
Abstract
Background
More than 300,000 children are newly infected with HIV each year, predominantly through mother-to-child transmission (HIV MTCT). Identification of host genetic traits associated with transmission may more clearly explain the mechanisms of HIV MTCT and further the development of a vaccine to protect infants from infection. Associations between transmission and a selection of genes or single nucleotide polymorphisms (SNP)s may give an incomplete picture of HIV MTCT etiology. Thus, this study employed a genome-wide association approach to identify novel variants associated with HIV MTCT.
Methods
We conducted a nested case-control study of HIV MTCT using infants of HIV(+) mothers, drawn from a cohort study of malaria and HIV in pregnancy in Blantyre, Malawi. Whole genome scans (650,000 SNPs genotyped using Illumina genotyping assays) were obtained for each infant. Logistic regression was used to evaluate the association between each SNP and HIV MTCT.
Results
Genotype results were available for 100 HIV(+) infants (at birth, 6, or 12 weeks) and 126 HIV(-) infants (at birth, 6, and 12 weeks). We identified 9 SNPs within 6 genes with a P-value < 5 × 10-5 associated with the risk of transmission, in either unadjusted or adjusted by maternal HIV viral load analyses. Carriers of the rs8069770 variant allele were associated with a lower risk of HIV MTCT (odds ratio = 0.27, 95% confidence interval = 0.14, 0.51), where rs8069770 is located within HS3ST3A1, a gene involved in heparan sulfate biosynthesis. Interesting associations for SNPs located within or near genes involved in pregnancy and development, innate immunological response, or HIV protein interactions were also observed.
Conclusions
This study used a genome-wide approach to identify novel variants associated with the risk of HIV MTCT in order to gain new insights into HIV MTCT etiology. Replication of this work using a larger sample size will help us to differentiate true positive findings.
Similar content being viewed by others


Background
In sub-Saharan Africa, over 1,300,000 pregnant women were living with HIV in 2007, 73,000 of which were in the small southern country, Malawi, landlocked between Tanzania, Zambia, and Mozambique, just North of Zimbabwe [1]. More than 300,000 children were newly infected with HIV in 2007, predominantly through mother-to-child transmission (HIV MTCT) [2]. Much of the risk of HIV MTCT can be reduced by treatment with single dose nevirapine (NVP). However, in many areas, mothers and their infants do not receive such regimens, and even in the context of prophylactic treatment, some infants become infected whereas others remain free of infection. Furthermore, HIV transmission can occur during pregnancy, labor and delivery, or through breastfeeding, by mechanisms which remain to be elucidated.
There is evidence for genetic variability in the mother and/or infant to be associated with susceptibility to HIV MTCT. However, a larger wealth of research describes genetic associations with adult HIV transmission and progression to AIDS. The following paragraphs note pertinent findings for various modes of HIV transmission and disease progression.
Alteration of viral entry has been implicated for several genes. One mechanism of cell entry involves HIV-1 binding with the CD4 receptor and co-receptor chemokine (CC motif) receptor 5 (CCR5). The CCR5 co-receptor also binds with chemokines produced by CD8+ T cells, including RANTES (CCL5), and MIP (macrophage inflammatory protein) 1α (CCL3) and 1β (CCL4). Higher concentrations of these ligands have been associated with a lower risk of HIV-1 infection and progression to AIDS, likely through competition with R5 strains of HIV for binding with the CCR5 receptor, preventing HIV from entering the cell and replicating [3–8]. Genes that regulate ligands for chemokine receptor genes have been associated with the risk of HIV infection, a notable example existing for chemokine (C-C motif) ligand 3-like 1 (CCL3L1). CCL3L1 copy number lower than population average has been associated with an increased risk of HIV transmission through different modes of transmission (adult and perinatal) and across various ethnic groups [9–13]. CCL3L1 copy number variation has also been associated with HIV/AIDS progression in adults [10, 14–16].
Genes regulating co-receptor availability are also involved in HIV susceptibility. A prominent example in adults is the 32-base-pair deletion in the open reading frame of the CCR5 gene (CCR5-Δ32), where individuals homozygous for the Δ32 mutation are nearly resistant to infection by R5 strains [5–7, 17, 18]. However, the mutation does not always significantly alter susceptibility to maternal infection among infants [19]. The rarity of the Δ32 mutation in African populations [20], where HIV MTCT is more common, may account for this lack of association. It is possible that other CCR5 variations, such as the promoter polymorphisms 2459 (59029 or rs1799987) and 2135 (59353 or rs1799988), play stronger roles for HIV MTCT, when taking maternal HIV viral load into account [21]. CCR5-2132 (59356) has been noted for an increased risk of death among HIV-infected women, although the same study did not observe associations between CCR5 polymorphisms 2135 (59353), 2086 (59402 or rs1800023), and 2459 (59029 or rs1799987) and HIV MTCT [22].
Depending on the viral strain [23], HIV can use the CXC chemokine receptor 4 (CXCR4) as a co-receptor for CD4 for cell entry. Like CCR5, CXCR4 can be blocked by endogenous ligands [24, 25]. The natural ligand for CXCR4 is the stromal cell-derived factor 1 (SDF1) [26–28], encoded by SDF1 (CXCL12). SDF1-3-prime-A has been associated with a reduced risk of HIV-1 infection [24, 25], but not necessarily progression to AIDS [29, 30] or HIV MTCT in African or other ancestry groups [31, 32].
