
Abstract
Innate immunity plays a crucial role in the pathogenesis of type 2 diabetes and related complications. Since the toll-like receptors (TLRs) are central to innate immunity, it appears that they are important participants in the development and pathogenesis of the disease. Previous investigations demonstrated that TLR2 homodimers and TLR2 heterodimers with TLR1 or TLR6 activate innate immunity upon recognition of damage-associated molecular patterns (DAMPs). Several DAMPs are released during type 2 diabetes, so it may be hypothesized that TLR2 is significantly involved in its progression. Here, we review recent data on the important roles and status of TLR2 in type 2 diabetes and related complications.
Similar content being viewed by others

Background
Type 2 diabetes, also referred to as adult-onset diabetes or as non-insulin-dependent diabetes mellitus (NIDDM) is increasingly common [1, 2]. The heightened levels of blood glucose are associated with several complications, including nerve damage (neuropathy), which is caused by injury to the walls of the capillaries that nourish the nerves and results in tingling, numbness, burning or pain [3, 4]. Nephropathy is also associated with injuries to the glomeruli [5]. Eye damage or retinopathy is induced by glucose-related damage to the blood vessels of the retina [6–8]. Type 2 diabetes correlates significantly with cardiovascular disease, including heart attack, angina, stroke and atherosclerosis. Moreover, investigations have shown a link between type 2 diabetes and brain diseases, such as mild cognitive impairment, Alzheimer’s disease and vascular dementia [9]. Other complications include periodontitis [10, 11], cystic fibrosis [12], hypertension [13] and hearing impairment [14].
The parameters of innate immunity are associated with type 2 diabetes and its complications [15, 16]. Therefore, it has been hypothesized that the disease is immune dependent [16, 17]. For instance, in cases of type 2 diabetes, immune cells produce inappropriate levels of inflammatory cytokines, which may have a deleterious effect on the pathogenesis of type 2 diabetes [18]. Elevated serum levels of innate immunity inflammatory cytokines such as IL-6 [19], IL-18 [20] and TNF-α [19] have been reported for subjects with type 2 diabetes and related complications.
Several cross-sectional studies also confirmed the relationship between innate immunity and type 2 diabetes. For instance, studies were made of non-diabetic subjects and patients with type 2 diabetes determined as impaired glucose tolerance or impaired fasting glucose. They identified that innate immunity soluble molecules, such as C-reactive protein (CRP) and its related cytokines, are: positively associated with insulin resistance, plasma insulin concentration, circulating triglyceride level, and BMI and waist circumference measurements; and negatively correlated with HDL concentration [21]. Additionally, it has been documented that exercise can improve type 2 diabetes complications through its immunomodulatory effects [22].
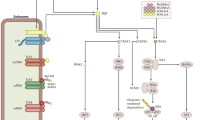
Although a significant relationship between innate immunity and type 2 diabetes has been confirmed, it has yet to be clarified if innate immunity activation is a major mechanism in the induction of type 2 diabetes or if the disease activates innate immunity. It seems that it is mutual (Fig. 1), so evaluation of innate immunity receptors and their signaling molecules should our knowledge of the condition and relationship.

There is a mutual avenue between type 2 diabetes and innate immunity. Type 2 diabetes can activate innate immunity and its pathogenesis can be accelerated by activated innate immunity. CRP: C-reactive protein, SAP: Serum amyloid protein
Toll-like receptors (TLRs) are the innate immune cell receptors. They play pivotal roles in the recognition of damage-associated molecular patterns (DAMPs), which occur during type 2 diabetes. Their recognition leads to several functions of innate immune cells, including phagocytosis [23], cytokine production [24], and expression of co-stimulatory molecules [25] and adhesion molecules [26, 27].
Toll-like receptor 2 (TLR2) plays significant roles in the induction of innate immune cells through a MYD88-dependent pathway [28, 29]. Interestingly, there is some evidence that confirms the suppressive roles of TLR2 and ligand interactions on immune responses [30]. Therefore, TLR2 may participate in the development and pathogenesis of immune-related diseases, including type 2 diabetes. Accordingly, the altered expression and functions of TLR2 and its intracellular molecular signaling may be associated with the mechanisms that result in the progression and pathogenesis of type 2 diabetes and its related complications.
