
Abstract
There has been considerable interest in cancer stem cells (CSCs) among cancer biologists and clinicians, most likely because of their role in the heterogeneity of cancer and their potential application in cancer therapeutics. Recent studies suggest that CSCs play a key role in liver carcinogenesis. A small subpopulation of cancer cells with CSC properties has been identified and characterized from hepatocellular carcinoma (HCC) cell lines, animal models and human primary HCCs. Considering the high mortality and ineffectiveness of current therapies for HCC, understanding the characteristics and function of CSCs is likely to lead to development of new therapies resulting in improvement of patient survival. This review summarizes recent progress in liver cancer stem cell research with regard to the identification, cell origin, regulation of self-renewal capacity, and therapeutic implications of liver CSCs.
Similar content being viewed by others

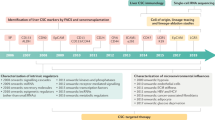

Background
Tumors are characterized by uncontrolled proliferation of abnormal cells that frequently display morphological and functional heterogeneity [1]. Two models have been proposed to explain the heterogeneity of tumors, namely the stochastic [2] and hierarchical models [3]. The stochastic model suggests that all cells within a tumor are biologically homogenous and, therefore, have equal capacity to regenerate the tumor. The hierarchical model, also known as the cancer stem cell model, proposes that only a small subset of tumor cells, designated cancer stem cells (CSCs) or tumor-initiating cells, within a tumor exhibit the capacity to initiate and sustain tumor growth. CSCs possess two important properties similar to those of normal stem cells, namely self-renewal and differentiation capacity [4]. Although CSCs were first identified 26 years ago in acute myelogenous leukemia with the cell surface marker CD34+CD38- [5], the isolation of these cells from solid tumors was accomplished only in 2003 [6]. This study showed that CD44+CD24- cells from breast cancer specimens are able to generate xenografts that phenotypically resemble the initial tumor. Furthermore, the xenografts could be serially passed to generate similar tumors, indicating the self-renewal ability of these cells. Following this seminal study, CSCs were isolated from various solid tumors such as brain, colon, pancreas, lung and liver. The isolation of CSCs was based on a series of cell surface markers, including CD133 [7, 8], CD44 [6, 9], CD90 [10], CD24 [11] and EpCAM [12, 13], as well as functional markers such as ALDH1 [14] and ABCB5 [15], and side population (SP) cells [16].
Hepatocellular carcinoma (HCC) is a deadly disease with no promising therapeutic options. It is the fifth most prevalent cancer in the world, and the third leading cause of cancer-related death, with an annual death rate exceeding 500,000. The incidence of HCC among the population between the ages of 45 and 60 years is rising dramatically, particularly in the western world, due to an increase in hepatitis C virus infection, and alcoholic and non-alcoholic fatty liver disease. While surgical resection/ablation and liver transplantation are potentially curative, the high mortality is due to diagnosis at an advanced stage [17, 18]. Although the multikinase inhibitor sorafenib has recently been approved for treatment of HCC, poor response of the late-stage cancer to this and almost all other available chemotherapeutic agents continues to be a major obstacle to successful HCC therapy. The 5-year survival rate for this cancer is only 5%, and the death rate is expected to rise in the next 20 years. Primary HCC, the most common malignant tumor, accounts for >90% of all primary liver cancer. Its development is a complex, multistep process. Elucidation of the molecular mechanisms of liver carcinogenesis is critical to determine the specific pathways involved in the initiation and progression of HCC that will eventually lead to identification of novel molecular targets for therapy. As observed in other solid tumors, recent studies suggested that CSCs may be involved in the development of liver cancer [10, 13, 19–21]. This review will summarize recent progress in the potential role of CSCs in hepatocarcinogenesis.
