
Abstract
Background
Adenosine is a purinergic signaling molecule with a wide range of physiological functions including anti- and pronociceptive properties. Adenosine receptors are expressed in the trigeminovascular system, and adenosine receptor antagonist, caffeine, relieves migraine headache. We performed a systematic review of the literature of preclinical data addressing the role of adenosine in migraine pathophysiology.
Methods
PubMed and EMBASE were searched for pre-clinical studies on the role of adenosine in migraine pathophysiology on September 5th, 2021.
Results
A total of 2510 studies were screened by title and abstract. Of these, thirteen pre-clinical studies evaluating adenosine, adenosine A1, A2A and A3 receptors were included.
These studies showed that adenosine signaling pathway is involved in controlling vascular tone. Furthermore, electrical stimulation of the trigeminal ganglion modulates the expression of adenosine A1 and A2A receptors in the trigeminal ganglion and trigeminal nucleus caudalis implicating adenosine signaling pathway in pain transmission.
Conclusion
Preclinical studies showed that adenosine has a dual effect on vasodilation and trigeminal pain pathway due to different receptor activation, suggesting a possible role of adenosine in migraine pathophysiology. Studies investigating pharmacological characteristics of subtypes of adenosine receptors are needed to further elucidate their role as a potential target for migraine treatment.
Similar content being viewed by others

Introduction
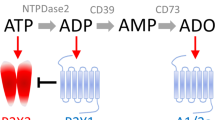
Adenosine, a vasoactive amine produced by the hydrolysis of adenosine monophosphate (AMP) or S-adenosylhomocysteine (SAH) [43], is involved in numerous physiological processes such as metabolism, inflammation, respiration and pain [39]. Adenosine binds to four G-protein coupled receptors (GPCR), A1, A2A, A2B and A3, with a unique profile of tissue distribution, signaling pathways and function (Table 1 & Fig. 1) [20, 21]. Adenosine receptors activate the mitogen-activated protein kinase (MAPK), leading to survival, cell growth, and differentiation [20], and modulate the activity of adenylate cyclase [11, 20], the enzyme that regulates intracellular concentration of cyclic adenosine monophosphate (cAMP) [38]. Adenosine A1 and A3 receptors are coupled to a Gαi subunit that downregulates cAMP by inhibiting adenylate cyclase [20, 38], while A2A and A2B receptors are coupled to a Gαs subunit which stimulates adenylate cyclase to upregulate cAMP [20, 38]. A1 and A3 receptors are considered to have anti-nociceptive effects, whereas, activation of A2A and A2B receptors induces nociception [11]. Adenosine A1 receptors are expressed at the trigeminovascular system (TVS), including the trigeminal ganglion (TG) and trigeminal nucleus caudalis (TNC) [6, 32], which is considered to be the anatomical and physiological substrate of migraine pain [2]. Stimulation of this receptor causes inhibition of the TVS by reducing neuronal firing from the trigeminal nucleus and decreasing the release of calcitonin gene-related peptide (CGRP) [6, 13]. A2A and A2B receptors are located in vascular smooth muscle cells [20, 38] and in pre- and postsynaptic nerve terminals [38], and stimulation of these receptors causes dural vasodilation [16], leading to stimulation of the TVS. Collectively, adenosine signaling pathways are complex and might be involved in headache and migraine pathophysiology.

Adenosine signaling pathway. Adenosine binds to G-protein coupled receptors (GPCR) resulting in either activation (adenosine A2A and A2B receptors) or inactivation (adenosine A1 and A3 receptors) of adenylyl cyclase (AC). Activation of AC increases the formation of cyclic adenosine monophosphate (cAMP) which binds to protein kinase A (PKA). Active PKA then phosphorylates and thereby modulates cellular responses. ATP: adenosine triphosphate
Here, we systematically review preclinical studies on the involvement of adenosine in trigeminal pain pathway, to make the case that adenosine signaling pathways may play a role in migraine and discuss adenosine receptors as potential target for future treatment of migraine (Table 2).
