132–41.
Kern S, Eichler H, Stoeve J, Kluter H, Bieback K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells. 2006;24:1294–301.
Jurgens WJ, Oedayrajsingh-Varma MJ, Helder MN, Zandiehdoulabi B, Schouten TE, Kuik DJ, Ritt MJ, van Milligen FJ. Effect of tissue-harvesting site on yield of stem cells derived from adipose tissue: implications for cell-based therapies. Cell Tissue Res. 2008;332:415–26.
Choudhery MS, Badowski M, Muise A, Pierce J, Harris DT. Donor age negatively impacts adipose tissue-derived mesenchymal stem cell expansion and differentiation. J Transl Med. 2014;12:8.
Mirsaidi A, Genelin K, Vetsch JR, Stanger S, Theiss F, Lindtner RA, von Rechenberg B, Blauth M, Muller R, Kuhn GA, Hofmann Boss S, Ebner HL, Richards PJ. Therapeutic potential of adipose-derived stromal cells in age-related osteoporosis. Biomaterials. 2014;35:7326–35.
Cho SW, Sun HJ, Yang JY, Jung JY, Choi HJ, An JH, Kim SW, Kim SY, Park KJ, Shin CS. Human adipose tissue-derived stromal cell therapy prevents bone loss in ovariectomized nude mouse. Tissue Eng Part A. 2012;18:1067–78.
Bieback K, Brinkmann I. Mesenchymal stromal cells from human perinatal tissues: From biology to cell therapy. World J Stem Cells. 2010;2:81–92.
Yen BL, Huang HI, Chien CC, Jui HY, Ko BS, Yao M, Shun CT, Yen ML, Lee MC, Chen YC. Isolation of multipotent cells from human term placenta. Stem Cells. 2005;23:3–9.
Sanvoranart T, Supokawej A, Kheolamai P, U-Pratya Y, Klincumhom N, Manochantr S, Wattanapanitch M, Issaragrisil S. Bortezomib enhances the osteogenic differentiation capacity of human mesenchymal stromal cells derived from bone marrow and placental tissues. Biochem Biophys Res Commun. 2014;447:580–5.
Karahuseyinoglu S, Cinar O, Kilic E, Kara F, Akay GG, Demiralp DO, Tukun A, Uckan D, Can A. Biology of stem cells in human umbilical cord stroma: in situ and in vitro surveys. Stem Cells. 2007;25:319–31.
Diao Y, Ma Q, Cui F, Zhong Y. Human umbilical cord mesenchymal stem cells: osteogenesis in vivo as seed cells for bone tissue engineering. J Biomed Mater Res A. 2009;91:123–31.
McElreavey KD, Irvine AI, Ennis KT, McLean WH. Isolation, culture and characterisation of fibroblast-like cells derived from the Wharton's jelly portion of human umbilical cord. Biochem Soc Trans. 1991;19:29s.
Wang HS, Hung SC, Peng ST, Huang CC, Wei HM, Guo YJ, Fu YS, Lai MC, Chen CC. Mesenchymal stem cells in the Wharton's jelly of the human umbilical cord. Stem Cells. 2004;22:1330–7.
Li X, Bai J, Ji X, Li R, Xuan Y, Wang Y. Comprehensive characterization of four different populations of human mesenchymal stem cells as regards their immune properties, proliferation and differentiation. Int J Mol Med. 2014;34:695–704.
Kang BJ, Ryu HH, Park SS, Koyama Y, Kikuchi M, Woo HM, Kim WH, Kweon OK. Comparing the osteogenic potential of canine mesenchymal stem cells derived from adipose tissues, bone marrow, umbilical cord blood, and Wharton's jelly for treating bone defects. J Vet Sci. 2012;13:299–310.
von Bahr L, Batsis I, Moll G, Hagg M, Szakos A, Sundberg B, Uzunel M, Ringden O, Le Blanc K. Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long-term engraftment and no ectopic tissue formation. Stem Cells. 2012;30:1575–8.