Toll-Like Receptor 2
TLR2 (TIL4, CD282) was identified and characterized in 1998 [31]. It is an innate immune cell receptor that recognizes several pathogen-associated molecular patterns (PAMPs) and DAMPs and subsequently activates MYD88-dependent intracellular signaling [29]. Chromosome 4 (4p32) is the location of the TLR2 gene. This molecule is a type I transmembrane protein that has a similar structure to other TLRs, consisting of the following domains from N-terminal to C-terminal: extracellular leucine-rich repeat (LRR) domains; a transmembrane domain; and a toll/interleukin-1 receptor (TIR) domain. TLR2 is expressed in several immune cell types, including macrophages and dendritic cells, and non-immune cell types, including endothelial cells, epithelial cell lines and hepatocytes [32, 33].
TLR2 recognizes its ligands in both homodimer and heterodimer (with TLR1 or 6) forms [34]. In its homodimer form, it recognizes lipopolysacharide (LPS), porins, lipoprotein, lipoteichoic acid, bacterial peptidoglycan, viral hemagglutinin and glycoproteins. In its TLR2/1 heterodimer form, it recognizes bacterial triacylated lipopeptides and synthetic triacylated lipopeptide (Pam3CSK4) [35], and in its TLR2/6 heterodimer, it recognizes bacterial diacylated lipopeptides and lipoteichoic acid [36]. As mentioned previously, TLR2 also recognizes some DAMPs as endogenous ligands, including human glycosaminoglycan hyaluronan [37], β-defensin-3, heat shock proteins and high mobility group box 1 protein, some of which are released during inflammatory diseases like type 2 diabetes [30, 38–40].
TLR2–ligand interactions lead to the activation of MAPK and MYD88-dependent signaling pathways (Fig. 2) [41, 42]. Although MYD88-dependent signaling pathway activation results in the phosphorylation and activation of pro-inflammatory transcription factors, such as IRF3, IRF7, AP-1 and NF-kB, it also induces the PI3K/AKT pathway, which leads to upregulation of IL-10, and activates SOCS proteins [43]. IL-10 is a major anti-inflammatory cytokine [44]. SOCS proteins also suppress MAPK and JAK–STAT signaling pathways [45]. Therefore, it appears that TLR2-ligand interaction leads to either activation or suppression of immune responses [46, 47].

Following TLR2-ligand interaction a signaling pathway is started using MYD88 as an adaptor molecule. The signaling pathway leads to the activation of several transcription factors, including NF-kB, MAPK and AP-1, and subsequently cell activation. Adapted from Bagheri et al. [29]
The principal mechanisms causing the activation or suppression of immune responses by TLR2 are unclear, but it has been hypothesized that the TLR2 ligand concentration may be the determining factor.
Type 2 Diabetes And TLR2
DAMPs are endogenous molecules that are produced and released by several cell systems during inflammation or infection [48, 49]. They can also be released during type 2 diabetes [50]. Both can be recognized by TLR2, leading to the either activation or suppression of immune cells. It has been documented that inflammation is a major cause of pancreatic beta cell dysfunction in type 2 diabetes [51, 52]. Therefore, the inflammatory effects of TLR2-ligand interaction may be an important factor in type 2 diabetes progression. Nackiewicz et al. showed that interaction between TLR2/6 and its related ligands results in the activation of macrophages and the production of IL-1 and IL-6 as pro-inflammatory cytokines that contribute to islet inflammation [51].
Several studies confirmed the important roles played by reactive oxygen species (ROS) in the pathogenesis of type 2 diabetes. Interestingly, activation of TLR2 by zymosan leads to ROS production by neutrophils in a manner dependent on TLR2 NADPH oxidase but not dependent on MAPK [53]. Hyperglycemia and chronic periodontitis also lead to upregulation of TLR2 in the gingival tissue of type 2 diabetes patients [54]. Interestingly, it has been demonstrated that insulin suppresses the expression of TLR2 at the mRNA level, possibly via downregulation of PU.1 [55]. Kuzmicki et al. revealed that the gestational diabetes patients had significantly higher TLR2 expression than pregnant women with normal glucose tolerance [56]. Interestingly, TLR2 was found to be upregulated in women who exhibited normal glucose tolerance but later developed gestational diabetes when compared to the women who remained normoglycemic [56]. Ahmad et al. demonstrated that the expression levels of TLR2 were upregulated in obese individuals [57]. Additionally, they showed that obese type 2 diabetes patients had higher expressions of TLR2 in comparison to obese patients without type 2 diabetes [57]. The phagocytic cells of type 2 diabetes patients have also upregulated TLR2 [58].
