
Abstract
X-chromosome inactivation occurs randomly for one of the two X chromosomes in female cells during development. Inactivation occurs when RNA transcribed from the Xist gene on the X chromosome from which it is expressed spreads to coat the whole X chromosome. In the first issue of Epigenetics and Chromatin, Nesterova and colleagues investigate the role of the RNA interference pathway enzyme Dicer in DNA methylation of the Xist promoter.
Similar content being viewed by others


X-chromosome inactivation is the transcriptional silencing of one X chromosome in female mammalian cells that equalizes dosage of gene products from the X chromosome between XX females and XY males [1–3]. X-chromosome inactivation in the embryo proper occurs early in development. The two X chromosomes have an equal probability of being silenced [4]. Silencing, once established, is stable: the same X chromosome remains inactivated in all subsequent cell generations. As a result, each female is a mosaic of cells in which either the maternally inherited or the paternally inherited X is silenced. Nesterova and colleagues in the first issue of Epigenetics and Chromatin shed new light on how this process is regulated [5].
An antisense pair of non-coding RNAs, encoded by Xist and Tsix (Figure 1), is important in the regulation of the random inactivation of mouse X chromosomes. Before the signal that initiates random X-chromosome inactivation is received, Xist and Tsix are transcribed from all active X chromosomes in each male and female cell [6]. Once inactivation is initiated, Xist and Tsix are differentially regulated on the X that will become the active X chromosome (XA) and the one that will become the inactive X chromosome (XI). On the X chromosome that will become the XI, Xist transcripts spread in cis from their site of synthesis to coat the entire X chromosome and establish transcriptional silencing. Concomitant with Xist RNA coating, Tsix is silenced on the XI. The expression of Xist and Tsix persists on the XA for a brief period after silencing of the XI is complete, and is eventually extinguished. Xist RNA continues to coat the XI throughout all subsequent cell divisions, where it contributes to the maintenance of silencing. These patterns of Xist and Tsix expression are also seen in mouse female embryonic stem (ES) cells, which have two XAs and which undergo X-chromosome inactivation when they are induced to differentiate in vitro. Thus, ES cells provide a useful model system to study X-chromosome inactivation.

Transcription of Xist and Tsix on the X chromosome. The coding sequences of Xist and Tsix overlap on opposite strands of the X-chromosome DNA.
Mutations in Xist or Tsixcan cause non-random X inactivation
Heterozygous mutation of Xist or Tsix causes non-random X-chromosome inactivation in female cells. When Xist expression is increased from one X chromosome in pre-X-chromosome-inactivation cells, that X chromosome always becomes the XI and the wild-type X always becomes the XA [7]. In female ES cells or embryos in which Xist is disrupted on one X chromosome, the mutant X chromosome always becomes the XA and the wild-type X chromosome always becomes the XI [8–10]. Disruption of Tsix has the opposite effect: the mutant X chromosome becomes the XI and the wild-type X chromosome is always the XA [11–13]. This is known as primary non-random X-chromosome inactivation because the X chromosomes are chosen as the XA and XI before silencing is initiated. A second cause of non-random X-chromosome inactivation is the selective death of cells that inactivate the incorrect number of X chromosomes: because the fates of the X chromosomes are not determined before silencing, this is known as secondary non-random X-chromosome inactivation [14]. Because Xist and Tsix mutations cause primary non-random X-chromosome inactivation, it is likely that these non-coding RNAs function in the choice of the XA and XI before silencing is initiated. Understanding how Xist and Tsix are regulated in pre-X-chromosome-inactivation cells is central to understanding how one X chromosome is randomly selected as the XA and the other as the XI in each cell.