Method
Data source
We conducted two searches on PubMed and Embase on September 5th, 2021. Firstly, we searched “(“adenosine”[MeSH Terms] OR “adenosine”[All Fields] OR “adenosin”[All Fields] OR “adenosine s”[All Fields] OR “adenosines”[All Fields]) AND (“migrain”[All Fields] OR “migraine disorders”[MeSH Terms] OR (“migraine”[All Fields] AND “disorders”[All Fields]) OR “migraine disorders”[All Fields] OR “migraine”[All Fields] OR “migraines”[All Fields] OR “migraine s”[All Fields] OR “migraineous”[All Fields] OR “migrainers”[All Fields] OR “migrainous”[All Fields])”. Secondly, we searched for “(“adenosine”[MeSH Terms] OR “adenosine”[All Fields] OR “adenosin”[All Fields] OR “adenosine s”[All Fields] OR “adenosines”[All Fields]) AND (“headache”[MeSH Terms] OR “headache”[All Fields] OR “headaches”[All Fields] OR “headache s”[All Fields])”. Both searches were restricted to English language.
Selection criteria and study inclusion
Studies were restricted to pre-clinical or clinical studies that investigated adenosine, adenosine agonist, adenosine antagonist, adenosine deaminase, adenosine deaminase inhibitor and adenosine reuptake inhibitors in headache and migraine pathophysiology. We excluded reviews, meta-analysis, conference proceedings and case reports.
Two investigators (J.T. and L.K.) screened all studies by title and abstract, followed by full text screening to confirm eligibility. References of the included studies were screened to find studies that were missed by the search. For each included study, both investigators (J.T. and L.K.) extracted hypothesis or purpose of the study, method, sample size, main outcome, conclusion, and limitations. Any disagreements were resolved through discussion by the two investigators (J.T. and L.K.). If the conflict remained, a third investigator (M.M.K) made the final decision.
Results
The database search identified 3209 citations of which 701 were duplicates (Fig. 2). An additional two studies were included through a manual search of identified primary articles. A total of 2510 studies were screened by title and abstract and 44 were full text screened. Of these, 20 studies were included – 13 preclinical (Table 3) and 7 clinical studies. Data for clinical studies has been published recently [45].

Flow chart of search strategy
Narrative summaries
Arulmani et al. [1]. In pigs, intravenous infusion of adenosine A1 receptor agonist, GR79236, was compared to vehicle prior to capsaicin infusion. Total carotid blood flow, conductance, and plasma CGRP concentrations in jugular vein were assessed at baseline and after the infusions. GR79236 dose-dependently attenuated the capsaicin-induced carotid hemodynamic changes but not the CGRP release, compared to vehicle infusion.
Carruthers et al. [6]. In rats, adenosine agonists and antagonist (A1 receptor agonist GR79236X, adenosine A1 receptor antagonist, DPCPX, adenosine A2A agonist, CGS21680, and A3 receptor agonist, 2-CI-IB-MECA) were applied to cultured trigeminal neurons in combination with forskolin or vehicle to induce release CGRP. Immunocytochemical studies and Western analysis assessed whether these pharmacological agents could modulate the forskolin induced CGRP release. GR79236X concentration-dependently inhibited forskolin-stimulated CGRP release, while DPCPX abolished GR79236X´s effect. CGS21680 and 2-CI-IB-MECA were unable to attenuate forskolin-induced CGRP secretion.
Faraci et al. [9]. Intravenous infusion of adenosine was administered to anesthetized dogs. Blood flow was measured with labelled, radioactive microspheres. Adenosine decreased aortic pressure along with blood flow and vascular resistance in the dura. Adenosine infusion did not alter cerebral blood flow.
Goadsby et al. [13]. Intravenous infusion of adenosine A1 receptor agonists were administered in anesthetized dogs, following electrical stimulation of the superior sagittal sinus (SSS). Jugular vein blood samples were taken at baseline, immediately after the SSS stimulation and following the A1 agonist infusion, for detection of CGRP levels. Both A1 receptor agonists, GR79236 and GR190178, inhibited SSS-induced activation in TNC and CGRP release in cranial circulation, in a dose-dependent manner. Moreover, adenosine A1 receptor antagonist, DPCPX, was able to reverse GR79236's inhibitory effect on TNC activation.
