
Abstract
Airway neuroepithelial bodies sense changes in inspired O2, whereas arterial O2 levels are monitored primarily by the carotid body. Both respond to hypoxia by initiating corrective cardiorespiratory reflexes, thereby optimising gas exchange in the face of a potentially deleterious O2 supply. One unifying theme underpinning chemotransduction in these tissues is K+ channel inhibition. However, the transduction components, from O2 sensor to K+ channel, display considerable tissue specificity yet result in analogous end points. Here we highlight how emerging data are contributing to a more complete understanding of O2 chemosensing at the molecular level.
Similar content being viewed by others


Introduction
Aerobic metabolism requires an adequate supply of O2, and rapid adaptation to changes in the partial pressures of inspired atmospheric gases is crucial to survival. During episodes of compromised O2 availability, numerous chemosensory systems, acting in concert, rapidly modulate pulmonary ventilation and perfusion to optimize the supply of O2 from alveolus to metabolising tissues. This review focuses on two key systems involved in this homeostatic response: the carotid bodies (CBs) and neuroepithelial bodies (NEBs), representative chemoreceptors of the arterial circulation and the airway, respectively [1,2]. So far, CBs and NEBs, together with pulmonary smooth muscle (which will not be examined in great depth here), have been the most extensively studied of O2-sensitive tissues, and recent investigations have provided major new insights into the expression and interactions of molecular components that link a decreased partial pressure of oxygen (pO2) to appropriate cellular responses in the circulation and respiratory systems.
CBs are highly vascularised organs, located at the bifurcations of the common carotid arteries, that rapidly initiate increased activity in afferent chemosensory fibres of the carotid sinus nerve in response to systemic hypoxaemia. There is widespread agreement that the sensory elements of the CB are the type I (glomus) cells, which contain numerous transmitters and lie in synaptic contact with afferent sensory neurones [1,3]. Type I cells release catecholamines, acetylcholine and ATP in response to hypoxia to initiate afferent discharge [4]. Commonly located at airway bifurcations are NEBs, tight clusters of neurone-derived, transmitter-containing cells that synapse with branches of both afferent and efferent neurones. They evoke appropriate responses to airway hypoxia (as opposed to hypoxaemia) by initiating afferent information to the respiratory centres [5] and releasing peptides and amine modulators [particularly 5-hydroxytryptamine (serotonin)] [6] into the local pulmonary circulation [2]. The prominence of NEBs in neonatal lungs and the association of pathological conditions, such as apnoea of prematurity and sudden infant death syndrome, with NEB cell hyperplasia strongly suggest that NEBs are involved in both the initiation of breathing at birth and cardiorespiratory control postnatally [7].
Although the specific details of the signal transduction mechanisms that link a decreased pO2 to transmitter release in CBs and NEBs exhibit significant differences, the unifying response elements in both are pO2-sensitive K+ channels [8]. Thus, decreasing pO2 causes, sequentially, K+ channel inhibition [9,10], membrane depolarization [11,12], activation of voltage-gated Ca2+ channels and Ca2+-dependent transmitter release [13]. This is not generally agreed to be so in pulmonary arterioles; there is still controversy about the relative roles of capacitative/voltage-independent Ca2+ entry [14] and O2-sensitive K+ channels in hypoxic pulmonary vasoconstriction (HPV) [15,16].
Investigations into the nature of O2 sensing in CBs and NEBs, from sensor to effector, have had surprisingly similar aetiologies. As more detailed dissection of the signal transduction pathways was required, the use of isolated, cultured and cellular models of CBs and NEBs emerged. Thus, the precise mechanistic perspectives that are now available have been derived from the whole gamut of techniques ranging from human studies through intact CB/sinus nerve and lung slice preparations to cellular and molecular studies in PC12 cells (a rat phaeochromocytoma cell line, a model for CBs), H146 cells (a human small cell carcinoma of the lung cell line, a model for NEBs) and, most recently, knockout and recombinant experiments.