CSCs in hepatocellular carcinoma
In an early attempt to isolate liver-cancer-specific CSCs, SP cells with CSC characteristics were separated from a total population of HCC cells [19]. SP sorting was initially used to detect hematopoietic stem cells exploiting the ability of these cells to efflux Hoechst 33342 through an ATP-binding cassette membrane transporter. This property contributes to multidrug resistance of these cells, and this is a key feature of CSCs. Only a very small proportion of HCC cells (0.25% in Huh7 and 0.8% in PLC/RPL5 cells) possess a SP phenotype; these show higher proliferative and tumorigenic potential compared with non-SP cells. Indeed, 1 × 103 SP cells were sufficient for tumor formation in xenograft transplantation while at least 1 × 106 unsorted HCC cells were required to form a tumor, indicating enrichment of CSCs in SP cells. Despite these key observations, it remains uncertain whether SP cells are authentic CSCs, since Hoechst 33342 is cytotoxic, and non-SP cells are unable to grow in the presence of this dye. Consequently, differential resistance to Hoechst 33342 rather than the intrinsic difference in the stem cell properties between SP and non-SP cells could contribute to the difference in tumor formation between these cell types.
In the ensuing several years, significant efforts were made to identify CSCs in HCC cell lines, mouse models and primary HCC (Table 1). CD133, also known as prominin-1 (Prom1), encoding a pentaspan transmembrane glycoprotein [22, 23], has been used as a marker for both somatic stem cells [24–26] and CSCs in different tissues, including brain [7], colon [8], pancreas [27] and skin [28]. Recently, several studies have shown that CD133+ cells isolated from various liver cancer cell lines [20, 21, 29] exhibit the properties of CSCs and only a small fraction of CD133+ cells produce hepatocellular carcinoma in severe combined immunodeficiency mice. Although CD133 has been used to identify CSCs from HCC cell lines and mouse models, some studies have suggested that CD133- cells can form tumors in immunodeficient mice at higher cell number [21], indicating that CD133 may not be an effective CSC marker in HCC. To identify CSCs in human primary HCC specimens, Yang et al. [10] used CD90 cell surface markers and found that, unlike CD90- cells, CD90+ cells from HCC specimens as well as patient blood samples exhibited tumorigenic potential. This study suggested that CD90 is a potent marker of liver CSCs and that circulating CSCs probably contributed to tumor metastasis.
Since CSCs share key features with normal stem cells, including self-renewal and differentiation, it is conceivable that these cells may possess similar cell surface markers. Indeed, some markers, such as EpCAM [13] and OV6 [30], that were used to isolate hepatic stem cells have also been shown to function as specific markers for CSCs in HCC.
Cellular origin of liver CSCs
It has been proposed that CSCs may arise through mutations accrued in normal tissue stem cells or more differentiated tissue progenitor cells [31]. Recently, crypt cells expressing Lgr5 in small intestine and colon tissues have been identified as long-lived stem cells [32]. Apc deletion in these cells was necessary and sufficient to produce tumors in vivo [32]. Another study showed that CD133 marks the intestinal stem cells, and overproduction of β-catenin can transform CD133+ cells into CSCs and generate tumors [33]. Those studies suggested that normal stem cells may be the origin of CSCs. In contrast, introduction of MLL-AF9 into committed granulocyte macrophage progenitors produced leukemia stem cells [34], suggesting that CSCs could also be derived from committed progenitors.
The existence of stem cells in adult liver has been debated for many years. Recent studies have suggested that hepatic progenitor cells (HPCs) or oval cells, a subset of small epithelial cells with an oval nucleus located in the bile ductules and canals of Hering in adult liver [35], may function as stem cells in adult liver. HPCs can be activated to proliferate and differentiate into hepatocytes and biliary lineage in response to severe liver damage [36–41], such as that induced by treatment of rats with 2-acetylaminofluorene after partial hepatectomy (AAF/PH model) [42]. Under this condition, hepatocytes were unable to proliferate to replace the lost tissues. HPCs are, therefore, considered to be the bipotent liver stem cells.