Another study also identified that TLR2 not only participates in the development of type 2 diabetes but is also involved in the pathogenesis of related vascular complications [59]. Moreover, via upregulation of IL-6 and osteopontin, TLR2 causes impaired insulin-mediated brain activities, which are an early step in the development toward type 2 diabetes [60]. Duarte et al. revealed higher mRNA levels of TLR2 in gingival biopsies from type 2 diabetes patients with chronic periodontitis in comparison to periodontally healthy patients [61]. Thus, it appears that the upregulation of TLR2 is a marker of type 2 diabetes rather than a marker of periodontitis. Rojo-Botello et al. also confirmed this result [62]. Another study identified that free fatty acids and high glucose levels upregulate the expression of TLR2 and TLR6, which resulted in increased activity of monocytes and increased production of superoxides, which are released in an NF-kB-dependent manner [63]. Ehses et al. also reported that a high-fat diet was unable to induce insulin resistance and beta cell dysfunction in TLR2-deficient mice [64]. Free fatty acids also play important roles in the induction of inflammation in pancreatic beta cells via TLR2 [65]. Interestingly, another study showed that not only TLR2 has been more highly expressed on the immune cells of type 2 diabetes patients than on those of healthy subjects, but also the levels of TLR2 ligands, including hyaluronan, HSP60, HSP70, HMGB1 and endotoxin, were higher [66]. TLR2 inhibition using a TLR2 antisense oligonucleotide (ASON) leads to recovery of insulin sensitivity and signaling in muscle and white adipose tissue of mice that were fed a high-fat diet [67]. It has also been documented that oxidized LDL, which is produced during type 2 diabetes, induced expression of TLR2 in macrophages [68]. The expression of TLR2 on the monocytes of obese women is also higher [68].
TLR2 also recognizes PAMPs such as exogenous microbial ligands, so it may be hypothesized that microbial infections could be important factors in the development of type 2 diabetes and may also participate in the pathogenesis of the disease. Interestingly, a study by Ajuwon et al. revealed that peptidoglycan derived from Staphylococcus aureus resulted in elevated TLR2 expression on the 3 T3-L1 adipocytes cell lines [69]. Chen et al. identified that treatments with agents that improve glycemic control are associated with decreased expressions of TLR2 and its related intracellular signaling molecules [70]. Previous studies demonstrated that decreased methylation is associated with higher expression of the genes [71]. Accordingly, CpGs methylation in the promoter of the TLR2 gene was significantly decreased in type 2 diabetes patients in comparison to the levels for the controls [72].
As mentioned in the previous section, TLR2 is also able to suppress immune cells through unknown mechanisms. A few studies demonstrated that TLR2 expression decreased during type 2 diabetes. For instance, a study by Cortez-Espinosa et al. showed that the percentage of TLR2-positive monocytes decreased in type 2 diabetes patients with poor glycemic control when compared to patients with appropriate glycemic control [73]. Another study revealed that although type 2 diabetes patients had higher serum levels of IL-6, the mRNA levels of TLR2 were lower than in healthy subjects [74]. Interestingly, their results, which were obtained under in vitro conditions, demonstrate that high glucose levels lead to reduced expression of TLR2 [74]. High doses of insulin (100 IU/ml), such as those seen in cases of type 2 diabetes, also increased IL-6 and decreased TLR2 expression [74]. In an in vitro study, it was shown that TLR4 but not TLR2 interaction with corresponded ligands leads to pancreatic beta cell apoptosis [75]. S6K1 plays key roles in driving insulin resistance and the induction of type 2 diabetes [76]. Kim et al. found that upregulation of S6K1 causes a significant reduction in NF-kB and AP-1 activities, which are induced by TLR2–ligand interactions [77].
Since more than 90 % of studies reported that TLR2 is positively associated with type 2 diabetes and its complications, it seems that the inflammatory effects of TLR2 during the condition are predominant. Collectively, it appears that TLR2 plays remarkable roles in the development of type 2 diabetes and related complications. Therapeutic approaches involving modulation of the expression of TLR2 and related signaling molecules could be considered as novel approaches for the treatment of type 2 diabetes.
There have not been many studies on the variation of the TLR2 gene in type 2 diabetes are rare. Liu et al. reported on the low frequency of TLR2 Arg677Trp and Arg753Gln polymorphisms in type 2 diabetes patients in the Chinese Han population [78]. Another study showed that the TLR2 R753Q polymorphism was not associated with type 2 diabetes in Mexican population [79]. Further studies need to be done to find the relationship between genetic variations and the diseases.