Intermediary receptors on dendritic or endothelial cells can be used by HIV-1 [33, 34], and altered susceptibility to infection may result from polymorphisms in the genes regulating such receptors. This includes Dendritic cell-specific ICAM-grabbing non-integrin (DC-SIGN) [35–38] and syndecan genes such as SDC-2 [39]. High levels of DC-SIGN mRNA in the human placenta suggests a role for DC-SIGN for in utero transmission of HIV, even in the context of low maternal viral load [34]. Syndecans may be less important alone as they are when connected with other factors such as chemokine receptors or heparan sulfate. For example, the SDC-4/CXCR4 complex binds with SDF-1 [40], which can alter HIV binding. The syndecan protein bound with heparan sulfate (proteoglycan) can also bind with gp120 of HIV-1 [41], which may facilitate HIV-1 cell entry [42] or cell-free transport [43]. There are multiple genes encoding syndecans and heparan sulfate proteglycans that remain to be clearly described in relation to HIV MTCT.
Finally, genes involved in the host immune response can play a role in HIV/AIDS susceptibility. The valine to isoleucine substitution at codon 64 in the chemokine co-receptor 2b gene (CCR2-V64I) demonstrates linkage disequilibrium with the CCR5 promoter region [44] and is common in populations of African ancestry [44–46]. The natural ligand of CCR2 is CCL2 (MCP-1), which does not bind with CCR5 or CXCR4 [47]. CCR2-V64I is associated with delayed disease progression in adults, but with variable replication [44, 48–50]. It is possible that the CCR2 gene does not individually influence HIV progression to AIDS, but rather, acts in combination with other gene polymorphisms such as the variants of CCR5, CXCR4, and possibly human leukocyte antigen (HLA) gene variants [51] in promoting or preventing infection. It has been suggested that activation of the immune system rather than receptor blockage explains the association with HIV/AIDS [47].
A variety of HLA gene variants are associated with susceptibility to HIV/AIDS in adults. This includes HLA complex P5 (HCP5) rs2395029 (in strong linkage disequilibrium with HLA-B*5701) and HLA-C rs926942 associated with HIV viral set point [52] in a genome-wide association study, HLA-Bw4 associated with a lower risk of heterosexual HIV transmission [53], and HLA-B*35 alone [54, 55] or in combination with HLA-Cw*04 [56] associated with disease progression. An epistatic interaction between HLA-B Bw4-80I and activating killer immunoglobulin-like receptors (KIR) variant KIR3DS1 has also been associated with a protection from rapid progression to AIDS [57, 58], likely through increases in natural killer cell activity, cell lysis, and subsequent reduction in viral load [57].
More pertinent to HIV MTCT are HLA variants evaluated in pregnant women or maternal-fetal polymorphism mismatches in HLA variants, which can protect infants from infection. One study found that mothers with HLA-B variants (*1302, *3501, *3503, *4402, *5001) transmitted HIV to their infant even in the context of low viral loads, whereas mothers with other variants (*4901, *5301) did not transmit the virus despite high viral loads [59]. Furthermore, mother-infant pairs discordant with regards to the HLA-G variants 3743C/T, 634C/G, or 714insG/G have been shown to experience a lower risk of HIV MTCT compared to concordant mother-child pairs [60].
The MBL2 gene plays a role in the innate immune responses to infection and encodes the mannose-binding lectin (MBL) protein [61–64]. Several MBL2 polymorphisms can result in MBL deficiency, which has been associated with increased risk of HIV MTCT [65]. Apolipoprotein B mRNA Editing Catalytic Polypeptide 3 g (APOBEC3G), inhibits HIV-1 replication [66] and is associated with disease progression in children [67]. However, the association between APOBEC3G variants in the risk of HIV MTCT has not been established.
It is possible that the genetic risk factors involved in HIV infection and disease progression in adults do not directly overlap with the HIV MTCT phenotype and that the mechanisms with genetic underpinnings for HIV MTCT await discovery. It is also likely that what we know about HIV MTCT genetic risk factors is only one piece of the puzzle. To uncover new genes associated with HIV MTCT, we conducted a whole genome scan for fetal susceptibility to maternal HIV infection using information from consenting mother-infant pairs receiving antenatal care in Blantyre, Malawi, a population with a high burden of HIV/AIDS.
Because HIV MTCT is a rare phenotype, it is difficult to ascertain thousands of cases in order to obtain adequate power for a typical genome-wide association study. However, genome-wide approaches for such a phenotype can still be fruitful for furthering our understanding of HIV MTCT etiology and for generating hypotheses. Where possible, we also report the effects of SNPs within genes known to be associated with HIV/AIDS, for the purposes of replication in our study population.