Seebach E, Freischmidt H, Holschbach J, Fellenberg J, Richter W. Mesenchymal stroma cells trigger early attraction of M1 macrophages and endothelial cells into fibrin hydrogels, stimulating long bone healing without long-term engraftment. Acta Biomater. 2014;10:4730–41.
Galderisi U, Helmbold H, Squillaro T, Alessio N, Komm N, Khadang B, Cipollaro M, Bohn W, Giordano A. In vitro senescence of rat mesenchymal stem cells is accompanied by downregulation of stemness-related and DNA damage repair genes. Stem Cells Dev. 2009;18:1033–42.
Tsai CC, Yew TL, Yang DC, Huang WH, Hung SC. Benefits of hypoxic culture on bone marrow multipotent stromal cells. Am J Blood Res. 2012;2:148–59.
Xu L, Sun X, Cao K, Wu Y, Zou D, Liu Y, Zhang X, Zhang X, Wang G, Huang Q, Jiang X. Hypoxia induces osteogenesis in rabbit adipose-derived stem cells overexpressing bone morphogenic protein-2. Oral Dis. 2014;20:430–9.
Liu TM, Ng WM, Tan HS, Vinitha D, Yang Z, Fan JB, Zou Y, Hui JH, Lee EH, Lim B. Molecular basis of immortalization of human mesenchymal stem cells by combination of p53 knockdown and human telomerase reverse transcriptase overexpression. Stem Cells Dev. 2013;22:268–78.
He X, Dziak R, Yuan X, Mao K, Genco R, Swihart M, Sarkar D, Li C, Wang C, Lu L, Andreadis S, Yang S. BMP2 genetically engineered MSCs and EPCs promote vascularized bone regeneration in rat critical-sized calvarial bone defects. PLoS One. 2013;8:e60473.
Fierro FA, Kalomoiris S, Sondergaard CS, Nolta JA. Effects on proliferation and differentiation of multipotent bone marrow stromal cells engineered to express growth factors for combined cell and gene therapy. Stem Cells. 2011;29:1727–37.
Kim D, Cho SW, Her SJ, Yang JY, Kim SW, Kim SY, Shin CS. Retrovirus-mediated gene transfer of receptor activator of nuclear factor-kappaB-Fc prevents bone loss in ovariectomized mice. Stem Cells. 2006;24:1798–805.
Egermann M, Baltzer AW, Adamaszek S, Evans C, Robbins P, Schneider E, Lill CA. Direct adenoviral transfer of bone morphogenetic protein-2 cDNA enhances fracture healing in osteoporotic sheep. Hum Gene Ther. 2006;17:507–17.
Kumar S, Ponnazhagan S. Bone homing of mesenchymal stem cells by ectopic alpha 4 integrin expression. Faseb j. 2007;21:3917–27.
Lien CY, Chih-Yuan Ho K, Lee OK, Blunn GW, Su Y. Restoration of bone mass and strength in glucocorticoid-treated mice by systemic transplantation of CXCR4 and cbfa-1 co-expressing mesenchymal stem cells. J Bone Miner Res. 2009;24:837–48.
Sterling JA, Guelcher SA. Biomaterial scaffolds for treating osteoporotic bone. Curr Osteoporos Rep. 2014;12:48–54.
Muller CW, Hildebrandt K, Gerich T, Krettek C, van Griensven M, Balmayor ER. BMP-2-transduced human bone marrow stem cells enhance neo-bone formation in a rat critical-sized femur defect. J Tissue Eng Regen Med. 2015; doi:10.1002/term.2015.
Zhang Y, Cheng N, Miron R, Shi B, Cheng X. Delivery of PDGF-B and BMP-7 by mesoporous bioglass/silk fibrin scaffolds for the repair of osteoporotic defects. Biomaterials. 2012;33:6698–708.