Other TLRs like TLR3 and 4 also play key roles in the induction of this disease. We discussed the roles played by TLR3 in its development in our previous review [80]. Our research confirmed that TLR4 can also participate in its pathogenesis [81]. TLR3 and TLR4 use another signaling pathway, the TRIF-dependent pathway, which may be important in the development of type 2 diabetes. Thus, it seems that TLR2 is not a unique innate immunity receptor involved in the development of the disease.
Conclusion
TLR2 plays crucial roles in the initiation and pathogenesis of type 2 diabetes and related complications. Downregulation of TLR2 can be considered as a novel approach for the treatment of these conditions. Of particular interest in such research are these points:
TLR2 is upregulated in several tissues that are affected by adult-onset type 2 diabetes and the gestational version of the disease.
-
1.
TLR2 induces the production of several molecules including ROS and pro-inflammatory cytokines, which contribute to the worsening type 2 diabetes and related complications.
-
2.
The expression of TLR2 positively correlates with elevated serum levels of free fatty acids and glucose as well as obesity.
-
3.
Several exogenous and endogenous ligands induce the activation of immune cells and the dysfunction of pancreatic beta cells in a TLR2-initiated pathway-dependent manner.
-
4.
Different genetic variations and methylation may play key roles in the upregulation of TLR2 in type 2 diabetes patients.
-
5.
PAMPs participate in the activation of TLR2-initiated pathways, so it may be hypothesized that infection can be considered a crucial candidate for the development of type 2 diabetes and related complications.
-
6.
Upregulation of insulin is an important mechanism that suppresses the expression of TLR2.
Abbreviations
AP-1, activator protein 1; CRP, C-reactive protein; DAMPs, damage-associated molecular patterns; IRF, interferon regulatory factor; JAK-STAT, Janus kinase-signal transducer and activator of transcription; LDL, low-density lipoprotein; LRRs, leucine-rich repeats; MAPK, mitogen-activated protein kinase; MYD88, myeloid differentiation primary response; NADPH, nicotinamide adenine dinucleotide phosphate; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; NIDDM, non-insulin-dependent diabetes mellitus; PAMP, pathogen-associated molecular pattern; PBMC, peripheral blood mononuclear cell; ROS, reactive oxygen species; SOCS, suppressor of cytokine signaling proteins; TLR, toll-like receptor; TIR, toll/interleukin-1 receptor
References
Steyn NP, Lambert EV, Tabana H. Nutrition interventions for the prevention of type 2 diabetes. Proc Nutr Soc. 2008;10:1–16.
Joost HG. Diabetes and cancer: Epidemiology and potential mechanisms. Diab Vasc Dis Res. 2014;11:390–4.
Debresser J, Reijmer YD, Vandenberg E, Breedijk MA, Kappelle LJ, Viergever MA, et al. Microvascular determinants of cognitive decline and brain volume change in elderly patients with type 2 diabetes. Dement Geriat Cogn Dis. 2010;30:381–6.
Hartemann A, Attal N, Bouhassira D, Dumont I, Gin H, Jeanne S, et al. Diabetology WGotDFftF-sSo. Painful diabetic neuropathy: diagnosis and management. Diabet Metab. 2011;37:377–88.
Lemley KV. Diabetes and chronic kidney disease: lessons from the Pima Indians. Pediat Nephr. 2008;23:1933–40.
Noda K, Nakao S, Zandi S, Sun D, Hayes K, Hafezi-Moghadam A. Retinopathy in a novel model of metabolic syndrome and type 2 diabetes: new insight on the inflammatory paradigm. FASEB J. 2014;28:2038–46.
Gholamhossein Y, Behrouz H, Asghar Z. Diabetic retinopathy risk factors: plasma erythropoietin as a risk factor for proliferative diabetic retinopathy. Korean J Ophthalmol. 2014;28:373–8.
Arababadi MK, Nosratabadi R, Hassanshahi G, Yaghini N, Pooladvand V, Shamsizadeh A, et al. Nephropathic complication of type-2 diabetes is following pattern of autoimmune diseases? Diabetes Res Clin Pract. 2009;87:33–7.
Lee JH, Choi Y, Jun C, Hong YS, Cho HB, Kim JE, et al. Neurocognitive changes and their neural correlates in patients with type 2 diabetes mellitus. Endocrinol Metab. 2014;29:112–21.
Longo PL, Artese HP, Rabelo MS, Kawamoto D, Foz AM, Romito GA, et al. Serum levels of inflammatory markers in type 2 diabetes patients with chronic periodontitis. J Appl Oral Sci. 2014;22:103–8.
Southerland JH, Taylor GW, Moss K, Beck JD, Offenbacher S. Commonality in chronic inflammatory diseases: periodontitis, diabetes, and coronary artery disease. Periodontol. 2006;2000(40):130–43.