O2sensor and signal transduction
It has been clear for some time that putative O2 sensors would be drawn from a pool of proteins that naturally underwent oxido-reductive transitions. Candidates included plasma membrane bound enzymes, cytosolic enzymes and mitochondrial complexes that contained, as key elements in the proposed redox mechanism, one or more transition metals. Thus, iron-containing haem proteins, including cytochromes and NADPH oxidases, were proposed some time ago as potential O2 sensors in a variety of cellular systems. In NEBs, a number of lines of evidence point towards a significant, if not exclusive, involvement of NADPH oxidase in airway O2 sensing [17,18,19]. The NADPH oxidase model for O2 chemoreception suggests that, under normoxic conditions, the oxidase tonically generates superoxide (O2- .) from O2 which is rapidly converted to H2O2 by several enzymes including superoxide dismutase and catalase. This H2O2 is believed to promote channel activity. Thus, native, isolated and cultured NEB cells express a number of important proteins that together constitute the multimeric functional NADPH oxidase enzyme complex, including gp91phox and p22phox [17]. Hypoxia caused decreased fluorescence of rhodamine 123 (indicative of decreased free radical formation) and K+ channel inhibition, effects that were suppressed by the relatively non-selective NADPH oxidase inhibitor, diphenylene iodonium ('DPI') [17]. Furthermore, H2O2 (a product of the oxidase activity) was able to stimulate K+ channels [17].
The suggestion that NADPH oxidase acted as a O2 sensor and transduced the signal via changes in the intracellular redox potential was tested in the human NEB model, H146 cells [12], by exploiting the fact that NADPH oxidase activity can be regulated by the protein kinase C (PKC)-dependent phosphorylation of two components of the complex, p67phox and p47phox [20]. H146 cells express these proteins, hypoxia suppresses H2O2 levels, H2O2 activates 4-aminopyridine-insensitive K+ currents, and hypoxic K+ channel inhibition is suppressed by PKC activation [19]. These results provide direct functional evidence to support a role for NADPH oxidase in this important process and also suggest that PKC might modulate chemoreception by altering the affinity of the oxidase for O2. Recently, the involvement of this oxidase has received further reinforcement by the observation that NEB cell K+ currents recorded from gp91phox knockout mouse lung slices were acutely insensitive to acute hypoxia [18].
In contrast, the idea that NADPH oxidase provides the upstream signal for K+ channel inhibition has been thoroughly investigated and largely discounted by most investigators in the CB field; the haem hypothesis has gained greater credence since the observation that hypoxic inhibition of K+ channels can be completely reversed upon the application of carbon monoxide [21]. Similarly, the involvement of NADPH oxidase as an O2 sensor in the pulmonary circulation has essentially been discounted by the recent report that HPV is maintained in pulmonary arterioles isolated from gp91phox knockout mice [22].
The generation of reactive oxygen species (ROS) from mitochondria, as demonstrated in a number of cell types, has been suggested as one mechanism by which hypoxia can induce a cellular response [23]. However, results from most of these studies are inconsistent with mitochondrial ROS production being the major mechanism for rapid O2 sensing, such as that seen in CBs and NEBs, because ROS are not significantly elevated during the first 10 min of the hypoxic challenge and do not become maximal for up to 2 h [24]. Mitochondrial ROS production is therefore more likely to underlie responses to chronic hypoxia, which exerts effects at the level of the gene. This does not in itself discount mitochondrial involvement in rapid O2 sensing, because specific inhibitors of mitochondrial complexes mimic the actions of hypoxia in isolated type I CB cells [25], suggesting a potential interaction of different ROS-generating systems acting on different timescales.
Identity of the O2-sensing K+channels
An interesting parallel has arisen in CB and NEB studies relating to the specific identity of the K+ channels involved in the hypoxic response downstream of the sensor. In both tissues, voltage-dependent and voltage-independent channels have been implicated, and controversy still exists about the physiological contribution of each in the overall cellular response to hypoxia. Studies on CB have been further complicated by genuine species variation [26] (a factor that has not yet been thoroughly investigated for NEBs). In the rat CB, iberiotoxin-sensitive, high-conductance, Ca2+-activated K+ (maxi-K) channels were first proposed as being the O2-sensitive channel [27], but several years later this was brought into question with the identification of a low-conductance, acid-sensitive background K+ channel that was proposed to be TWIK-related, acid-sensitive K2P channel-1 (TASK1; TWIK refers to 'tandem of P-domains, weakly inward rectifying K2P channel') — a member of the newly emerging gene family of voltage-insensitive tandem P-domain K+ (K2P) channels [28].