Haanes et al. [15]. Adenosine was applied to pre-contracted middle meningeal artery (MMA) segments isolated from rats. RT-PCR was used to characterize the expression of purinergic receptor and myography to access the vascular effects. Notably, all purinergic receptor mRNAs were detected in the trigeminal ganglion and MMA. Adenosine caused dilation of MMA, which was reversed by SCH58261 (A2A receptor antagonist) and caffeine (adenosine receptor antagonist).
Haanes et al. [16]. Adenosine and caffeine were administered intravenously to six rats. Adenosine resulted in dural vasodilation and decrease in blood pressure. However, pre-treatment with caffeine inhibited adenosine´s effect. Caffeine caused an increase in blood pressure and a non-significant dilation of dural arteries. Secondly, intravenous infusions of different adenosine A2A receptor antagonists (JNJ compounds) were given following intravenous administration of adenosine A2A receptor agonist, CGS21680, or periarterial electrical stimulation (mode of CGRP-release), in rats. The closed cranial window was used to evaluate the antagonists´ effect on the CGS21680 and CGRP -induced dural dilation. CGS21680 caused vasodilation and decrease in arterial blood pressure. All A2A receptor antagonists blocked CGS21680-induced dural vasodilation with a more potent respond with A2A over A1 selectivity, while they did not affect electrical stimulated neurogenic vasodilation.
Hardebo et al. [17]. Adenosine, cAMP, ADP and ATP were applied in segments of middle cerebral artery and extracranial arteries of feline and humans. The dissected vessels were pre-constricted by prostaglandin F2a (PGF2a) or 5-hydroxytryptamine (5-HT). The tension was measured with force displacement transducers and recorded on a Grass polygraph. All adenine compounds dilated feline pial arteries, however the dilatory response was less pronounced when extracellular K+ concentration increased. Adenine compounds did not influence the diameter of human and feline extracranial arteries.
Honey et al. [19]. In rats, intravenous infusion of adenosine A1-receptor agonist, GR79236, was compared to saline infusion in models of neurogenic dural vasodilation. Vasodilation was induced by either electrical stimulation of perivascular trigeminal nerves or intravenous CGRP. Cranial window was used to evaluate the vascular responses. GR79236 inhibited electrically induced neurogenic vasodilation in a dose-dependent manner but had no effect on vasodilation caused by CGRP. Selective A1 receptor antagonist, DPCPX, inhibited the effect of GR79236 on electrically evoked vasodilation, compared to vehicle.
Jenkins et al. [22]. Adenosine deaminase was applied in cultured rat trigeminal neurons. Application of prostaglandin E2 (PGE2) led to CGRP release from the cultured cells. Adenosine deaminase did not alter baseline or PGE2-evoked CGRP levels.
Lindquist et al. [31]. Accumulation of adenosine following spreading depolarization (SD) was investigated in brain slices of mice and in vivo. Amperometric recordings from adenosine-sensitive enzyme-linked electrochemical were made in brain slices and applied in vivo. SD generated transient adenosine accumulation in vivo which could reliably report underlying metabolic status in brain slices.
Lu et al. [32]. Εlectrical stimulation of the trigeminal ganglion (ESTG) or sham operation was performed in rats to investigate its effect on CGRP, adenosine A1 receptor and adenosine A2A receptor expression. RT-qPCR and Western analysis was used for detection and quantification of the proteins. In the trigeminal nucleus caudalis (TNC) and ipsilateral trigeminal ganglion (TG), CGRP and A2A expression increased following ESTG, while A1 decreased. Interestingly, pretreatment with hinese medicine Tianshu capsule (TSC) decreased CGRP and A2A expression and increased A1 receptor expression.
Wei et al. [46]. In cats, the effect of topical application of adenosine and adenosine diphosphate was investigated before and following application of CGRP receptor antagonist, CGRP8-37. Cranial window was used to evaluate the vascular responses. CGRP8-37 was not able to reverse vasodilating effect of adenosine and adenosine diphosphate. Moreover, guanylate cyclase inhibitor, LY83583, had no effect on adenosine-induced vasodilation.