Perano S, Rayner CK, Couper J, Martin J, Horowitz M. Cystic fibrosis related diabetes—a new perspective on the optimal management of postprandial glycemia. J Diabetes Complications. 2014;28:904–11.
Castrotorres Y, Katholi RE. Novel treatment approaches in hypertensive type 2 diabetic patients. World J Diabetes. 2014;5:536–45.
Calvin D, Watley SR. Diabetes and Hearing Loss Among Underserved Populations. Nurs Clin N Am. 2015;50:449–56.
Nathanson D, Nystrom T. Hypoglycemic pharmacological treatment of type 2 diabetes: Targeting the endothelium. Mol Cell Endocrinol. 2008;21:21.
Jin C, Henao-Mejia J, Flavell RA. Innate immune receptors: key regulators of metabolic disease progression. Cell Metab. 2013;17:873–82.
Cruz M, Maldonado-Bernal C, Mondragon-Gonzalez R, Sanchez-Barrera R, Wacher NH, Carvajal-Sandoval G, et al. Glycine treatment decreases proinflammatory cytokines and increases interferon-gamma in patients with type 2 diabetes. J Endocrinol Invest. 2008;31:694–9.
Giulietti A, van Etten E, Overbergh L, Stoffels K, Bouillon R, Mathieu C. Monocytes from type 2 diabetic patients have a pro-inflammatory profile. 1,25-Dihydroxyvitamin D(3) works as anti-inflammatory. Diabetes Res Clin Pract. 2007;77:47–57.
Pickup JC, Chusney GD, Thomas SM, Burt D. Plasma interleukin-6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci. 2000;67:291–300.
Skopinski P, Rogala E, Duda-Krol B, Lipinska A, Sommer E, Chorostowska-Wynimko J, et al. Increased interleukin-18 content and angiogenic activity of sera from diabetic (Type 2) patients with background retinopathy. J Diabetes Complications. 2005;19:335–8.
Müller S, Martin S, Koenig W, Hanifi-Moghaddam P, Rathmann W, Haastert B, Giani, G, Illig, T, Thorand, B. and Kolb, H. Impaired glucose tolerance is associated with increased serum concentrations of interleukin 6 and co-regulated acute-phase proteins but not TNF-α or its receptors. Diabetologia. 2002;45:805–12.
Dipenta JM, Green-Johnson JM, Murphy RJ. Type 2 diabetes mellitus, resistance training, and innate immunity: is there a common link? Appl Physiol Nutr Metab. 2007;32:1025–35.
Hirayama T, Tamaki Y, Takakubo Y, Iwazaki K, Sasaki K, Ogino T, Goodman, S.B., Konttinen, Y.T and Takagi, M. Toll-like receptors and their adaptors are regulated in macrophages after phagocytosis of lipopolysaccharide-coated titanium particles. J Orthop Res. 2011;29:984–92.
Bhatelia K, Singh K, Singh R. TLRs: Linking inflammation and breast cancer. Cell Signal. 2014;26:2350–7.
Sioud M, Fløisand Y. TLR agonists induce the differentiation of human bone marrow CD34+ progenitors into CD11c + CD80/86+ DC capable of inducing a Th1-type response. Eur J Immunol. 2007;37:2834–46.
Takebayashi K, Hokari R, Kurihara C, Okada Y, Okudaira K, Matsunaga H, Komoto, S, Watanabe, C, Kawaguchi, A. and Nagao, S. Oral Tolerance Induced by Enterobacteria Altered the Process of Lymphocyte Recruitment to Intestinal Microvessels: Roles of Endothelial Cell Adhesion Molecules, TGF-beta and Negative Regulators of TLR Signaling. Microcirculation. 2009;16:251–64.
Nguyen-Pham TN, Lim MS, Nguyen TA, Lee YK, Jin CJ, Lee HJ, and Le, J.J. Type I and II interferons enhance dendritic cell maturation and migration capacity by regulating CD38 and CD74 that have synergistic effects with TLR agonists. Cell Mol Immunol. 2011;8:341–7.
Rusz A, Pilatz A, Wagenlehner F, Linn T, Diemer T, Schuppe H, Lohmeyer, J, Hossain, H. and Weidner, W. Influence of urogenital infections and inflammation on semen quality and male fertility. World J Urol. 2012;30:23–30.
Bagheri V, Askari A, Arababadi MK, Kennedy D. Can Toll-Like Receptor (TLR) 2 be considered as a new target for immunotherapy against hepatitis B infection? Hum Immunol. 2014;75:549–54.