HPCs play an important role in carcinogenesis in at least some subtypes of HCC. The number of HPCs in human chronic liver diseases, such as hepatitis B and C infection and alcoholic liver disease, are directly related to disease severity, suggesting that oval cell proliferation is associated with increased risk of development of HCC in chronic liver disease [41]. A recent study has shown that certain subtypes of primary human HCCs with poor prognosis may be derived from HPCs [43], based on the identification of human HCCs that exhibited gene expression profiles identical to that of hepatoblasts. Further, this subtype of HCC can be distinguished from other types by the markers of HPCs. HPCs are also activated in several mouse models of hepatocarcinogenesis, suggesting their potential role in the initiation and progression of liver cancer [44–48]. Fang et al. [44] found that oval cell proliferation was prominent from the initial liver injury stage to the occurrence and development of HCC in a HCC model fed 3,3'-diaminobenzidine. More importantly, blocking the expansion of oval cells by targeting c-Kit with imatinib mesylate significantly reduced liver tumor formation in mice fed a choline-deficient, ethionine-supplemented diet [49]. Similarly, TNF was upregulated during oval cell proliferation induced by a choline-deficient, ethionine-supplemented diet, and was produced by oval cells [50]. Oval cell proliferation and tumorigenesis were substantially impaired in TNF receptor type 1 knockout mice. These two studies suggested that oval cells may be directly involved in the initiation and progression of liver cancer in this mouse model. More direct evidence for the involvement of oval cells in hepatocarcinogenesis was revealed in a study that demonstrated tumor formation in nude mice upon subcutaneous injection of p53 null oval cells [46]. Further studies showed that CD133+CD45- oval cells isolated from Mat1a (methionine adenosyltransferase 1a) [51] and Pten (phosphatase and tensin homolog deleted on chromosome 10) [52] knockout mice exhibited greater tumorigenic capacity. Furthermore, these oval cells exhibited high levels of resistance to chemotherapeutic agents, consistent with cancer stem cell characteristics. These findings, together with the notion that CSCs may arise from adult stem cells or progenitor cells, suggest that liver CSCs are probably derived from HPCs.
However, it should be noted that not all HCCs arise from HPCs, since oval cell proliferation is very rare in diethylnitrosamine-induced HCC. It is widely accepted that the HCC induced by this carcinogen develops from hepatocytes [53]. Based on these observations, the exact origin of CSCs in the liver appears to depend on the etiology of the cancer.
Signaling pathways involved in maintaining the characteristics of liver CSCs
The maintenance of self-renewal and tumorigenic capacity of CSCs involves several cancer-related signaling pathways, including Bmi-1 [54], Wnt/β-catenin [55], Notch [56], and Sonic Hedgehog [55, 57] (Figure 1). Bmi-1, a polycomb group (PcG) gene, has previously been shown to regulate the self-renewal of hematopoietic stem cells [58]. Overexpression of Bmi-1 enhanced self-renewal of both hematopoietic stem cells and neural stem cells, indicating that Bmi-1 may function as a universal determinant of stem cell self-renewal. Overexpression of Bmi-1 in HPCs dramatically promoted the self-renewal of these cells, and transplantation of Bmi-1-transduced HPCs produced tumors in vivo, indicating the important role of Bmi-1 in regulating the self-renewal of normal or cancer stem cells in the liver [54]. Notably, upregulation of Bmi-1 has also frequently been observed in human primary HCC [59].

Proposed model for the cell origin and transformation of liver cancer stem cells. Dysregulation of signaling pathways involved in self-renewal and proliferation of normal stem cells (hepatic progenitor cells, HPCs) is likely to cause uncontrolled proliferation and increased self-renewal capacity of HPCs, resulting in the transformation of HPCs into cancer stem cells (CSCs). HCC, hepatocellular carcinoma.
The Wnt/β-catenin signaling pathway can play a critical role in stem cell proliferation and potency maintenance [60], as well as differentiation of stem cells in a variety of tissues [61]. The activation of Wnt/β-catenin has been shown to be associated with hepatocyte proliferation in liver development, regeneration and tumor growth [62]. As observed in Bmi-1, mutation in β-catenin and consistent activation of this signaling pathway significantly enhanced the self-renewal of HPCs and promoted cancer initiation [54].