Methods
Study design and population
The study participants were a subset of a larger prospective cohort study of malaria and HIV in pregnancy [68, 69]. The cohort was conducted from 2000 to 2004 and included 3,825 consenting pregnant women admitted to Queen Elizabeth Central Hospital in Blantyre, Malawi, as previously described [69]. HIV-infected women and their infants received a single dose (200 mg) of NVP at the onset of labor or at the time of delivery, respectively. A total of 1,157 women tested positive for HIV, 884 of which delivered at Queen Elizabeth Central Hospital, resulting in 807 singleton live births. At delivery, 751 infants were tested for HIV, identifying 65 HIV positive infants at birth. Of the 686 HIV negative infants, 179 were lost to follow-up. The remaining 507 HIV negative infants were tested for HIV at 6 and 12 weeks, resulting in 89 additional HIV positive infants. Based on mother reports, 98.4% and 96.5% of infants were breast fed at 6 and 12 weeks postpartum, respectively.
In order to evaluate infant susceptibility to maternal HIV infection, a nested case control was conducted, focusing on infants of HIV positive mothers. Given that all such infants were HIV-exposed, cases were defined as infants who became HIV positive at birth, 6 weeks, or 12 weeks. Controls were defined as infants who remained HIV negative at all visits. Genotyping was performed for as many cases as possible. We first evaluated samples for sufficient DNA for genome-wide genotyping, which was obtainable for 115 of the 154 cases. Funding and supplies were only available to test an approximately 1:1 case:control ratio. We selected controls in a slightly higher than 1:1 case:control ratio, anticipating loss of samples due to insufficient DNA. A total of 203 of the 418 controls were selected using simple random selection in STATA version 10 [70], 153 of which had sufficient DNA. The controls had a similar distribution across time of enrollment as the cases. The total sample size subjected to genotyping was 268 infants (115 cases + 153 controls) of HIV positive mothers. Because the control status of subjects was designated at the beginning of sample selection for the nested case control, this study was analyzed as a case-cohort study [71]. Mothers of infants could not be genotyped as the original institutional review board approval did not include this. It was not possible to return to study participants in order to obtain informed consent for maternal genotyping. Thus, no test of transmission disequilibrium or analyses involving mother-infant pairs could be conducted. The focus was infant genomic susceptibility to HIV infection, given an HIV positive mother. The original cohort study obtained consent from study participants to collect and use samples for biological measurements including but not limited to diagnosis of disease and for genotyping. Written informed consent forms were available in both English and Chichewa, the predominant language in Malawi. This study was approved by the Malawi College of Medicine Research and Ethics Committee and by the institutional review board at the University of North Carolina at Chapel Hill. Modification of the original institutional review board approval was obtained to ensure the approval of large-scale genotyping of SNPs across the genome.
Power analysis
Power was calculated based on a genome-wide scan of approximately 587,000 SNPs, as over 68,000 SNPs were removed due to quality control. Per specifications of the software Quanto [72], power was computed using a log-additive model, varying allele frequency (10 to 30%), a baseline risk of 25% (to approximate the proportion of infants that became infected with HIV from HIV positive mothers in the genome wide association study population), a case to control ratio of 1:1, and an Bonferroni adjusted P-value of 0.05/600,000 SNPs = 1 × 10-8 to account for multiple testing. Power was estimated for varying relative risks (1.25 to 3.25).
Genotyping
Infant genotyping was performed at Duke University Genotyping Core Laboratories, by using Illumina's HumanHap650Y Genotyping BeadChip. This BeadChip enables whole-genome genotyping of over 655,000 tagSNPs derived from the International HapMap Project [73] and over 100,000 tag SNPs selected based on the Yoruban Nigerian HapMap population. The BeadChip contains over 4,300 SNPs with copy number polymorphism regions of the genome, 8,000 non-synonymous SNPs, 1,800 tag SNPs in the major histocompatibility complex important for immunological relevance, 177 mitochondrial SNPs, and 11 Y-chromosome SNPs.
Quality control
The quality control for genotyping error was performed at Duke University Genomic Laboratories as previously described [52]. Briefly, all samples were brought into a BeadStudio data file and clustering of samples was evaluated in order to determine random clustering of SNPs. Samples with very low call rates (<95%) or insufficient DNA concentration were excluded. Subsequent reclustering of undeleted SNPs and additional exclusion by call rate was performed [52]. SNPs with a Het Excess value between -1.0 to -0.1 and 0.1 to 1.0 were evaluated to determine if raw and normalized data indicated clean calls for the genotypes [52].
Statistical quality control was performed at the University of North Carolina at Chapel Hill. Individuals missing more than 10% of marker data, SNPs missing more than 10% of genotypes, SNPs with a minor allele frequency (MAF) ≤ 0.01, and SNPs out of Hardy-Weinberg equilibrium (HWE) (P < 0.001) in the control group were excluded. Gender verification was completed for all subjects to ensure that gender recorded in the covariate dataset matched with gender based on genetic data. For mismatched or missing gender, gender was imputed based on the X chromosome (N = 9). Related individuals were identified by first estimating identity by descent (IBD). A minimal list of individuals with estimated genome-wide IBD proportions > 0.05 with at least one included subject were removed (N = 5). Statistical quality control was performed in PLINK version 1.05 [74]. Analyses were run without exclusions due to HWE in order to assess the difference in results.
Statistical analysis
Assuming an additive genetic model, logistic regression was performed where the outcome of interest was HIV status of the infant (positive or negative). The null hypothesis was that the SNP of interest was not associated with HIV MTCT: Ho: β1 = 0, compared to the alternative hypothesis, that the SNP was associated with HIV MTCT: Ha: β1 ≠ 0. All SNPs were assumed to be independent, and Bonferroni correction was used to adjust for multiple testing. Odds ratios (ORs) were obtained to approximate the risk ratios. These statistical analyses were conducted in PLINK version 1.05 [74] and the results were visualized in WGAViewer version 1.26F [75].