Yegutkin et al. [48]. The effects of adenine nucleotides were assessed in meninges of rats and cultured trigeminal cells following application of CGRP or placebo. Bioluminescent and fluorometric techniques were used to measure purine levels in trigeminal ganglion cells, and electrophysiology to record the nociceptive spikes in the meningeal trigeminal nerves, before and after pre-treatment with CGRP. CGRP decreased adenosine levels in cultured cells but not in the meninges, while adenosine was not able to activate nociceptive firing in the meningeal nerves. Moreover, basal levels of adenosine and AMP where higher compared to ATP and ADP in trigeminal cells.
Discussion
The main findings of the present systematic review are that adenosine receptors modulate pain transmission through the TVS. While A1 receptor has an inhibitory effect, stimulation of A2A receptor causes vasodilation and activation of trigeminal pain pathway.
In rats, electrical stimulation of the TG decreased adenosine A1 receptor expression and increased adenosine A2A receptor expression in ipsilateral TG and TNC [32]. The former is suggested to be involved in migraine attack initiation, while upregulation of adenosine A1 receptors or activation of this receptor might block migraine attacks [32]. In support, one study found that treatment with adenosine A1 receptor agonists, GR79236 and GR190178, inhibited TVS activation after electrical stimulation of the superior sagittal sinus in cats [13]. Upregulation of both adenosine A2A and CGRP receptors following electrical stimulation implies that when combined, the two receptors activate trigeminal pain transmission and cause migraine [32]. Overall, these findings implicate both adenosine A1 and A2A receptors in pain regulation and transmission during migraine [13, 32].
Three studies showed that adenosine caused prominent dilation of pre-contracted middle meningeal artery, dural and pial arteries in vitro [15,16,17]. Another study showed that adenosine caused dural vasodilation in dogs [9]. Pretreatment with caffeine or adenosine A2A receptor antagonist, SCH58261, was able to block adenosine-induced dilation in vitro [15, 16], suggesting that adenosine-induced vasodilation might be mainly dependent on adenosine A2A receptor [15].
Adenosine A1 receptor agonist, GR79236, inhibited electrically-induced vasodilation and capsaicin-induced hemodynamic changes in carotid artery [1, 19]. Pretreatment with DPCPX prevented the inhibition following GR79236, indicating that its inhibitory or vasoconstricting effect is mediated through adenosine A1 receptor [19]. The same agonist, GR79236, inhibited CGRP release induced by adenylate cyclase activator, forskolin [6] without any effect on CGRP-induced vasodilation in rats [19]. These data indicate that GR79236 inhibits CGRP release via a pre-junctional inhibition, and that adenosine A1 receptors are present on CGRP-positive neurons [6, 13, 19]. Together with its vasoconstricting ability, it is suggested that GR79236 and adenosine A1 receptors hold anti-migraine potential [1, 6, 19].
In contrast to GR79236, adenosine A2A receptor agonist, CGS21680, had no effect on forskolin-induced CGRP release [6]. However, CGS21680 caused dural vasodilation that was blocked by adenosine A2A receptor antagonists (JNJ-compounds) [16]. The study showed that the lower the selectivity for A2A receptor over A1 receptors, the higher the potential to attenuate the CGS21680 induced vasodilation. It was suggested that blocking both A1 and A2A receptors might be necessary to completely attenuate dural vasodilation [16].
Of note, 2-CI-IB-MECA, an adenosine A3 receptor agonist, had no effect on forskolin-induced CGRP secretion in rats [6]. The involvement of adenosine A3 receptor in migraine has not been further investigated, however, adenosine A3 receptor agonist exhibited anti-nociceptive properties in models of chronic pain in rats and mice [24].
While CGRP antagonist, CGRP(8–37), and guanylate cyclase inhibitor, LY83583, inhibited CGRP and nitroglycerine induced vasodilation, both compounds did not alter adenosine-induced vasodilation [46]. This finding demonstrates that adenosine induced dilation is not dependent on activation of CGRP receptors or an increase in cyclic guanosine monophosphate. Another study showed that pretreatment of trigeminal ganglion cells with CGRP is followed by decreased adenosine levels compared to baseline [48]. It is suggested that the finding might be a part of migraine sensitization but due to other contradicting findings (i.e., no change in nociceptive firing), further investigation on adenosine’s mechanisms was recommended [48].