Marie PJ. Targeting integrins to promote bone formation and repair. Nat Rev Endocrinol. 2013;9:288–95.
van Wijnen AJ, van de Peppel J, van Leeuwen JP, Lian JB, Stein GS, Westendorf JJ, Oursler MJ, Im HJ, Taipaleenmaki H, Hesse E, Riester S, Kakar S. MicroRNA functions in osteogenesis and dysfunctions in osteoporosis. Curr Osteoporos Rep. 2013;11:72–82.
Guo D, Li Q, Lv Q, Wei Q, Cao S, Gu J. MiR-27a targets sFRP1 in hFOB cells to regulate proliferation, apoptosis and differentiation. PLoS One. 2014;9:e91354.
Wang Q, Cai J, Cai XH, Chen L. miR-346 regulates osteogenic differentiation of human bone marrow-derived mesenchymal stem cells by targeting the Wnt/beta-catenin pathway. PLoS One. 2013;8:e72266.
Hu W, Ye Y, Zhang W, Wang J, Chen A, Guo F. miR1423p promotes osteoblast differentiation by modulating Wnt signaling. Mol Med Rep. 2013;7:689–93.
Zhang JF, Fu WM, He ML, Xie WD, Lv Q, Wan G, Li G, Wang H, Lu G, Hu X, Jiang S, Li JN, Lin MC, Zhang YO, Kung HF. MiRNA-20a promotes osteogenic differentiation of human mesenchymal stem cells by co-regulating BMP signaling. RNA Biol. 2011;8:829–38.
Huang J, Zhao L, Xing L, Chen D. MicroRNA-204 regulates Runx2 protein expression and mesenchymal progenitor cell differentiation. Stem Cells. 2010;28:357–64.
Deng Y, Wu S, Zhou H, Bi X, Wang Y, Hu Y, Gu P, Fan X. Effects of a miR-31, Runx2, and Satb2 regulatory loop on the osteogenic differentiation of bone mesenchymal stem cells. Stem Cells Dev. 2013;22:2278–86.
Zhang JF, Fu WM, He ML, Wang H, Wang WM, Yu SC, Bian XW, Zhou J, Lin MC, Lu G, Poon WS, Kung HF. MiR-637 maintains the balance between adipocytes and osteoblasts by directly targeting Osterix. Mol Biol Cell. 2011;22:3955–61.
Nawa K, Ikeno H, Matsuhashi N, Ogasawara T, Otsuka E. Discovering small molecules that inhibit adipogenesis and promote osteoblastogenesis: unique screening and Oncostatin M-like activity. Differentiation. 2013;86:65–74.
Zhang JF, Li G, Chan CY, Meng CL, Lin MC, Chen YC, He ML, Leung PC, Kung HF. Flavonoids of Herba Epimedii regulate osteogenesis of human mesenchymal stem cells through BMP and Wnt/beta-catenin signaling pathway. Mol Cell Endocrinol. 2010;314:70–4.
Ho ML, Chen YH, Liao HJ, Chen CH, Hung SH, Lee MJ, Fu YC, Wang YH, Wang GJ, Chang JK. Simvastatin increases osteoblasts and osteogenic proteins in ovariectomized rats. Eur J Clin Invest. 2009;39:296–303.
Darcy A, Meltzer M, Miller J, Lee S, Chappell S, Ver Donck K, Montano M. A novel library screen identifies immunosuppressors that promote osteoblast differentiation. Bone. 2012;50:1294–303.
Chen PY, Sun JS, Tsuang YH, Chen MH, Weng PW, Lin FH. Simvastatin promotes osteoblast viability and differentiation via Ras/Smad/Erk/BMP-2 signaling pathway. Nutr Res. 2010;30:191–9.
Acknowledgments
This project was supported by Mahidol University.
Authors’ contributions
JP and TS: conception and drafting of manuscript; KN, NN, and KS: drafting of manuscript. VP: approval of manuscript. AS: conception and final approval of manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Phetfong, J., Sanvoranart, T., Nartprayut, K. et al. Osteoporosis: the current status of mesenchymal stem cell-based therapy. Cell Mol Biol Lett 21, 12 (2016). https://doi.org/10.1186/s11658-016-0013-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11658-016-0013-1