Logistic regression was adjusted for maternal viral load (MVL), as it is a key risk factor for HIV MTCT. MVL could not be evaluated for effect measure modification because of the small sample size. Logistic regression results were presented for both unadjusted and MVL adjusted analyses. We also investigated maternal syphilis for significant confounding, although the number of infants of HIV positive mothers who also had syphilis was small (N = 20). We did not evaluate maternal malaria for confounding as it was not associated with the outcome, HIV MTCT [68, 69]. In order to evaluate population stratification, principal components analysis was performed by using EIGENSOFT version 2.0 [76, 77]. Principal component(s) (PCs) were then evaluated for association for SNPs associated with HIV MTCT. PCs were determined to represent potential confounders if they were associated with both the SNP of interest and HIV MTCT. If necessary, logistic regression was repeated adjusting for confounding PCs.
In order to evaluate the consistency of associations by mode of transmission, we evaluated each SNP for association with intrauterine and intrapartum transmission. Intrauterine transmission was estimated by infant HIV status at birth. Intrapartum transmission was assigned to infants who were HIV negative at birth but who became HIV positive at week 6. Transmission through breastfeeding was estimated at week 12. For each mode of transmission, the results for SNPs within key genes previously associated with HIV/AIDS were summarized.
Results
Quality control and power analysis
A total of 246 infants (114 cases, 132 controls; 116 males, 121 females, 9 with imputed gender) passed laboratory quality control. Statistical quality control removed 15 individuals for low genotyping and 5 who had estimated genome-wide IBD proportions > 0.05 with at least one included subject. This resulted in a total of 226 individuals (100 cases, 126 controls; 112 males, 114 females). Of the 655,352 SNPs tested, 68,671 failed statistical quality control due to HWE P < 0.001 in the controls (N = 425), low genotyping rate (N = 21,589), or for MAF <0.01 (N = 53,477), where some overlap of SNPs across exclusion criteria existed. Results are summarized for 586,681 SNPs.
No evidence of population stratification was present (Eigen value range: 0.817 to 1.20, mean = 0.995, genomic inflation factor based on median χ2 = 1.023, mean χ2 = 1.013). The power analyses estimated that with a P-value of 1 × 10-8, a baseline risk of 25%, and an allele frequency of 10%, our power was ≤32% and 58% for a relative risk (RR) of ≤3.0 and 3.5, respectively. For an allele frequency of 20%, this changed to 10%, 50%, 85%, and 97%, for RR = 2.0, 2.5, 3.0, and 3.5, respectively. And for an allele frequency of 30%, this changed to 22%, 75%, 96%, and 99%, for RR = 2.0, 2.5, 3.0, and 3.5, respectively. This implies that our genome-wide association dataset with a sample size of 226 is powered to detect large effects of very common variants, but underpowered to detect small effects of rare variants. Because additional cases could not be obtained, we were unable to increase sample size in order to boost power. Rather, limited genome-wide statistical significance was noted.
Association results
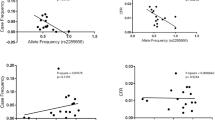
Although no genome-wide significant SNPs were detected (P < 1 × 10-7), we identified nine SNPs within six genes with a P-value < 5 × 10-5 in either unadjusted analyses and/or analyses adjusted by MVL (Table 1). Adjustment by maternal syphilis made little impact on the effect estimates or statistical significance (data not shown). Several of the 50 most significant SNPs were located within interesting genes, including 7 SNPs near or within genes involved in pregnancy and development (Table 2). An additional 7 SNPs were located near or within genes with immunological function and/or HIV-1 protein interactions (Table 3). One of the top SNPs corresponding to functional interest was rs8069770, located within the gene heparan sulfate (glucosamine) 3-O-sulfotransferase 3A1 (HS3ST3A1; Figure 1). Analyses run including SNPs out of HWE in the control group gave similar results (data not shown). None of the ten PCs evaluated were associated with rs8069770 (P = 0.763, 0.977, 0.715, 0.447, 0.320, 0.714, 0.523, 0.958, 0.696, 0.902). Thus, subsequent adjustment by PCs was not necessary.

Map of the HS3ST3A1 gene on chromosome 17. Position and -log(p) of SNPs in the region are displayed, including the SNP rs8069770 with the highest -log(p). Triangle display of linkage disequilibrium across SNPs corresponds to r2 estimates. Plot constructed using WGAViewer software version 1.26F.
For the top 20 most significant SNPs summarized in Table 1, we evaluated the effect estimates and statistical significance for intrauterine and intrapartum HIV transmission (Additional file 1). We were unable to include results for transmission through breastfeeding because the outcome was too rare. For all SNPs described, the direction of effect (higher risk or lower risk of HIV transmission) was consistent across mode of transmission (Additional file 1). The results for SNPs within 10 kb of key genes of interest were also reported (Additional file 2). We were unable to report results specific to the marker for the CCL3L1 copy number variation, rs72248989, but we report the effects of SNPs in this region (Additional file 2).