Oliveira-Nascimento L, Massari P, Wetzler LM. The Role of TLR2 in Infection and Immunity. Front Immunol. 2012;3:79.
Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci. 1998;95:588–93.
Preiss S, Thompson A, Chen X, Rodgers S, Markovska V, Desmond P, Visvanathan, K, Li, K, Locarnini, S. and Revill, P. Characterization of the innate immune signalling pathways in hepatocyte cell lines. J Viral Hepat. 2008;15:888–900.
Flo TH, Halaas Ĝ, Torp S, Ryan L, Lien E, Dybdahl B, et al. Differential expression of Toll-like receptor 2 in human cells. J Leukocyte Biol. 2001;69:474–781.
Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809.
Tsolmongyn B, Koide N, Jambalganiin U, Odkhuu E, Naiki Y, Komatsu T, Yoshida, T and Yokochi, T. A toll-like receptor 2 ligand, Pam3CSK4, augments interferon-gamma-induced nitric oxide production via a physical association between MyD88 and IFN-gamma receptor in vascular endothelial cells. Immunology. 2013;140:352–61.
Leoni V, Gianni T, Salvioli S, Campadelli-Fiume G. Herpes simplex virus glycoproteins gH/gL and gB bind Toll-like receptor 2, and soluble gH/gL is sufficient to activate NF-kappaB. J Virol. 2012;86:6555–62.
Abe T, Fukuhara T, Wen X, Ninomiya A, Moriishi K, Maehara Y, et al. CD44 participates in IP-10 induction in cells in which hepatitis C virus RNA is replicating, through an interaction with Toll-like receptor 2 and hyaluronan. J Virol. 2012;86:6159–70.
Ertugrul A, Dikilitas A, Sahin H, Alpaslan N, Bozoglan A, Tekin Y. Gingival crevicular fluid levels of human beta-defensins 1 and 3 in subjects with periodontitis and/or type 2 diabetes mellitus: a cross-sectional study. J Period Res. 2013;48:475–82.
Morteza A, Nakhjavani M, Larry M, Nargesi AA, Esteghamati A. Heat shock protein 70 and albuminuria in patients with type 2 diabetes: a matched case control study. Cell Stress Chaperones. 2013;18:815–9.
Zhao D, Wang Y, Tang K, Xu Y. Increased serum HMGB1 related with HbA1c in coronary artery disease with type 2 diabetes mellitus. Int J Cardiol. 2013;168:1559–60.
Liang B, Chen R, Wang T, Cao L, Liu Y, Yin F, et al. Myeloid differentiation factor 88 promotes growth and metastasis of human hepatocellular carcinoma. Clin Cancer Res. 2013;19:2905–016.
Maeda S. NF-kB, JNK, and TLR signaling pathways in hepatocarcinogenesis. Gastroent Res Pract. 2010;2010:367694.
Gerber PA, Buhren BA, Steinhoff M, Homey B. Rosacea: The cytokine and chemokine network. J Investig Dermatol Symp Proc. 2011;15:40–7.
Hakimi H, Zare-Bidaki M, Zainodini N, Assar S, Arababadi MK. Significant Roles Played by IL-10 in Chlamydia Infections. Inflammation. 2014;37:818–23.
Ryu JH, Yoo JY, Kim MJ, Hwang SG, Ahn KC, Ryu JC, Choi, M.K, Joo, J.H, Kim, C.H, Lee, S.N, Lee, W.J, Kim, J., Shin, D.M, Kweon, M.N, Bae, Y.S. and Yoon, J.H. Distinct TLR-mediated pathways regulate house dust mite-induced allergic disease in the upper and lower airways. J Allergy Clin Immunol. 2013;131:549–61.
Kohanawa M, Zhao S, Ozaki M, Haga S, Nan G, Kuge Y, Y. and Tamaki, N. Contribution of Toll-Like Receptor 2 to the Innate Response against Staphylococcus aureus Infection in Mice. Plos One. 2013;8, e74287.
Szomolanyi-Tsuda E, Liang X, Welsh RM, Kurt-Jones EA, Finberg RW. Role for TLR2 in NK cell-mediated control of murine cytomegalovirus in vivo. J Virol. 2006;80:4286–91.
Samy RP, Lim LH. DAMPs and influenza virus infection in ageing. Age Res Rev. 2015;24:83–97.
Shin JJ, Lee EK, Park TJ, Kim W. Damage-associated molecular patterns and their pathological relevance in diabetes mellitus. Age Res Rev. 2015;24:66–76.