Although the Notch signaling pathway plays an important role in regulating stem cell self-renewal and differentiation [3], it may function as a tumor suppressor or oncogene in different tissues depending on other signaling pathways [63]. In breast tissue, the Notch signaling pathway was shown to be upregulated, and inhibition of the Notch pathway reduced the formation of mammosphere, an indicator of stem-like cells [56]. Similarly, inhibition of the Notch pathway selectively depleted glioblastoma CSCs [64]. The role of the Notch signaling pathway in HCC is controversial. One study found that activation of Notch1 signaling inhibits growth of human HCC [65], suggesting that it may function as a tumor suppressor. In contrast, another study showed that the Notch1 signaling pathway is constitutively activated in HCC [66], indicating a potential oncogenic role of this pathway in HCC.
A variety of tumors exhibit activating mutations or upregulation of Sonic Hedgehog (HH) pathway components, such as PTCH1, SMO and GLI1[67]. Recent studies showed that the HH pathway also regulates the self-renewal ability of CSCs in breast cancer [68], glioma [69] and multiple myeloma [70]. Although the role of the HH pathway in liver CSCs has not been well characterized, this pathway is known to be activated in HCC [71]. It would be of interest to investigate its potential role in regulating the self-renewal ability of liver CSCs.
Therapeutic implications of liver CSCs
Recent studies have shown that CSCs are more resistant to chemotherapy [11, 72] and radiotherapy [73] compared with non-CSCs within the same tumor, indicating that CSCs may be responsible for the recurrence and eventual metastasis of these tumors. It is conceivable that targeting liver CSCs would provide new alternatives to treat HCC, since HCC is more resistant to traditional therapies. Drugs targeting signaling pathways involved in regulating the self-renewal ability of CSCs may prove to be effective in HCC therapy. For example, cyclopamine, which targets the HH pathway, significantly reduces the self-renewal of glioma [74], whereas gamma secretase inhibitor, which targets the Notch pathway, decreases self-renewal of breast CSCs [75]. It is worth investigating whether such drugs can also effectively eliminate liver CSCs. Another strategy to target CSCs is to induce differentiation of liver CSCs. In this context, Yin et al. [76] have shown that the forced expression of HNF4α, a central regulator of the differentiated hepatocyte phenotype, in CSCs can induce the differentiation of CSCs into mature hepatocytes and abolish their tumorigenesis in mice, implicating a novel strategy for HCC therapy.
Conclusions
Although significant progress has been made in liver cancer stem cell research in HCC, many issues remain to be resolved, such as the identity and origin of liver CSCs. Several markers are currently used to isolate CSCs. Although CSCs thus identified are known to form tumors in a xenograft model, it is not known whether these markers coexist or whether they are present in different cells. Additionally, the origin of liver CSCs needs to be studied by a lineage tracing study. The development of single cell sorting and in vivo imaging technology, as well as the use of mouse HCC models and syngeneic transplantation, will facilitate the identification of unique and specific liver CSC markers. It is critical to elucidate the detailed mechanisms underlying the transformation of liver CSCs. Comparing the gene expression profiles between normal stem cells (HPCs) and CSCs by microarray analysis or next-generation sequencing may prove to be useful to identify the dysregulated oncogenes or tumor suppressors and signaling pathways that are involved in the transformation of liver CSCs. Finally, these findings are likely to lead to the development of new diagnostic markers and therapeutic targets for HCC.
Abbreviations
- CSC:
-
cancer stem cell
- HCC:
-
hepatocellular carcinoma
- HH:
-
Sonic Hedgehog
- HPC:
-
hepatic progenitor cell
- SP:
-
side population
- TNF:
-
tumor necrosis factor.
References
Visvader JE, Lindeman GJ: Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008, 8: 755-768. 10.1038/nrc2499.
Nowell PC: The clonal evolution of tumor cell populations. Science. 1976, 194: 23-28. 10.1126/science.959840.