Discussion
We conducted a genome-wide association study to identify genetic variants that may influence HIV MTCT. Although limited by sample size and the power to detect genome-wide statistical significance, we were powered to detect large genetic effects for common variants (effect estimate >3.0, MAF >20% or effect estimate >2.5, allele frequency >25%). No such genome-wide statistically significant genetic effects were detected. Nonetheless, several findings were notable and may offer supportive data for other studies of the genetics of HIV MTCT.
Several SNPs with biological significance were noted. One of these is the SNP rs8069770, located within the gene HS3ST3A1. This gene encodes the enzyme 3-O-sulfotransferase, which catalyzes the biosynthesis of a specific subtype of heparan sulfate (HS), 3-O-sulfated heparan sulfate. This HS subtype has specific functional significance for herpes simplex virus-1 [78, 79]. Although HS has been shown to be involved in HIV infection [80–83], to our knowledge, no sub-type-specific investigations of HS have been conducted for association with HIV MTCT. Furthermore, HIV-1 virus [41, 84] and the chemokine RANTES [41, 85, 86] have been noted to bind to syndecans, which are core transmembrane proteins capable of carrying HS [87]. It is possible that specific or multiple components of HS proteoglycans, which consist of the bound core protein attached to HS, are involved in HIV MTCT. We suggest two possible mechanisms: the attachment of HS proteoglycans to HIV could prevent the virus from crossing the placenta and possibly facilitate viral sequestration in the placenta; or, HS proteoglycans binding with RANTES could leave CCR5 receptors available to bind with HIV virus and facilitate transmission across the placenta. The former mechanism would agree with the direction of effect we observed for rs8069770. However, much more research is needed in order to more clearly develop mechanistic hypotheses involving HS, at both the genetic level regulating the biosynthesis of HS subtypes, and at the protein level. We observed that the frequency of the minor allele of rs8069770 among cases/controls was similar across transmission type: case/control frequencies were 0.07/0.19, 0.07/0.16, and 0.09/0.18 for cumulative HIV MTCT, intrauterine transmission, and intrapartum transmission, respectively. The direction of effect was also consistent across transmission category (Additional file 1), suggesting that the mechanism may not be specifically localized to the placenta.
Two SNPs were located within genes involved in embryonic development in animal models [88]: rs12306 (P = 3.29 × 10-5) within the WD repeats and SOCS box-containing 1 (WSB1) gene, and rs1433666 (P = 0.0001) within the Glutamate receptor, ionotropic, delta 2 (GRID2) gene. The role of WSB1 in human embryonic development or in the risk of HIV MTCT is not well described. GRID2 has been noted as a large region of genomic instability (fragile site) and has been associated with cancer and neural development [89, 90]. Subsequent studies of these genes in humans would be valuable, in particular for probing roles in viral infection.
There were two SNPs (rs216743 and rs216744) with P-values < 7 × 10-5 identified in the cAMP response element binding protein 5 gene (CREB5). The CREB5 product is part of the CRE (cAMP response element)-binding protein family. One member of this family, CRE-BP1, is involved in mediating the adenovirus E1A-induced trans-activation [91]. CREB5 has also been noted to serve as an integration site for xenotropic murine leukemia virus-related virus (XMRV) in prostate cancer tissue from patients homozygous for a reduced activity variant of the antiviral enzyme RNase L [92]. Another SNP, rs1358594 (P = 0.0003), was of interest as it is within IL8, which mediates inflammatory response to HIV-1 infection [88]. Six other SNPs were found within genes that play a role in HIV infection. This may be suggestive of similar roles for such genes in HIV MTCT.
The Illumina 650Y BeadChip methodology provides genotypes of predominantly biallelic SNPs that are approximately evenly spaced across the genome rather than selected based on known functional significance. This limited our ability to replicate associations between some regions of interest (that is, CCR5) and HIV MTCT in this study. We were also unable to directly evaluate some key copy number variations (that is, CCL3L1) for association with HIV MTCT. However, we do describe the results for SNPs within 10 kb of the key genes associated with HIV/AIDS, including the association between SNPs close to the marker for the CCL3L1 copy number variation rs71148989 (Additional file 2). Our small sample size may also have limited our ability to detect statistically significant associations in some regions of interest, in particular for small effects.
We did not describe the most statistically significant SNPs (potentially different sets of top SNPs) by mode of transmission because of the small number of cases by transmission type. Rather, we compare the results for top SNPs from cumulative HIV MTCT analyses across other modes of transmission (intrauterine/intrapartum; Additional file 1) to assess consistency. Because the number of transmission events through breastfeeding was very rare (N = 10), we were unable to report the associations specific to postpartum transmission. We observed consistent direction of effects (higher/lower risk of HIV MTCT) across mode of transmission, which suggests that the effects of the top SNPs are not specific to biological events taking place in utero. However, for some SNPs, the strength of effect differed across transmission type. For example, rs5934013 of FERM and PDZ domain containing 4 (FRMPD4) was associated with a higher risk of HIV MTCT (MVL-adjusted OR = 4.09, 95% confidence interval (CI) = 2.08, 8.06), also found for intrauterine transmission (MVL-adjusted OR = 1.83, 95% CI = 0.96, 3.47), and intrapartum transmission (MVL-adjusted OR = 3.39, 95% CI = 1.46, 7.85). The stronger effect size for intrapartum compared to intrauterine transmission is interesting, possibly useful for developing mechanistic hypotheses, but warrants caution with interpretation due to sample size.