Collectively, studies showed adenosine receptors expression in the trigeminal pain pathway and indicated that adenosine-induced pronociceptive effect is mediated through A2A receptor activation, whilst A1 receptor mediates antinociception. A1 receptor agonist, GR79236, inhibits activation of the TVS and vasodilation, while A2A receptor antagonist, SCH58261, attenuated adenosine-induced vasodilation [13, 15, 19], designating adenosine A1 and A2A receptors as possible targets in the treatment of migraine.
Limitations and future perspective
The major limitations of the studies included, were differences in methodological approaches including designs, subjects, substances, and sampling sources. Additionally, concentrations and types of adenosine A1 receptor agonists applied, differed across the studies [6, 13, 19]. Different CGRP releasing mechanisms were applied throughout the studies, potentially affecting the potency of adenosine receptor agonists and antagonists in modulating the CGRP release [1, 6, 13, 22].
Human studies are needed to elucidate the headache inducing effect of adenosine in patients with migraine. A specific focus on adenosine A1 receptor agonists and A2A receptors antagonists would be of great interest because of their potentially opposite effects based on current knowledge. Several adenosine receptor agonists and antagonist are currently available for research purpose only, while only one adenosine receptor antagonist, istradefylline, is currently U.S. Food and Drug Administration (FDA) approved as treatment for Parkinson’s disease (JF and RA 2020). To our best knowledge, no studies have been conducted on the adenosine A2B receptor in migraine and adenosine A3 has only once been investigated in migraine [6]. This leaves a huge gap in our knowledge that needs to be explored in both clinical and pre-clinical setting.
Conclusion
Preclinical data demonstrated that adenosine caused vasodilation and modulated CGRP release. We suggest that the adenosine A1 receptor and adenosine A2A receptor could be potential targets for migraine treatment.
Availability of data and materials
Not applicable.
Abbreviations
- A1R:
-
A1 receptor
- A2AR:
-
A2A receptor
- AC:
-
Adenylyl cyclase
- AMP:
-
Adenosine monophosphate
- ADP:
-
Adenosine 5’-diphosphate
- ATP:
-
Adenosine triphosphate
- cAMP:
-
Cyclic adenosine monophosphate
- CGRP:
-
Calcitonin gene related peptide
- DAG:
-
Diacylglycerol
- ES:
-
Electrical stimulation
- ESTG:
-
Electrical stimulation of the trigeminal ganglion
- FDA:
-
U.S. Food and Drug Administration
- GPCR:
-
G-protein coupled receptors
- ICHD:
-
International Classification of Headache Disorders
- IP3 :
-
Inositol 1,4,5-triphosphate
- MAPK:
-
Mitogen-activated protein kinase
- MMA:
-
Meningeal media artery
- MO:
-
Migraine without aura
- mRNA:
-
Messenger RNA
- NR:
-
Not reported
- PKA:
-
Protein kinase A
- PCR:
-
Polymerase chain reaction
- PGE2 :
-
Prostaglandin E2
- PGF2a :
-
Prostaglandin F2a
- PIP2 :
-
Phosphatidylinositol 4,5-biphosphate
- PLC:
-
Phospholipase C
- PLD:
-
Phospholipase D
- SAH:
-
S-adenosylhomocysteine
- SD:
-
Spreading depression
- TG:
-
Trigeminal ganglion
- TNC:
-
Trigeminal nucleus caudalis
- TSC:
-
Tianshu capsule
- TVS:
-
Trigeminovascular system
References
Arulmani U, Heiligers JPC, Centurión D et al (2005) Lack of effect of the adenosine A1 receptor agonist, GR79236, on capsaicin-induced CGRP release in anaesthetized pigs. Cephalalgia. https://doi.org/10.1111/j.1468-2982.2005.00967.x
Ashina M (2020) Migraine. N Engl J Med 383:1866–1876. https://doi.org/10.1056/NEJMra1915327