Singh, K., Agrawal, N.K., Gupta, S.K., Mohan, G., Chaturvedi, S. and Singh, K. Increased expression of endosomal members of toll-like receptor family abrogates wound healing in patients with type 2 diabetes mellitus. Int. Wound. J. (2015) DOI: 10.1111/iwj.12411.
Nackiewicz D, Dan M, He W, Kim R, Salmi A, Rütti S, et al. TLR2/6 and TLR4-activated macrophages contribute to islet inflammation and impair beta cell insulin gene expression via IL-1 and IL-6. Diabetologia. 2014;57(8):1–10.
Yoshida H, Watanabe W, Oomagari H, Tsuruta E, Shida M, Kurokawa M. Citrus flavonoid naringenin inhibits TLR2 expression in adipocytes. J Nutrit Biochem. 2013;24:1276–84.
Fagundes-Netto F, Anjos P, Volpe C, Nogueira-Machado J. The production of reactive oxygen species in TLR-stimulated granulocytes is not enhanced by hyperglycemia in diabetes. Int Immunopharm. 2013;17:924–9.
Promsudthi A, Poomsawat S, Limsricharoen W. The role of Toll-like receptor 2 and 4 in gingival tissues of chronic periodontitis subjects with type 2 diabetes. J Periodontal Res. 2014;49:346–54.
Ghanim H, Mohanty P, Deopurkar R, Sia CL, Korzeniewski K, Abuaysheh S, Chaudhuri, A. and Dandona, P. Acute modulation of toll-like receptors by insulin. Diabetes Care. 2008;31:1827–31.
Kuzmicki M, Telejko B, Wawrusiewicz-Kurylonek N, Lipinska D, Pliszka J, Wilk J, Zielinska, A, Skibicka, J, Szamatowicz, J. and Kretowski, A. The expression of genes involved in NF-kB activation in peripheral blood mononuclear cells of patients with gestational diabetes. Eur J Endocrin. 2013;168:419–27.
Ahmad R, Al-Mass A, Atizado V, Al-Hubail A, Al-Ghimlas F, Al-Arouj M, et al. Elevated expression of the Toll-like receptors 2 and 4 in obese individuals: its significance for obesity-induced inflammation. J Inflamm (Lond). 2012;9:48.
Morris J, Williams N, Rush C, Govan B, Sangla K, Norton R, et al. Burkholderia pseudomallei triggers altered inflammatory profiles in a whole-blood model of Type 2 diabetes-melioidosis comorbidity. Infect Immun. 2012;80:2089–99.
Jialal I, Kaur H. The role of toll-like receptors in diabetes-induced inflammation: implications for vascular complications. Curr Diab Rep. 2012;12:172–9.
Sartorius T, Lutz SZ, Hoene M, Waak J, Peter A, Weigert C, et al. Toll-like receptors 2 and 4 impair insulin-mediated brain activity by interleukin-6 and osteopontin and alter sleep architecture. FASEB J. 2012;26:1799–809.
Duarte PM, Szeremeske-Miranda T, Lima JA, Dias-Gonçalves TE, Santos VR, Bastos MF, et al. Expression of immune-inflammatory markers in sites of chronic periodontitis in patients with type 2 diabetes. J Periodontol. 2012;83:426–34.
Rojo-Botello N, García-Hernández A, Moreno-Fierros L. Expression of toll-like receptors 2, 4 and 9 is increased in gingival tissue from patients with type 2 diabetes and chronic periodontitis. J Periodont Res. 2012;47:62–73.
Dasu MR, Jialal I. Free fatty acids in the presence of high glucose amplify monocyte inflammation via Toll-like receptors. Am J Physio-Endocrinol Metab. 2011;300:E145–54.
Ehses J, Meier D, Wueest S, Rytka J, Boller S, Wielinga P, Schraenen A, Lemaire K, Debray S, Van Lommel L. Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high-fat diet. Diabetol. 2010;53:1795–806.
Boni-Schnetzler M, Boller S, Debray S, Bouzakri K, Meier DT, Prazak R, et al. Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology. 2009;150:5218–29.
Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care. 2010;33:861–8.
Caricilli AM, Nascimento PH, Pauli JR, Tsukumo DM, Velloso LA, Carvalheira JB, et al. Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. J Endocrinol. 2008;199:399–406.
Holvoet P. Relations between metabolic syndrome, oxidative stress and inflammation and cardiovascular disease. Verhandelingen-Koninklijke Acad Geneeskunde Belgie. 2007;70:193–219.