We previously noted that all mothers in the study received NVP, in accordance with the HIVNET 012 protocol [93]. This may limit the generalizability of our findings to populations with different drug treatment or with no drug treatment during pregnancy or after delivery. It may also have limited our ability to replicate or identify novel SNP associations with HIV MTCT that are only present in the absence of treatment. However, because NVP treatment was administered to all subjects, this study may more clearly illustrate the genetic effects that are strong enough to maintain association with HIV MTCT even in the context of NVP. Such effects may be of greater interest for therapeutic applications or for pharmacogenomic research efforts.
Due to the nature and frequency of this rare HIV MTCT phenotype, we were unable to ascertain a sufficient number of cases to be powered to establish genome-wide statistical significance. However, this study did provide some new insights into the genetics of HIV MTCT and aims to facilitate future genetic studies for this phenotype.
Conclusions
This study evaluated over 586,000 SNPs for association with HIV MTCT in a set of HIV-exposed infants from Blantyre, Malawi. Although we were unable to detect genome-wide statistically significant effects, several SNPs with P-values < 5 × 10-5 with biological significance were noted. Replication of this work using a larger sample size will help us to differentiate true positive findings.
Abbreviations
- CI:
-
confidence interval
- CRE-BP:
-
cAMP response element-binding protein
- HLA:
-
human leukocyte antigen
- HS:
-
heparan sulfate
- HWE:
-
Hardy-Weinberg equilibrium
- IBD:
-
identity by descent
- MAF:
-
minor allele frequency
- MTCT:
-
mother-to-child transmission
- MVL:
-
maternal viral load
- NVP:
-
nevirapine
- OR:
-
odds ratio
- RANTES:
-
regulated upon activation, normal T cell expressed and secreted
- PC:
-
principal component
- RR:
-
relative risk
- SNP:
-
single nucleotide polymorphism.
References
Sub-Saharan Africa: Comprehensive Indicator Report. [http://hivinsite.ucsf.edu/global?page=cr09-00-00&post=20&cid=AOX]
UNAIDS: Report on the Global AIDS Epidemic: 2008. 2008, Geneva: Joint United Nations programme on HIV/IADS (UNAIDS) and the World Health Organization
Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA: CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996, 272: 1955-1958. 10.1126/science.272.5270.1955.
Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J: The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996, 85: 1135-1148. 10.1016/S0092-8674(00)81313-6.
Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR: Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996, 381: 661-666. 10.1038/381661a0.
Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW: A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996, 85: 1149-1158. 10.1016/S0092-8674(00)81314-8.
Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA: HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996, 381: 667-673. 10.1038/381667a0.
Samson M, Labbe O, Mollereau C, Vassart G, Parmentier M: Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry. 1996, 35: 3362-3367. 10.1021/bi952950g.
Arenzana-Seisdedos F, Parmentier M: Genetics of resistance to HIV infection: Role of co-receptors and co-receptor ligands. Semin Immunol. 2006, 18: 387-403. 10.1016/j.smim.2006.07.007.
Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, Murthy KK, Rovin BH, Bradley W, Clark RA, Anderson SA, O'Connell RJ, Agan BK, Ahuja SS, Bologna R, Sen L, Dolan MJ, Ahuja SK: The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005, 307: 1434-1440. 10.1126/science.1101160.
Kuhn L, Schramm DB, Donninger S, Meddows-Taylor S, Coovadia AH, Sherman GG, Gray GE, Tiemessen CT: African infants' CCL3 gene copies influence perinatal HIV transmission in the absence of maternal nevirapine. Aids. 2007, 21: 1753-1761. 10.1097/QAD.0b013e3282ba553a.
Meddows-Taylor S, Donninger SL, Paximadis M, Schramm DB, Anthony FS, Gray GE, Kuhn L, Tiemessen CT: Reduced ability of newborns to produce CCL3 is associated with increased susceptibility to perinatal human immunodeficiency virus 1 transmission. J Gen Virol. 2006, 87: 2055-2065. 10.1099/vir.0.81709-0.
Shostakovich-Koretskaya L, Catano G, Chykarenko ZA, He W, Gornalusse G, Mummidi S, Sanchez R, Dolan MJ, Ahuja SS, Clark RA, Kulkarni H, Ahuja SK: Combinatorial content of CCL3L and CCL4L gene copy numbers influence HIV-AIDS susceptibility in Ukrainian children. AIDS. 2009, 23: 679-688. 10.1097/QAD.0b013e3283270b3f.
Dolan MJ, Kulkarni H, Camargo JF, He W, Smith A, Anaya JM, Miura T, Hecht FM, Mamtani M, Pereyra F, Marconi V, Mangano A, Sen L, Bologna R, Clark RA, Anderson SA, Delmar J, O'Connell RJ, Lloyd A, Martin J, Ahuja SS, Agan BK, Walker BD, Deeks SG, Ahuja SK: CCL3L1 and CCR5 influence cell-mediated immunity and affect HIV-AIDS pathogenesis via viral entry-independent mechanisms. Nat Immunol. 2007, 8: 1324-1336. 10.1038/ni1521.