Baxter GF, Hale SL, Miki T et al (2000) Adenosine A1 agonist at reperfusion trial (AART): Results of a three-center, blinded, randomized, controlled experimental infarct study. Cardiovasc Drugs Ther 14:607–614. https://doi.org/10.1023/A:1007850527878
Borea PA, Gessi S, Merighi S et al (2018) Pharmacology of Adenosine Receptors: The State of the Art. Physiol Rev 98:1591–1625. https://doi.org/10.1152/physrev.00049.2017.-Adenosine
Carley DW, Hagan RM, Sheehan M et al (1997) Adenosine A1 receptor agonist GR79236 suppresses apnea during all sleep stages in the rat. Sleep 20:1093–1098. https://doi.org/10.1093/sleep/20.12.1093
Carruthers AM, Sellers LA, Jenkins DW et al (2001) Adenosine A1 receptor-mediated inhibition of protein kinase A-induced calcitonin gene-related peptide release from rat trigeminal neurons. Mol Pharmacol 59:1533–1541. https://doi.org/10.1124/mol.59.6.1533
Chou SY, Lee YC, Chen HM et al (2005) CGS21680 attenuates symptoms of Huntington’s disease in a transgenic mouse model. J Neurochem 93:310–320. https://doi.org/10.1111/j.1471-4159.2005.03029.x
Dastjerdi MN, Rarani MZ, Valiani A, Mahmoudieh M (2016) The effect of adenosine A1 receptor agonist and antagonist on p53 and caspase 3, 8, and 9 expression and apoptosis rate in MCF-7 breast cancer cell line. Res Pharm Sci 11:303–310. https://doi.org/10.4103/1735-5362.189301
Faraci FM, Kadel KA, Heistad DD (1989) Vascular responses of dura mater. Am J Physiol - Hear Circ Physiol 257:H157-61. https://doi.org/10.1152/ajpheart.1989.257.1.h157
Faudone G, Arifi S, Merk D (2021) The Medicinal Chemistry of Caffeine. J Med Chem 64:7156–7178. https://doi.org/10.1021/ACS.JMEDCHEM.1C00261
Fried NT, Elliott MB, Oshinsky ML (2017) The Role of Adenosine Signaling in Headache: A Review. Brain Sci 7:30. https://doi.org/10.3390/brainsci7030030
Giffin NJ, Kowacs F, Libri V et al (2003) Effect of the adenosine A1 receptor agonist GR79236 on trigeminal nociception with blink reflex recordings in healthy human subjects. Cephalalgia 23:287–292. https://doi.org/10.1046/j.1468-2982.2003.00511.x
Goadsby PJ, Hoskin KL, Storer RJ et al (2002) Adenosine A1 receptor agonists inhibit trigeminovascular nociceptive transmission. Brain 125:1392–1401. https://doi.org/10.1093/brain/awf141
González-Fernández E, Sánchez-Gómez MV, Pérez-Samartín A et al (2014) A3 Adenosine receptors mediate oligodendrocyte death and ischemic damage to optic nerve. Glia 62:199–216. https://doi.org/10.1002/glia.22599
Haanes KA, Edvinsson L (2014) Expression and characterization of purinergic receptors in rat middle meningeal artery-potential role in migraine. PLoS One 9(9):e108782–e108782. https://doi.org/10.1371/journal.pone.0108782
Haanes KA, Labastida-Ramírez A, Chan KY et al (2018) Characterization of the trigeminovascular actions of several adenosine A2A receptor antagonists in an in vivo rat model of migraine. J Headache Pain 19:1–10. https://doi.org/10.1186/s10194-018-0867-x
Hardebo JE, Edvinsson L (1979) Adenine compounds: Cerebrovascular effects In Vitro with reference to their possible involvement in migraine. Stroke. https://doi.org/10.1161/01.STR.10.1.58
Heseltine L, Webster JM, Taylor R (1995) Adenosine effects upon insulin action on lipolysis and glucose transport in human adipocytes. Mol Cell Biochem 144:147–151. https://doi.org/10.1007/BF00944394
Honey AC, Bland-Ward PA, Connor HE et al (2002) Study of an adenosine A1 receptor agonist on trigeminally evoked dural blood vessel dilation in the anaesthetized rat. Cephalalgia 22:260–264. https://doi.org/10.1046/j.1468-2982.2002.00345.x
Jacobson KA, Gao ZG (2006) Adenosine receptors as therapeutic targets. Nat Rev Drug Discov 5:247–264. https://doi.org/10.1038/nrd1983
Jacobson KA, Tosh DK, Jain S, Gao ZG (2019) Historical and current adenosine receptor agonists in preclinical and clinical development. Front Cell Neurosci 13:1–17. https://doi.org/10.3389/fncel.2019.00124