In addition to having opposing roles in random choice, Xist and Tsix also negatively regulate each other in ES cells. Xist and Tsix are transcribed from overlapping regions on opposite strands of the X-chromosome DNA (Figure 1). Deletion of Tsix promoter sequences or a mutation that blocks Tsix transcription before it reaches Xist RNA coding sequences abolishes Tsix transcription and causes a roughly ten-fold increase in Xist RNA levels from the mutant X chromosome [11–13]. Thus, transcription of Tsix across Xist is necessary for Tsix to negatively regulate Xist. In the Tsix truncation mutant the Tsix promoter has histone modification patterns that are generally associated with transcriptional silencing [15]. These epigenetic marks also characterize the XI, and their recruitment to the XI requires transcription of Xist [16–18]. Together, these results suggest that the increase in Xist RNA that occurs on Tsix mutant chromosomes represses Tsix. Consistent with the possibility that Xist negatively regulates Tsix, Tsix RNA levels are increased from Xist mutant X chromosomes [10, 19]. Insights into the nature of factors that are involved in the mutual regulation of Xist and Tsix in pre-X-chromosome-inactivation cells are likely to be important in developing an understanding of how these non-coding RNAs ensure that the two X chromosomes have an equal probability of being silenced in each cell.
The role of DNA methylation
The mechanisms underlying the mutual regulation of Xist and Tsix in pre-X-chromosome-inactivation cells are not well characterized. An interesting new study by Nesterova and colleagues suggests that DNA methylation may be involved in this mutual negative regulation [5]. Nesterova et al. demonstrate a correlation between Xist promoter DNA methylation and Xist expression in ES cells. In XY ES cells (in which the single X chromosome remains active), two regions flanking the Xist transcription start site show high levels of DNA methylation. Two XY ES cell lines bearing Xist promoter mutations that result in increased Xist expression showed DNA hypomethylation at these sites. In addition, a mutation that truncates Tsix transcription before it traverses Xist also resulted in increased Xist expression and DNA hypomethylation at these sites. These results establish a clear correlation between the levels of DNA methylation at Xist and expression of Xist in ES cells. It remains to be established whether the increase in Xist expression triggers demethylation or vice versa. In addition, Xist and Tsix negatively regulate each other, raising the possibility that Tsix also has a role in regulation of Xist DNA methylation.
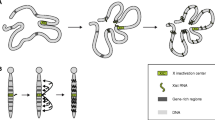
Tsix has also been implicated in the direct regulation of DNA methylation. The de novo DNA methyltransferase Dnmt3a can be immunoprecipitated with Tsix RNA using an RNA-chromatin immunoprecipitation procedure [20]. Furthermore, Dnmt3a can de novo methylate Xist [21, 22]. Together, these data suggest a model in which Tsix RNA directs Dnmt3a to Xist in ES cells (Figure 2a). Thus, the hypomethylation of Xist DNA in the Tsix truncation line may occur because Dnmt3a cannot act on Xist when Tsix RNA is not present to recruit it there.

Models for the coordinate regulation of Xist DNA methylation and expression by Tsix, de novo DNA methyltransferases and Dicer. De novo DNA methyltransferases (Dnmt) promote methylation of Xist DNA. Increased Xist expression, as is seen in the Xist promoter mutants, could trigger Xist DNA hypomethylation (a) indirectly by affecting Tsix RNA levels, if Tsix is necessary to direct de novo DNA methyltransferases to the Xist gene, or (b) directly, if Xist RNA can interfere with de novo DNA methyltransferase activity locally. (c) Because Dicer deficiency causes a global decrease in levels of de novo DNA methyltransferases, Dicer must lie directly upstream of the de novo DNA methyltransferases and need not function through either Xist or Tsix to regulate Xist DNA methylation. (The DNA is shown as methylated in a, b and c (bottom), although in a and b if the inhibitory interactions between Xist and Tsix RNA (a) or Dmt (b) prevail, the DNA will be hypomethylated.)