Reya T, Morrison SJ, Clarke MF, Weissman IL: Stem cells, cancer, and cancer stem cells. Nature. 2001, 414: 105-111. 10.1038/35102167.
Jordan CT, Guzman ML, Noble M: Cancer stem cells. N Engl J Med. 2006, 355: 1253-1261. 10.1056/NEJMra061808.
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE: A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994, 367: 645-648. 10.1038/367645a0.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF: Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003, 100: 3983-3988. 10.1073/pnas.0530291100.
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB: Identification of human brain tumour initiating cells. Nature. 2004, 432: 396-401. 10.1038/nature03128.
O'Brien CA, Pollett A, Gallinger S, Dick JE: A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007, 445: 106-110.
Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, Weissman IL, Clarke MF, Ailles LE: Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA. 2007, 104: 973-978. 10.1073/pnas.0610117104.
Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, Chu PW, Lam CT, Poon RT, Fan ST: Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008, 13: 153-166. 10.1016/j.ccr.2008.01.013.
Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM: Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67: 1030-1037. 10.1158/0008-5472.CAN-06-2030.
Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, Shelton AA, Parmiani G, Castelli C, Clarke MF: Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007, 104: 10158-10163. 10.1073/pnas.0703478104.
Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, Jia H, Ye Q, Qin LX, Wauthier E, Reid LM, Minato H, Honda M, Kaneko S, Tang ZY, Wang XW: EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009, 136: 1012-1024. 10.1053/j.gastro.2008.12.004.
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G: ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007, 1: 555-567. 10.1016/j.stem.2007.08.014.
Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, Zhan Q, Jordan S, Duncan LM, Weishaupt C, Fuhlbrigge RC, Kupper TS, Sayegh MH, Frank MH: Identification of cells initiating human melanomas. Nature. 2008, 451: 345-349. 10.1038/nature06489.
Wu C, Wei Q, Utomo V, Nadesan P, Whetstone H, Kandel R, Wunder JS, Alman BA: Side population cells isolated from mesenchymal neoplasms have tumor initiating potential. Cancer Res. 2007, 67: 8216-8222. 10.1158/0008-5472.CAN-07-0999.
Bruix J, Boix L, Sala M, Llovet JM: Focus on hepatocellular carcinoma. Cancer Cell. 2004, 5: 215-219. 10.1016/S1535-6108(04)00058-3.
El-Serag HB, Rudolph KL: Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007, 132: 2557-2576. 10.1053/j.gastro.2007.04.061.
Chiba T, Kita K, Zheng YW, Yokosuka O, Saisho H, Iwama A, Nakauchi H, Taniguchi H: Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology. 2006, 44: 240-251. 10.1002/hep.21227.
Yin S, Li J, Hu C, Chen X, Yao M, Yan M, Jiang G, Ge C, Xie H, Wan D, Yang S, Zheng S, Gu J: CD133-positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int J Cancer. 2007, 120: 1444-1450. 10.1002/ijc.22476.
Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, Zheng BJ, Guan XY: Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007, 132: 2542-2556. 10.1053/j.gastro.2007.04.025.
Weigmann A, Corbeil D, Hellwig A, Huttner WB: Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc Natl Acad Sci USA. 1997, 94: 12425-12430. 10.1073/pnas.94.23.12425.
Corbeil D, Roper K, Hellwig A, Tavian M, Miraglia S, Watt SM, Simmons PJ, Peault B, Buck DW, Huttner WB: The human AC133 hematopoietic stem cell antigen is also expressed in epithelial cells and targeted to plasma membrane protrusions. J Biol Chem. 2000, 275: 5512-5520. 10.1074/jbc.275.8.5512.
Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, Olweus J, Kearney J, Buck DW: AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997, 90: 5002-5012.
Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL: Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci USA. 2000, 97: 14720-14725. 10.1073/pnas.97.26.14720.
Schmelzer E, Zhang L, Bruce A, Wauthier E, Ludlow J, Yao HL, Moss N, Melhem A, McClelland R, Turner W, Kulik M, Sherwood S, Tallheden T, Cheng N, Furth ME, Reid LM: Human hepatic stem cells from fetal and postnatal donors. J Exp Med. 2007, 204: 1973-1987. 10.1084/jem.20061603.
Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C: Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007, 1: 313-323. 10.1016/j.stem.2007.06.002.
Monzani E, Facchetti F, Galmozzi E, Corsini E, Benetti A, Cavazzin C, Gritti A, Piccinini A, Porro D, Santinami M, Invernici G, Parati E, Alessandri G, La Porta CA: Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur J Cancer. 2007, 43: 935-946. 10.1016/j.ejca.2007.01.017.
Zhu Z, Hao X, Yan M, Yao M, Ge C, Gu J, Li J: Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int J Cancer. 2010, 126: 2067-2078. 10.1002/ijc.24728.
Yang W, Yan HX, Chen L, Liu Q, He YQ, Yu LX, Zhang SH, Huang DD, Tang L, Kong XN, Chen C, Liu SQ, Wu MC, Wang HY: Wnt/β-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008, 68: 4287-4295. 10.1158/0008-5472.CAN-07-6691.
Akhtar K, Bussen W, Scott SP: Cancer stem cells - from initiation to elimination, how far have we reached?. Int J Oncol. 2009, 34: 1491-1503.
Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H: Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009, 457: 608-611. 10.1038/nature07602.
Zhu L, Gibson P, Currle DS, Tong Y, Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW, Gilbertson RJ: Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. 2009, 457: 603-607. 10.1038/nature07589.
Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, Golub TR, Armstrong SA: Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006, 442: 818-822. 10.1038/nature04980.
Theise ND: Gastrointestinal stem cells. III. Emergent themes of liver stem cell biology: niche, quiescence, self-renewal, and plasticity. Am J Physiol Gastrointest Liver Physiol. 2006, 290: G189-G193. 10.1152/ajpgi.00041.2005.
Dabeva MD, Alpini G, Hurston E, Shafritz DA: Models for hepatic progenitor cell activation. Proc Soc Exp Biol Med. 1993, 204: 242-252.
Wang X, Foster M, Al-Dhalimy M, Lagasse E, Finegold M, Grompe M: The origin and liver repopulating capacity of murine oval cells. Proc Natl Acad Sci USA. 2003, 100 (Suppl 1): 11881-11888. 10.1073/pnas.1734199100.
Kofman AV, Morgan G, Kirschenbaum A, Osbeck J, Hussain M, Swenson S, Theise ND: Dose- and time-dependent oval cell reaction in acetaminophen-induced murine liver injury. Hepatology. 2005, 41: 1252-1261. 10.1002/hep.20696.
Suzuki A, Zheng Y, Kondo R, Kusakabe M, Takada Y, Fukao K, Nakauchi H, Taniguchi H: Flow-cytometric separation and enrichment of hepatic progenitor cells in the developing mouse liver. Hepatology. 2000, 32: 1230-1239. 10.1053/jhep.2000.20349.
Alison MR, Golding M, Sarraf CE, Edwards RJ, Lalani EN: Liver damage in the rat induces hepatocyte stem cells from biliary epithelial cells. Gastroenterology. 1996, 110: 1182-1190. 10.1053/gast.1996.v110.pm8613008.
Lowes KN, Brennan BA, Yeoh GC, Olynyk JK: Oval cell numbers in human chronic liver diseases are directly related to disease severity. Am J Pathol. 1999, 154: 537-541. 10.1016/S0002-9440(10)65299-6.
Evarts RP, Nagy P, Marsden E, Thorgeirsson SS: A precursor-product relationship exists between oval cells and hepatocytes in rat liver. Carcinogenesis. 1987, 8: 1737-1740. 10.1093/carcin/8.11.1737.
Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, Mikaelyan A, Roberts LR, Demetris AJ, Sun Z, Nevens F, Roskams T, Thorgeirsson SS: A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006, 12: 410-416. 10.1038/nm1377.
Fang CH, Gong JQ, Zhang W: Function of oval cells in hepatocellular carcinoma in rats. World J Gastroenterol. 2004, 10: 2482-2487.