Jenkins DW, Feniuk W, Humphrey PPA (2001) Characterization of the prostanoid receptor types involved in mediating calcitonin gene-related peptide release from cultured rat trigeminal neurones. Br J Pharmacol 134:1296–1302. https://doi.org/10.1038/sj.bjp.0704357
Chen JF, Cunha RA (2020) The belated US FDA approval of the adenosine A 2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal 16:167–174. https://doi.org/10.1007/S11302-020-09694-2
Little JW, Ford A, Symons-Liguori AM et al (2015) Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 138:28–35. https://doi.org/10.1093/BRAIN/AWU330
Kelion AD, Webb TP, Gardner MA et al (2002) Does a selective adenosine A1 receptor agonist protect against exercise induced ischaemia in patients with coronary artery disease? Heart 87:115–120. https://doi.org/10.1136/heart.87.2.115
Kirsch GE, Codina J, Birnbaumer L, Brown AM (1990) Coupling of ATP-sensitive K+ channels to A1 receptors by G proteins in rat ventricular myocytes
Kleppisch T, Nelson MT (1995) Adenosine activates ATP-sensitive potassium channels in arterial myocytes via A2 receptors and cAMP-dependent protein kinase (vasodilation/glibenclamide/hypoxia/P1 receptors)
Kunduri S, Dick G, Nayeem M, Mustafa S (2013) Adenosine A(1) receptor signaling inhibits BK channels through a PKCα-dependent mechanism in mouse aortic smooth muscle. Physiol Rep 1:e00037. https://doi.org/10.1002/phy2.37
Li M, Kang R, Shi J et al (2013) Anticonvulsant Activity of B2, an Adenosine Analog, on Chemical Convulsant-Induced Seizures. PLoS ONE 8:67060. https://doi.org/10.1371/journal.pone.0067060
Lin Z, Yin P, Reierstad S et al (2010) Adenosine A1 receptor, a target and regulator of estrogen receptorα action, mediates the proliferative effects of estradiol in breast cancer. Oncogene 29:1114–1122. https://doi.org/10.1038/onc.2009.409
Lindquist BE, Shuttleworth CW (2014) Spreading depolarization-induced adenosine accumulation reflects metabolic status in vitro and in vivo. J Cereb Blood Flow Metab 34:1779–1790. https://doi.org/10.1038/jcbfm.2014.146
Lu W, Li B, Chen J et al (2016) Expression of calcitonin gene-related peptide, adenosine A2a receptor and adenosine A1 receptor in experiment rat migraine models. Biomed Reports 4:379–383. https://doi.org/10.3892/br.2016.591
Mediero A, Wilder T, Perez-Aso M, Cronstein BN (2015) Direct or indirect stimulation of adenosine A2Areceptors enhances bone regeneration as well as bone morphogenetic protein-2. FASEB J 29:1577–1590. https://doi.org/10.1096/fj.14-265066
Nowaczewska M, Wiciński M, Kaźmierczak W (2020) The Ambiguous Role of Caffeine in Migraine Headache: From Trigger to Treatment. Nutrients 12:1–16. https://doi.org/10.3390/NU12082259
Ocaña M, Baeyens JM (1994) Role of ATP-sensitive K+ channels in antinociception induced by R-PIA, an adenosine A1 receptor agonist. Naunyn Schmiedebergs Arch Pharmacol 350:57–62. https://doi.org/10.1007/BF00180011
Paterniti I, Melani A, Cipriani S et al (2011) Selective adenosine A2Areceptor agonists and antagonists protect against spinal cord injury through peripheral and central effects. J Neuroinflammation 8:1–14. https://doi.org/10.1186/1742-2094-8-31
Ragozzino D, Limatola C, Sobrero F et al (2021) Hippocampal Neurons Neuroprotection and Neuromodulation in CL1-Mediated 3 Required for CX Activity of Adenosine Receptors Type 1 Is. J Immunol Ref 180:7590–7596. https://doi.org/10.4049/jimmunol.180.11.7590
Sachdeva S, Gupta M (2013) Adenosine and its receptors as therapeutic targets: An overview. Saudi Pharm J 21:245–253. https://doi.org/10.1016/j.jsps.2012.05.011
Sawynok J (2016) Adenosine receptor targets for pain. Neuroscience 338:1–18