This model explains the hypomethylation of Xist DNA in the Tsix truncation line, but how does it account for the hypomethylation in the Xist promoter mutation lines? As in the Tsix truncation line, the Xist promoter mutation lines show increased Xist expression. In contrast to the truncation line, which does not produce Tsix RNA, the Xist promoter mutation lines continue to express Tsix RNA. However, Tsix RNA levels have not been quantitated in these cell lines, so it is not possible to establish a correlation between Tsix expression levels and Xist DNA methylation. One possibility is that the increase in Xist expression causes a decrease in Tsix RNA levels and a corresponding decrease in Dnmt3a activity at Xist DNA. There is also an alternative possibility: it may be that Xist RNA (or an epigenetic modification induced by Xist RNA) interferes with the activity of Dnmt3a or other de novo methyltransferases (Figure 2b). Indeed, the Xist RNA-coated XI shows overall lower levels of DNA methylation than the XA, consistent with Xist RNA interfering with DNA methylation [23]. Because Xist RNA accumulates only locally in ES cells, this activity would be restricted to the Xist locus and perhaps nearby genes. Analysis of Xist DNA methylation in Xist and combined Xist + Tsix mutant ES cells will be required to distinguish between these possibilities.
X inactivation and Dicer deficiency
Nesterova and colleagues have further investigated the role of de novo methyltransferases in regulation of Xist expression in an analysis of Dicer mutant male ES cells. Dicer is an RNAse III enzyme that is central to the RNA interference (RNAi) pathway. RNAi regulates many aspects of gene expression and involves the production of antisense RNA complementary to sequences in the mRNA of the gene that is being regulated [24]. The formation of sense-antisense double-stranded RNA can trigger transcriptional or post-transcriptional gene silencing. Given that Tsix RNA contains sequences complementary to Xist RNA, an obvious question is whether the RNAi pathway has a role in X-chromosome inactivation. Nesterova et al. show that several independently derived Dicer-deficient male ES cell lines show Xist DNA hypomethylation and upregulation of Xist expression. They also find that the two imprinted loci H19 and Igf2rAir show hypomethylation in Dicer-deficient cells. Hypomethylation of Xist, H19 and Igf2rAir seems to be the consequence of changes in the levels of the de novo methyltransferases Dnmt3a, Dnmt3b and DnmtL, all of which were downregulated upon deletion of Dicer. This decrease in de novo methyltransferase activity in Dicer-deficient cells was also seen in two other studies of independently derived Dicer mutant ES cell lines [25, 26]. In these studies Dicer mutant ES cells show hypomethylation of subtelomeric repeats or of Oct4, Tsp50 and Sox30 promoters, which are normally methylated. The downregulation of the de novo methyltransferases could be attributed to an increase in levels of the repressor Rbl2, which is negatively regulated by the miR-290 microRNA cluster [25, 26]. Together, these results provide a compelling argument that the change in Xist DNA methylation seen in Dicer mutant ES cells is an indirect consequence of the loss of de novo methyltransferase activity (Figure 2c).
Does the change in Xist DNA methylation in pre-X-chromosome-inactivation cells affect the fate of the X chromosomes after inactivation is initiated? To answer this question Nesterova et al. analyzed Dicer mutant embryos. Dicer mutants die shortly after implantation, between embryonic day (E)7.5 and E8.5. X-chromosome inactivation is initiated at approximately E5.5, providing a brief window in which X-chromosome inactivation can be assayed in Dicer mutants. The cells of male and female Dicer-deficient E6.5 embryos and their wild-type littermates did not show any appreciable difference in either Xist or Tsix expression. These results indicate that one X chromosome can be selected as the inactive X and Xist RNA can coat that X chromosome in Dicer mutant embryos. Thus, X-chromosome inactivation seems unaffected by Dicer deficiency in vivo.