The importance of maxi-K in transducing hypoxic stimuli into CB transmitter release had been contested until the recent observation that iberiotoxin (the selective maxi-K channel inhibitor) could, like acute hypoxia, evoke catecholamine secretion from type I cells in a novel thin slice preparation of CBs [29]. However, the contribution of TASK1 to the overall hypoxic response cannot be discounted, and awaits clarification in a preparation in situ. Similarly, a number of K+ channels have been implicated in HPV but recent recombinant studies point toward a voltage-activated shaw K+ channel (KCNC1), Kv3.1b, as the primary pulmonary arteriolar effector [16].
In NEBs, a similar controversy has arisen, in part owing to the vexed nature of consistently isolating native NEB cells from airway. At present, hypoxic inhibition of both Ca2+-sensitive and Ca2+-insensitive K+currents has been demonstrated in NEBs, both isolated [10] and in situ [30], but there has been a paucity of further information on the channels that underlie these currents, because of the unsuitability of primary cultured cells and lung slices for detailed molecular characterisation. A recent approach to this problem has been to establish the H146 cell as an appropriate model in which to study O2 sensing in human NEB-derived cells [12,19,31]. Employing this model, it has been possible to verify that O2-sensitive channels are insensitive to Ca2+ [12], but the contribution of Ca2+-activated channels still remains to be investigated robustly in native human cells and lung slices. Notwithstanding that H146 cells and native cells show some differences, it is clear that the Ca2+-insensitive components in the two species are almost certainly identical because their pharmacologies and biophysical natures are essentially indistinguishable. On the basis of these observations, debate still rages about the molecular identification of the Ca2+-insensitive K+ channel: a voltage-activated shaw K+ channel (KCNC3), Kv3.3, is proposed in native NEBs [17] and a TASK-like conductance is suggested in H146 cells [31].
Screening, by reverse-transcriptase-mediated polymerase chain reaction, for all the known human homologues of the K2P gene family has indicated that only TWIK1 and TWIK-related, arachidonic acid-sensitive K2P channel ('TRAAK') are not expressed in H146 cells [32]. Importantly, however, in situ hybridisation and immunohistochemical studies have now exclusively localised TASK to mouse NEB cells in lung, and recent antisense knock-down experiments in the H146 cell model have shown a high correlation between quantitative TASK expression and functional hypoxic sensitivity [33]. This antisense approach could not distinguish between TASK1 and TASK3 because they share such high identity in their open reading frame sequences; of considerable import, however, is the recent demonstration that recombinant TASK1 and TASK3 are exquisitely sensitive to decreased pO2 when expressed in HEK 293 cells [34]. Further pharmacological dissection (using Zn2+ as a discriminating blocker) has now lent support to the notion that the O2-sensitive channel is TASK3, although heterodimerism in H146 cells cannot at present be excluded (PJ Kemp, GJ Searle and C Peers, unpublished data).
Conclusion
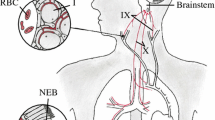
O2 sensing in NEBs and CBs therefore exhibits diverse yet convergent mechanistic features; these are summarised in Figure 1. Upstream, the main O2 sensors in the two tissues are clearly different, although a contribution by mitochondrial ROS generation might be shared. Transduction of the hypoxic signal almost certainly converges, as a unifying theme, on a K2P channel, but how different K+ channels interact to evoke transmitter release and a full physiological response to hypoxia in CBs and NEBs is still debated fiercely and integrative approaches might again be crucial in resolving this important issue.

Schematic flow diagram illustrating the diverse but converging transduction pathways linking hypoxia to transmitter release from arterial (carotid body) and airway (neuroepithelial body) chemoreceptors. Kv3.3 channel, voltage-activated shaw K+ channel (KCNC3); Maxi K, high-conductance, Ca2+-activated K+ channel (KCMA1); ROS, reactive oxygen species; TASK, TWIK-related, acid-sensitive K2P channel; TWIK, tandem of P-domains, weakly inward rectifying K2P channel.