Shen WT, Huang YJ, Zhang Q et al (2020) SCH58261, the antagonist of adenosine A2A receptor, alleviates cadmium-induced preeclampsia via sirtiun-1/hypoxia-inducible factor-1a pathway in rats. Eur Rev Med Pharmacol Sci 24:10941–10953. https://doi.org/10.26355/eurrev_202011_23577
Sneyd RJ, Langton JA, Allan LG et al (2007) Multicentre evaluation of the adenosine agonist GR79236X in patients with dental pain after third molar extraction. Br J Anaesth 98:672–676. https://doi.org/10.1093/bja/aem075
Szopa A, Poleszak E, Bogatko K et al (2018) DPCPX, a selective adenosine A1 receptor antagonist, enhances the antidepressant-like effects of imipramine, escitalopram, and reboxetine in mice behavioral tests. Naunyn Schmiedebergs Arch Pharmacol 391:1361. https://doi.org/10.1007/s00210-018-1551-z
Tabrizchi R, Bedi S (2001) Pharmacology of adenosine receptors in the vasculature. Pharmacol Ther 91:133–147
Teoh LKK, Grant R, Hulf JA et al (2002) The effect of preconditioning (ischemic and pharmacological) on myocardial necrosis following coronary artery bypass graft surgery. Cardiovasc Res 53:175–180. https://doi.org/10.1016/S0008-6363(01)00435-7
Thuraiaiyah J, Kokoti L, Al-Karagholi MA, Ashina M (2022) Involvement of adenosine signaling pathway in migraine pathophysiology: A systematic review of clinical studies. Cephalalgia. https://doi.org/10.1177/03331024221077665.
Wei EP, Moskowitz MA, Boccalini P, Kontos HA (1992) Calcitonin gene-related peptide mediates nitroglycerin and sodium nitroprusside-induced vasodilation in feline cerebral arterioles. Circ Res 70:1313–1319. https://doi.org/10.1161/01.RES.70.6.1313
Yang Y, Zhang H, Lu Q et al (2021) Suppression of adenosine A2a receptors alleviates bladder overactivity and hyperalgesia in cyclophosphamide-induced cystitis by inhibiting TRPV1. Biochem Pharmacol 183:114340. https://doi.org/10.1016/j.bcp.2020.114340
Yegutkin GG, Guerrero-Toro C, Kilinc E et al (2016) Nucleotide homeostasis and purinergic nociceptive signaling in rat meninges in migraine-like conditions. Purinergic Signal 12:561–574. https://doi.org/10.1007/s11302-016-9521-8
Yuan K, Bai GY, Park WH et al (2008) Stimulation of ANP secretion by 2-Cl-IB-MECA through A3 receptor and CaMKII. Peptides 29:2216–2224. https://doi.org/10.1016/j.peptides.2008.09.003
Zhou Y, Tong L, Chu X et al (2017) The Adenosine A1 Receptor Antagonist DPCPX Inhibits Tumor Progression via the ERK/JNK Pathway in Renal Cell Carcinoma. Cell Physiol Biochem 43:733–742. https://doi.org/10.1159/000481557
Acknowledgements
Not applicable.
Funding
M.A. was supported by the Lundbeck Foundation Professor Grant (R310-2018–3711).
Author information
Authors and Affiliations
Contributions
JT did the search, screening of articles, data extraction and drafted first manuscript. LK did the screening of articles and data extraction. MMK and MA initiated, designed, supervised, and revised the paper. All authors reviewed and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
JT, LK and MMK report no conflict of interest. MA has received consulting fees and honoraria for lectures/presentations from AbbVie, Allergan, Amgen, Eli Lily, Lundbeck, Novartis and Teva. MA has also received personal payments for participating on data Safety Monitoring Board or Advisory Board for AbbVie, Amgen, Eli Lily, Lundbeck and Novartis.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Thuraiaiyah, J., Kokoti, L., Al-Karagholi, M.AM. et al. Involvement of adenosine signaling pathway in migraine pathophysiology: a systematic review of preclinical studies. J Headache Pain 23, 43 (2022). https://doi.org/10.1186/s10194-022-01412-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10194-022-01412-0