Kulkarni H, Agan BK, Marconi VC, O'Connell RJ, Camargo JF, He W, Delmar J, Phelps KR, Crawford G, Clark RA, Dolan MJ, Ahuja SK: CCL3L1-CCR5 genotype improves the assessment of AIDS Risk in HIV-1-infected individuals. PLoS One. 2008, 3: e3165-10.1371/journal.pone.0003165.
Shalekoff S, Meddows-Taylor S, Schramm DB, Donninger SL, Gray GE, Sherman GG, Coovadia AH, Kuhn L, Tiemessen CT: Host CCL3L1 gene copy number in relation to HIV-1-specific CD4+ and CD8+ T-cell responses and viral load in South African women. J Acquir Immune Defic Syndr. 2008, 48: 245-254. 10.1097/QAI.0b013e31816fdc77.
Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, Goedert JJ, Buchbinder SP, Vittinghoff E, Gomperts E, Donfield S, Vlahov D, Kaslow R, Saah A, Rinaldo C, Detels R, O'Brien SJ: Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996, 273: 1856-1862. 10.1126/science.273.5283.1856.
Zimmerman PA, Buckler-White A, Alkhatib G, Spalding T, Kubofcik J, Combadiere C, Weissman D, Cohen O, Rubbert A, Lam G, Vaccarezza M, Kennedy PE, Kumaraswami V, Giorgi JV, Detels R, Hunter J, Chopek M, Berger EA, Fauci AS, Nutman TB, Murphy PM: Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk. Mol Med. 1997, 3: 23-36.
Contopoulos-Ioannidis DG, O'Brien TR, Goedert JJ, Rosenberg PS, Ioannidis JP: Effect of CCR5-delta32 heterozygosity on the risk of perinatal HIV-1 infection: a meta-analysis. J Acquir Immune Defic Syndr. 2003, 32: 70-76.
Gonzalez E, Dhanda R, Bamshad M, Mummidi S, Geevarghese R, Catano G, Anderson SA, Walter EA, Stephan KT, Hammer MF, Mangano A, Sen L, Clark RA, Ahuja SS, Dolan MJ, Ahuja SK: Global survey of genetic variation in CCR5, RANTES, and MIP-1alpha: impact on the epidemiology of the HIV-1 pandemic. Proc Natl Acad Sci USA. 2001, 98: 5199-5204. 10.1073/pnas.091056898.
Pedersen BR, Kamwendo D, Blood M, Mwapasa V, Molyneux M, North K, Rogerson SJ, Zimmerman P, Meshnick SR: CCR5 haplotypes and mother-to-child HIV transmission in Malawi. PLoS ONE. 2007, 2: e838-10.1371/journal.pone.0000838.
John GC, Bird T, Overbaugh J, Nduati R, Mbori-Ngacha D, Rostron T, Dong T, Kostrikis L, Richardson B, Rowland-Jones SL: CCR5 promoter polymorphisms in a Kenyan perinatal human immunodeficiency virus type 1 cohort: association with increased 2-year maternal mortality. J Infect Dis. 2001, 184: 89-92. 10.1086/321006.
Doranz BJ, Grovit-Ferbas K, Sharron MP, Mao SH, Goetz MB, Daar ES, Doms RW, O'Brien WA: A small-molecule inhibitor directed against the chemokine receptor CXCR4 prevents its use as an HIV-1 coreceptor. J Exp Med. 1997, 186: 1395-1400. 10.1084/jem.186.8.1395.
Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA: The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996, 382: 829-833. 10.1038/382829a0.
Cohen OJ, Kinter A, Fauci AS: Host factors in the pathogenesis of HIV disease. Immunol Rev. 1997, 159: 31-48. 10.1111/j.1600-065X.1997.tb01005.x.
De Clercq E, Schols D: Inhibition of HIV infection by CXCR4 and CCR5 chemokine receptor antagonists. Antivir Chem Chemother. 2001, 12 (Suppl 1): 19-31.
Brelot A, Heveker N, Montes M, Alizon M: Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J Biol Chem. 2000, 275: 23736-23744. 10.1074/jbc.M000776200.
Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, Virelizier JL, Baggiolini M, Sykes BD, Clark-Lewis I: Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997, 16: 6996-7007. 10.1093/emboj/16.23.6996.
Winkler C, Modi W, Smith MW, Nelson GW, Wu X, Carrington M, Dean M, Honjo T, Tashiro K, Yabe D, Buchbinder S, Vittinghoff E, Goedert JJ, O'Brien TR, Jacobson LP, Detels R, Donfield S, Willoughby A, Gomperts E, Vlahov D, Phair J, O'Brien SJ: Genetic restriction of AIDS pathogenesis by an SDF-1 chemokine gene variant. ALIVE Study, Hemophilia Growth and Development Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC). Science. 1998, 279: 389-393. 10.1126/science.279.5349.389.
Dezzutti CS, Guenthner PC, Green TA, Cohen OJ, Spira TJ, Lal RB: Stromal-derived factor-1 chemokine gene variant is associated with the delay of HIV-1 disease progression in two longitudinal cohorts. Aids. 2000, 14: 894-896. 10.1097/00002030-200005050-00018.
Mangano A, Kopka J, Batalla M, Bologna R, Sen L: Protective effect of CCR2-64I and not of CCR5-delta32 and SDF1-3'A in pediatric HIV-1 infection. J Acquir Immune Defic Syndr. 2000, 23: 52-57.