Ajuwon KM, Banz W, Winters TA. Stimulation with Peptidoglycan induces interleukin 6 and TLR2 expression and a concomitant downregulation of expression of adiponectin receptors 1 and 2 in 3 T3-L1 adipocytes. J Inflam. 2009;6:8.
Chen L, Klein TS, Leung P. Effects of combining linagliptin treatment with bi-38335, a novel sglt2 inhibitor, on pancreatic islet function and inflammation in db/db mice. Current Mol Med. 2012;12:995–1004.
Rozenberg JM, Tesfu DB, Musunuri S, Taylor JM, Mack CP. DNA Methylation of a GC Repressor Element in the Smooth Muscle Myosin Heavy Chain Promoter Facilitates Binding of the Notch-Associated Transcription Factor, Recombination Signal Binding Protein for Immunoglobulin k J Region/CSL1. Arterio Thromb Vasc Biol. 2014;34:2624–31.
Remely M, Aumueller E, Jahn D, Hippe B, Brath H, Haslberger A. Microbiota and epigenetic regulation of inflammatory mediators in type 2 diabetes and obesity. Benefic Microb. 2014;5:33–43.
Cortez-Espinosa N, García-Hernández MH, Reynaga-Hernández E, Cortés-García J, Corral-Fernández NE, Rodríguez-Rivera JG, et al. Abnormal expression and function of Dectin-1 receptor in type 2 diabetes mellitus patients with poor glycemic control (HbA1c > 8 %). Metabolism. 2012;61:1538–46.
Bernal-Lopez M, Llorente-Cortes V, Calleja F, Lopez-Carmona D, Mayas M, Gomez-Huelgas R, et al. Effect of different degrees of impaired glucose metabolism on the expression of inflammatory markers in monocytes of patients with atherosclerosis. Acta Diabetol. 2013;50:553–62.
Lee SM, Choi SE, Lee JH, Lee JJ, Jung IR, Lee SJ, et al. Involvement of the TLR4 (Toll-like receptor4) signaling pathway in palmitate-induced INS-1 beta cell death. Mol Cell Biochem. 2011;354:207–17.
Younis H, Hirakawa B, Scott W, Tran P, Bhat G, Affolter T, et al. Antisense inhibition of S6 kinase 1 produces improved glucose tolerance and is well tolerated for 4 weeks of treatment in rats. Pharmacology. 2010;87:11–23.
Kim SY, Baik KH, Baek KH, Chah KH, Kim KA, Moon G, Jung, E. Kim, S.T. Shim, J.H. and Greenblatt, M.B. S6K1 Negatively Regulates TAK1 Activity in the Toll-Like Receptor Signaling Pathway. Mol Cell Biol. 2014;34:510–21.
Liu F, Lu W, Qian Q, Qi W, Hu J, Feng B. Frequency of TLR 2, 4, and 9 gene polymorphisms in Chinese population and their susceptibility to type 2 diabetes and coronary artery disease. BioMed Res Int. 2012;2012:373945.
Maldonado-Bernal C, Trejo-Dela OA, Sánchez-Contreras M, Wacher-Rodarte N, Torres J, Cruz M. Low frequency of Toll-like receptors 2 and 4 gene polymorphisms in Mexican patients and their association with Type 2 diabetes. Int J Immunogen. 2011;38:519–23.
Sepehri Z, Kiani Z, Javadian F, Akbar NA, Kohan F, Sepehrikia S, Javan SS, Aali H, Daneshvar H, Kennedy D. TLR3 and its roles in the pathogenesis of type 2 diabetes. Cell Mol Biol. 2015;61:46. Noisy-le-Grand, France.
Sepehri Z, Kiani Z, Nasiri A, Mashhadi M, Javadian F, Haghighi A, Kohan F, Bahari A, Sargazi A. Human Toll-like receptor 4 gene expression of PBMCs in diabetes mellitus type 2 patients. Cell Mol Biol. 2014;61:92–5. Noisy-le-Grand, France.
Acknowledgments
This project was funded through the Zabol University of Medical Sciences.
Funding
This study was supported by Zabol University of Medical Sciences.
Authors’ contributions
ZS has collected the data from database. ZK has written the article, especially the conclusion. AAN has prepared the manuscript for submission, submitted the manuscript and help for preparing revised versions. FK has edited the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sepehri, Z., Kiani, Z., Nasiri, A.A. et al. Toll-like receptor 2 and type 2 diabetes. Cell Mol Biol Lett 21, 2 (2016). https://doi.org/10.1186/s11658-016-0002-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11658-016-0002-4