Gordon GJ, Coleman WB, Hixson DC, Grisham JW: Liver regeneration in rats with retrorsine-induced hepatocellular injury proceeds through a novel cellular response. Am J Pathol. 2000, 156: 607-619. 10.1016/S0002-9440(10)64765-7.
Dumble ML, Croager EJ, Yeoh GC, Quail EA: Generation and characterization of p53 null transformed hepatic progenitor cells: oval cells give rise to hepatocellular carcinoma. Carcinogenesis. 2002, 23: 435-445. 10.1093/carcin/23.3.435.
Becker R, Luthgens B, Oesch F, Dienes HP, Steinberg P: Ha-rasVal12 but not p53Ser247 leads to a significant neoplastic transformation rate of the putative rat liver stem cells (oval cell). Carcinogenesis. 1996, 17: 2635-2640. 10.1093/carcin/17.12.2635.
Yaswen P, Goyette M, Shank PR, Fausto N: Expression of c-Ki-ras, c-Ha-ras, and c-myc in specific cell types during hepatocarcinogenesis. Mol Cell Biol. 1985, 5: 780-786.
Knight B, Tirnitz-Parker JE, Olynyk JK: C-kit inhibition by imatinib mesylate attenuates progenitor cell expansion and inhibits liver tumor formation in mice. Gastroenterology. 2008, 135: 969-979. 10.1053/j.gastro.2008.05.077.
Knight B, Yeoh GC, Husk KL, Ly T, Abraham LJ, Yu C, Rhim JA, Fausto N: Impaired preneoplastic changes and liver tumor formation in tumor necrosis factor receptor type 1 knockout mice. J Exp Med. 2000, 192: 1809-1818. 10.1084/jem.192.12.1809.
Rountree CB, Senadheera S, Mato JM, Crooks GM, Lu SC: Expansion of liver cancer stem cells during aging in methionine adenosyltransferase 1A-deficient mice. Hepatology. 2008, 47: 1288-1297. 10.1002/hep.22141.
Rountree CB, Ding W, He L, Stiles B: Expansion of CD133-expressing liver cancer stem cells in liver-specific phosphatase and tensin homolog deleted on chromosome 10-deleted mice. Stem Cells. 2009, 27: 290-299. 10.1634/stemcells.2008-0332.
Scherer E, Hoffmann M, Emmelot P, Friedrich-Freksa M: Quantitative study on foci of altered liver cells induced in the rat by a single dose of diethylnitrosamine and partial hepatectomy. J Natl Cancer Inst. 1972, 49: 93-106.
Chiba T, Zheng YW, Kita K, Yokosuka O, Saisho H, Onodera M, Miyoshi H, Nakano M, Zen Y, Nakanuma Y, Nakauchi H, Iwama A, Taniguchi H: Enhanced self-renewal capability in hepatic stem/progenitor cells drives cancer initiation. Gastroenterology. 2007, 133: 937-950. 10.1053/j.gastro.2007.06.016.
Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY: Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008, 2: 333-344. 10.1016/j.stem.2008.02.009.
Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS: In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17: 1253-1270. 10.1101/gad.1061803.
Kasper M, Regl G, Frischauf AM, Aberger F: GLI transcription factors: mediators of oncogenic Hedgehog signalling. Eur J Cancer. 2006, 42: 437-445. 10.1016/j.ejca.2005.08.039.
Iwama A, Oguro H, Negishi M, Kato Y, Morita Y, Tsukui H, Ema H, Kamijo T, Katoh-Fukui Y, Koseki H, van Lohuizen M, Nakauchi H: Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 2004, 21: 843-851. 10.1016/j.immuni.2004.11.004.
Wang H, Pan K, Zhang HK, Weng DS, Zhou J, Li JJ, Huang W, Song HF, Chen MS, Xia JC: Increased polycomb-group oncogene Bmi-1 expression correlates with poor prognosis in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008, 134: 535-541. 10.1007/s00432-007-0316-8.
Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH: Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med. 2004, 10: 55-63. 10.1038/nm979.
Dravid G, Ye Z, Hammond H, Chen G, Pyle A, Donovan P, Yu X, Cheng L: Defining the role of Wnt/β-catenin signaling in the survival, proliferation, and self-renewal of human embryonic stem cells. Stem Cells. 2005, 23: 1489-1501. 10.1634/stemcells.2005-0034.
Sekine S, Gutierrez PJ, Lan BY, Feng S, Hebrok M: Liver-specific loss of beta-catenin results in delayed hepatocyte proliferation after partial hepatectomy. Hepatology. 2007, 45: 361-368. 10.1002/hep.21523.
Weng AP, Aster JC: Multiple niches for Notch in cancer: context is everything. Curr Opin Genet Dev. 2004, 14: 48-54. 10.1016/j.gde.2003.11.004.
Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, Nikkhah G, Dimeco F, Piccirillo S, Vescovi AL, Eberhart CG: NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010, 28: 5-16.
Qi R, An H, Yu Y, Zhang M, Liu S, Xu H, Guo Z, Cheng T, Cao X: Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003, 63: 8323-8329.
Ning L, Wentworth L, Chen H, Weber SM: Down-regulation of Notch1 signaling inhibits tumor growth in human hepatocellular carcinoma. Am J Transl Res. 2009, 1: 358-366.
Merchant AA, Matsui W: Targeting Hedgehog - a cancer stem cell pathway. Clin Cancer Res. 2010, 16: 3130-3140. 10.1158/1078-0432.CCR-09-2846.
Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS: Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66: 6063-6071. 10.1158/0008-5472.CAN-06-0054.
Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A: HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007, 17: 165-172. 10.1016/j.cub.2006.11.033.
Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR, Veelken H, Engelhardt M, Mertelsmann R, Kelleher JF, Schultz P, Warmuth M: Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med. 2007, 13: 944-951. 10.1038/nm1614.
Sicklick JK, Li YX, Jayaraman A, Kannangai R, Qi Y, Vivekanandan P, Ludlow JW, Owzar K, Chen W, Torbenson MS, Diehl AM: Dysregulation of the Hedgehog pathway in human hepatocarcinogenesis. Carcinogenesis. 2006, 27: 748-757. 10.1093/carcin/bgi292.
Todaro M, Perez Alea M, Scopelliti A, Medema JP, Stassi G: IL-4-mediated drug resistance in colon cancer stem cells. Cell Cycle. 2008, 7: 309-313. 10.4161/cc.7.3.5389.
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN: Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006, 444: 756-760. 10.1038/nature05236.
Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, Piccirillo S, Vescovi AL, DiMeco F, Olivi A, Eberhart CG: Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007, 25: 2524-2533. 10.1634/stemcells.2007-0166.
Pannuti A, Foreman K, Rizzo P, Osipo C, Golde T, Osborne B, Miele L: Targeting Notch to target cancer stem cells. Clin Cancer Res. 2010, 16: 3141-3152. 10.1158/1078-0432.CCR-09-2823.
Yin C, Lin Y, Zhang X, Chen YX, Zeng X, Yue HY, Hou JL, Deng X, Zhang JP, Han ZG, Xie WF: Differentiation therapy of hepatocellular carcinoma in mice with recombinant adenovirus carrying hepatocyte nuclear factor-4alpha gene. Hepatology. 2008, 48: 1528-1539. 10.1002/hep.22510.
Acknowledgements
We thank Dr Kalpana Ghoshal for critical review of the article and useful suggestions. The work done in the authors' laboratory was supported, in part, by NIH grants CA101956 and CA086978 (STJ).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
BW extensively reviewed the literature and prepared the first draft after discussion with SJ. SJ reviewed the draft, edited and helped to prepare the final draft.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Wang, B., Jacob, S.T. Role of cancer stem cells in hepatocarcinogenesis. Genome Med 3, 11 (2011). https://doi.org/10.1186/gm225
Published:
DOI: https://doi.org/10.1186/gm225