The results of Nesterova et al. contrast with those from another study of the role of Dicer in X-chromosome inactivation. Ogawa et al. [27] examined X-chromosome inactivation in Dicer mutant female ES cells and found that Xist RNA could not coat and silence an X chromosome on differentiation. These results indicate that Dicer is necessary for X-chromosome inactivation in vitro. Why do female ES cells and embryos differ in their requirements for Dicer during X-chromosome inactivation? One possibility is that maternal stores of Dicer persist long enough to promote X-chromosome inactivation in female Dicer mutant embryos. However, the homozygous Dicer mutant female ES cells used by Ogawa et al. contained a Dicer transgene that was expressed at less than 5% of wild-type levels (this was deployed to overcome the block to differentiation in Dicer mutants that would have otherwise interfered with analysis of X-chromosome inactivation), suggesting that small amounts of Dicer are not sufficient to promote random inactivation. A second possibility is that Dicer mutant female embryos fail to reverse imprinted X-chromosome inactivation in their embryonic compartment. In mice, the extraembryonic tissues undergo imprinted X-chromosome inactivation, in which there is exclusive silencing of the paternal X chromosome [28]. Imprinted X-chromosome inactivation is initiated in pre-implantation development and seems to occur in all cells of the early embryo. Imprinted X-chromosome inactivation is reversed in the cells that will go on to form the embryo proper, and these cells subsequently undergo random X-chromosome inactivation after implantation [29, 30]. Determining whether Dicer mutant female embryos show random or imprinted X-chromosome inactivation will establish whether Dicer is important to erase imprinted X-chromosome inactivation. Clearly much work remains to be done to determine how Dicer regulates Xist expression during development.
References
Heard E, Chaumeil J, Masui O, Okamoto I: Mammalian X-chromosome inactivation: an epigenetics paradigm. Cold Spring Harb Symp Quant Biol. 2004, 69: 89-102. 10.1101/sqb.2004.69.89.
Boumil RM, Lee JT: Forty years of decoding the silence in X-chromosome inactivation. Hum Mol Genet. 2001, 10: 2225-2232. 10.1093/hmg/10.20.2225.
Lyon MF: Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 1961, 190: 372-373. 10.1038/190372a0.
Wutz A, Gribnau J: X inactivation Xplained. Curr Opin Genet Dev. 2007, 17: 387-393. 10.1016/j.gde.2007.08.001.
Nesterova TB, Popova BC, Cobb BS, Norton S, Senner C, Tang YA, Spruce T, Rodriguez TA, Sado T, Merkenschlager M, Brockdorff N: Dicer regulates Xist promoter methylation in ES cells indirectly through transcriptional control of Dnmt3a. Epigenetics Chromatin. 2008, 1: 2-10.1186/1756-8935-1-2.
Mlynarczyk SK, Panning B: X inactivation: Tsix and Xist as yin and yang. Curr Biol. 2000, 10: R899-R903. 10.1016/S0960-9822(00)00847-2.
Nesterova TB, Johnston CM, Appanah R, Newall AE, Godwin J, Alexiou M, Brockdorff N: Skewing X chromosome choice by modulating sense transcription across the Xist locus. Genes Dev. 2003, 17: 2177-2190. 10.1101/gad.271203.
Marahrens Y, Loring J, Jaenisch R: Role of the Xist gene in X chromosome choosing. Cell. 1998, 92: 657-664. 10.1016/S0092-8674(00)81133-2.
Gribnau J, Luikenhuis S, Hochedlinger K, Monkhorst K, Jaenisch R: X chromosome choice occurs independently of asynchronous replication timing. J Cell Biol. 2005, 168: 365-373. 10.1083/jcb.200405117.
Sado T, Hoki Y, Sasaki H: Tsix defective in splicing is competent to establish Xist silencing. Development. 2006, 133: 4925-4931. 10.1242/dev.02670.
Lee JT, Lu N: Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell. 1999, 99: 47-57. 10.1016/S0092-8674(00)80061-6.
Luikenhuis S, Wutz A, Jaenisch R: Antisense transcription through the Xist locus mediates Tsix function in embryonic stem cells. Mol Cell Biol. 2001, 21: 8512-8520. 10.1128/MCB.21.24.8512-8520.2001.