Abbreviations
- CB:
-
carotid body
- HPV:
-
hypoxic pulmonary vasoconstriction
- K2P channel:
-
tandem P-domain K+ channel
- NEB:
-
neuroepithelial body
- po2 =:
-
partial pressure of oxygen
- PKC:
-
protein kinase C
- ROS:
-
reactive oxygen species
- TASK = TWIK-related:
-
acid-sensitive K2P channel
- TWIK = tandem of P-domains:
-
weakly inward rectifying K2P channel.
References
Gonzalez C, Almarez L, Obeso A, Rigual R: Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol Rev. 1994, 74: 829-898.
Cutz E, Jackson A: Neuroepithelial bodies as airway oxygen sensors. Respir Physiol. 1999, 115: 201-214. 10.1016/S0034-5687(99)00018-3.
Peers C, Buckler KJ: Transduction of chemostimuli by the type I carotid body cell. J Membr Biol. 1995, 144: 1-9.
Zhang M, Zhong H, Vollmer C, Nurse CA: Co-release of ATP and ACh mediates hypoxic signalling at rat carotid body chemoreceptors. J Physiol. 2000, 525: 143-158.
Lauweryns JM, VanLommel A: Ultrastructure of nerve endings and synaptic junctions in rabbit intrapulmonary neuroepithelial bodies. J Anat. 1987, 151: 65-65.
Lauweryns JM, Cokeleare M: Hypoxia sensitive neuroepithelial bodies intrapulmonary secretory neuroreceptors, modulated by CNS. Z Zellforsch Mikrosk Anat. 1973, 145: 521-540.
Gillan JE, Curran C, O'Rielly E, Cahalane SF, Unwin AR: Abnormal patterns of pulmonary neuroendocrine cells in victims of Sudden Infant Death Syndrome. Paediatrics. 1989, 84: 828-834.
Peers C: Oxygen-sensitive ion channels. Trends Pharmacol Sci. 1997, 18: 405-408. 10.1016/S0165-6147(97)01120-6.
Lopez-Barneo J, Lopez-Lopez JR, Urena J, Gonzalez C: Chemotransduction in the carotid body: K+ current modulated by pO2 in type I chemoreceptor cells. Science. 1988, 241: 580-582.
Youngson C, Nurse C, Yeger H, Cutz E: Oxygen sensing in airway chemoreceptors. Nature. 1993, 365: 153-155. 10.1038/365153a0.
Wyatt CN, Wright C, Bee D, Peers C: O2-sensitive K+ currents in carotid-body chemoreceptor cells from normoxic and chronically hypoxic rats and their roles in hypoxic chemotransduction. Proc Natl Acad Sci USA. 1995, 92: 295-299.
O'Kelly I, Peers C, Kemp PJ: Oxygen-sensitive K+ channels in neuroepithelial body-derived small cell carcinoma cells of the human lung. Am J Physiol. 1998, 275: L709-L716.
Urena J, Fernandez-Chacon R, Benot AR, Alvarez de Toledo G, Lopez-Barneo J: Hypoxia induces voltage-dependent Ca2+ entry and quantal dopamine secretion in carotid body glomus cells. Proc Natl Acad Sci USA. 1994, 91: 10208-10211.
Robertson TP, Hague D, Aaronson PI, Ward JP: Voltage-independent calcium entry in hypoxic pulmonary vasoconstriction of intrapulmonary arteries of the rat. J Physiol. 2000, 525: 669-680.
Archer SL, Weir EK, Reeve HL, Michelakis E: Molecular identification of O2 sensors and O2-sensitive potassium channels in the pulmonary circulation. Adv Exp Med Biol. 2000, 475: 219-240.
Osipenko ON, Tate RJ, Gurney AM: Potential role for kv3.1b channels as oxygen sensors. Circ Res. 2000, 86: 534-540.
Wang D, Youngson C, Wong V, Yeger H, Dinauer MC, Vega-Saenz de Miera E, Rudy B, Cutz E: NADPH-oxidase and hydrogen peroxide sensitive K+ channel may function as an oxygen sensor complex in airway chemoreceptors and small cell lung carcinoma cell lines. Proc Natl Acad Sci USA. 1996, 93: 13182-13187. 10.1073/pnas.93.23.13182.