Sado T, Wang Z, Sasaki H, Li E: Regulation of imprinted X-chromosome inactivation in mice by Tsix. Development. 2001, 128: 1275-1286.
McMahon A, Monk M: X-chromosome activity in female mouse embryos heterozygous for Pgk-1 and Searle's translocation, T(X; 16) 16H. Genet Res. 1983, 41: 69-83. 10.1017/S0016672300021078.
Navarro P, Pichard S, Ciaudo C, Avner P, Rougeulle C: Tsix transcription across the Xist gene alters chromatin conformation without affecting Xist transcription: implications for X-chromosome inactivation. Genes Dev. 2005, 19: 1474-1484. 10.1101/gad.341105.
Silva J, Mak W, Zvetkova I, Appanah R, Nesterova TB, Webster Z, Peters AH, Jenuwein T, Otte AP, Brockdorff N: Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 Polycomb group complexes. Dev Cell. 2003, 4: 481-495. 10.1016/S1534-5807(03)00068-6.
Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y: Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003, 300: 131-135. 10.1126/science.1084274.
Kohlmaier A, Savarese F, Lachner M, Martens J, Jenuwein T, Wutz A: A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biol. 2004, 2: E171-10.1371/journal.pbio.0020171.
Sado T, Hoki Y, Sasaki H: Tsix silences Xist through modification of chromatin structure. Dev Cell. 2005, 9: 159-165. 10.1016/j.devcel.2005.05.015.
Sun BK, Deaton AM, Lee JT: A transient heterochromatic state in Xist preempts X inactivation choice without RNA stabilization. Mol Cell. 2006, 21: 617-628. 10.1016/j.molcel.2006.01.028.
Sado T, Okano M, Li E, Sasaki H: De novo DNA methylation is dispensable for the initiation and propagation of X chromosome inactivation. Development. 2004, 131: 975-982. 10.1242/dev.00995.
Okano M, Bell DW, Haber DA, Li E: DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999, 99: 247-257. 10.1016/S0092-8674(00)81656-6.
Hellman A, Chess A: Gene body-specific methylation on the active X chromosome. Science. 2007, 315: 1141-1143. 10.1126/science.1136352.
Campbell TN, Choy FY: RNA interference: past, present and future. Curr Issues Mol Biol. 2005, 7: 1-6.
Sinkkonen L, Hugenschmidt T, Berninger P, Gaidatzis D, Mohn F, Artus-Revel CG, Zavolan M, Svoboda P, Filipowicz W: MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat Struct Mol Biol. 2008, 15: 259-267. 10.1038/nsmb.1391.
Benetti R, Gonzalo S, Jaco I, Muñoz P, Gonzalez S, Schoeftner S, Murchison E, Andl T, Chen T, Klatt P, Li E, Serrano M, Millar S, Hannon G, Blasco MA: A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat Struct Mol Biol. 2008, 15: 268-279. 10.1038/nsmb.1399.
Ogawa Y, Sun BK, Lee JT: Intersection of the RNA interference and X-inactivation pathways. Science. 2008, 320: 1336-1341. 10.1126/science.1157676.
Lyon MF: The X inactivation centre and X chromosome imprinting. Eur J Hum Genet. 1994, 2: 255-261.
Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E: Epigenetic dynamics of imprinted X inactivation during early mouse development. Science. 2004, 303: 644-649. 10.1126/science.1092727.
Mak W, Nesterova TB, de Napoles M, Appanah R, Yamanaka S, Otte AP, Brockdorff N: Reactivation of the paternal X chromosome in early mouse embryos. Science. 2004, 303: 666-669. 10.1126/science.1092674.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Panning, B. X-chromosome inactivation: the molecular basis of silencing. J Biol 7, 30 (2008). https://doi.org/10.1186/jbiol95
Published:
DOI: https://doi.org/10.1186/jbiol95