Fu XW, Wang D, Nurse C, Dinauer MC, Cutz E: NADPH oxidase is an O2 sensor in airway chemoreceptors: evidence from K+ current modulation in wild type and oxidase-deficient mice. Proc Natl Acad Sci USA. 2000, 97: 4374-4379. 10.1073/pnas.97.8.4374.
O'Kelly I, Lewis A, Peers C, Kemp PJ: O2 sensing by airway chemoreceptor-derived cells: protein kinase C activation reveals functional evidence for involvement of NADPH oxidase. J Biol Chem. 2000, 275: 7684-7692. 10.1074/jbc.275.11.7684.
Tauber AI: Protein kinase C and the activation of the human neutrophol NADPH-oxidase. Blood. 1987, 69: 711-720.
Lopez-Lopez JR, Gonzalez C: Time course of K+ current inhibition by low oxygen in chemoreceptor cells of adult rabbit carotid body. Effects of carbon monoxide. FEBS Lett. 1992, 299: 251-254. 10.1016/0014-5793(92)80126-2.
Archer SL, Reeve HL, Michelakis E, Puttagunta L, Waite R, Nelson DP, Dinauer MC, Weir EK: O2 sensing is preserved in mice lacking the gp91 phox subunit of NADPH oxidase. Proc Natl Acad Sci USA. 1999, 96: 7944-7949. 10.1073/pnas.96.14.7944.
Chandel NS, Schumacker PT: Cellular oxygen sensing by mitochondria: old questions, new insight. J Appl Physiol. 2000, 88: 1880-1889. 10.1063/1.1303764.
Duranteau J, Chandel NS, Kulisz A, Shao Z, Schumacker PT: Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J Biol Chem. 1998, 273: 11619-11624. 10.1074/jbc.273.19.11619.
Buckler KJ, Vaughan-Jones RD: Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. J Physiol. 1998, 513: 819-833.
Lopez-Lopez JR, Peers C: Electrical properties of chemoreceptor cells. In The Carotid Body Chemoreceptors. Edited by Gonza-lez C. Austin: Landes Bioscience;. 1997, 65-78.
Peers C: Hypoxic suppression of K+ currents in type I carotid body cells: selective effect on the Ca2+-activated K+ current. Neurosci Lett. 1990, 119: 253-256. 10.1016/0304-3940(90)90846-2.
Buckler KJ, Williams BA, Honore E: An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J Physiol. 2000, 525: 135-142.
Pardal R, Ludewig U, Garcia-Hirschfeld J, Lopez-Barneo J: Secretory responses of intact glomus cells in thin slices of rat carotid body to hypoxia and tetraethylammonium. Proc Natl Acad Sci USA. 2000, 97: 2361-2366. 10.1073/pnas.030522297.
Fu XW, Nurse C, Wang YT, Cutz E: Selective modulation of membrane currents by hypoxia in intact airway chemoreceptors from neonatal rabbit. J Physiol. 1999, 514: 139-150.
O'Kelly I, Stephens RH, Peers C, Kemp PJ: Potential identification of the O2-sensitive K+ current in a human neuroepithelial body-derived cell line. Am J Physiol. 1999, 276: L96-L104.
Kemp PJ, O'Kelly I, Lewis A, Peers C: Regulation of K+ currents in a human neuroepithelial body-derived cell line suggests that hTASK1 is an airway O2-sensitive K+channel. FASEB J. 2001,
Kemp PJ, Hartness ME, O'Kelly I, Lewis A, Peers C: Antisense depletion of a specific potassium channel in H146 cells indicates that hTASK-1 is an airway oxygen sensing channel. FASEB J. 2001,
Kemp PJ, Lewis A, Chapman CG, Meadows HJ, Peers C: Direct demonstration that hTASK1 is an O2 sensitive K+channel. FASEB J. 2001,
Acknowledgements
The authors' own studies are supported by The Wellcome Trust and the British Heart Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peers, C., Kemp, P.J. Acute oxygen sensing: diverse but convergent mechanisms in airway and arterial chemoreceptors. Respir Res 2, 145 (2001). https://doi.org/10.1186/rr51
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/rr51