Teglas JP, N'Go N, Burgard M, Mayaux MJ, Rouzioux C, Blanche S, Delfraissy JF, Misrahi M: CCR2B-64I chemokine receptor allele and mother-to-child HIV-1 transmission or disease progression in children. French pediatric HIV infection study group. J Acquir Immune Defic Syndr. 1999, 22: 267-271.
Soilleux EJ, Morris LS, Leslie G, Chehimi J, Luo Q, Levroney E, Trowsdale J, Montaner LJ, Doms RW, Weissman D, Coleman N, Lee B: Constitutive and induced expression of DC-SIGN on dendritic cell and macrophage subpopulations in situ and in vitro. J Leukoc Biol. 2002, 71: 445-457.
Soilleux EJ, Morris LS, Lee B, Pohlmann S, Trowsdale J, Doms RW, Coleman N: Placental expression of DC-SIGN may mediate intrauterine vertical transmission of HIV. J Pathol. 2001, 195: 586-592. 10.1002/path.1026.
Soilleux EJ, Coleman N: Transplacental transmission of HIV: a potential role for HIV binding lectins. Int J Biochem Cell Biol. 2003, 35: 283-287. 10.1016/S1357-2725(02)00132-2.
Lee B, Leslie G, Soilleux E, O'Doherty U, Baik S, Levroney E, Flummerfelt K, Swiggard W, Coleman N, Malim M, Doms RW: cis Expression of DC-SIGN allows for more efficient entry of human and simian immunodeficiency viruses via CD4 and a coreceptor. J Virol. 2001, 75: 12028-12038. 10.1128/JVI.75.24.12028-12038.2001.
Koizumi Y, Kageyama S, Fujiyama Y, Miyashita M, Lwembe R, Ogino K, Shioda T, Ichimura H: RANTES -28G delays and DC-SIGN - 139C enhances AIDS progression in HIV type 1-infected Japanese hemophiliacs. AIDS Res Hum Retroviruses. 2007, 23: 713-719. 10.1089/aid.2006.0225.
de Witte L, Bobardt M, Chatterji U, Degeest G, David G, Geijtenbeek TB, Gallay P: Syndecan-3 is a dendritic cell-specific attachment receptor for HIV-1. Proc Natl Acad Sci USA. 2007, 104: 19464-19469. 10.1073/pnas.0703747104.
Bobardt MD, Saphire AC, Hung HC, Yu X, Schueren Van der B, Zhang Z, David G, Gallay PA: Syndecan captures, protects, and transmits HIV to T lymphocytes. Immunity. 2003, 18: 27-39. 10.1016/S1074-7613(02)00504-6.
Hamon M, Mbemba E, Charnaux N, Slimani H, Brule S, Saffar L, Vassy R, Prost C, Lievre N, Starzec A, Gattegno L: A syndecan-4/CXCR4 complex expressed on human primary lymphocytes and macrophages and HeLa cell line binds the CXC chemokine stromal cell-derived factor-1 (SDF-1). Glycobiology. 2004, 14: 311-323. 10.1093/glycob/cwh038.
de Parseval A, Bobardt MD, Chatterji A, Chatterji U, Elder JH, David G, Zolla-Pazner S, Farzan M, Lee TH, Gallay PA: A highly conserved arginine in gp120 governs HIV-1 binding to both syndecans and CCR5 via sulfated motifs. J Biol Chem. 2005, 280: 39493-39504. 10.1074/jbc.M504233200.
Argyris EG, Acheampong E, Nunnari G, Mukhtar M, Williams KJ, Pomerantz RJ: Human immunodeficiency virus type 1 enters primary human brain microvascular endothelial cells by a mechanism involving cell surface proteoglycans independent of lipid rafts. J Virol. 2003, 77: 12140-12151. 10.1128/JVI.77.22.12140-12151.2003.
Bobardt MD, Chatterji U, Selvarajah S, Schueren Van der B, David G, Kahn B, Gallay PA: Cell-free human immunodeficiency virus type 1 transcytosis through primary genital epithelial cells. J Virol. 2007, 81: 395-405. 10.1128/JVI.01303-06.
Kostrikis LG, Huang Y, Moore JP, Wolinsky SM, Zhang L, Guo Y, Deutsch L, Phair J, Neumann AU, Ho DD: A chemokine receptor CCR2 allele delays HIV-1 disease progression and is associated with a CCR5 promoter mutation. Nat Med. 1998, 4: 350-353. 10.1038/nm0398-350.
Martinson JJ, Hong L, Karanicolas R, Moore JP, Kostrikis LG: Global distribution of the CCR2-64I/CCR5-59653T HIV-1 disease-protective haplotype. Aids. 2000, 14: 483-489. 10.1097/00002030-200003310-00003.
Williamson C, Loubser SA, Brice B, Joubert G, Smit T, Thomas R, Visagie M, Cooper M, Ryst van der E: Allelic frequencies of host genetic variants influencing susceptibility to HIV-1 infection and disease in South African populations. Aids. 2000, 14: 449-451. 10.1097/00002030-200003100-00020.
Modi WS, Goedert JJ, Strathdee S, Buchbinder S, Detels R, Donfield S, O'Brien SJ, Winkler C: MCP-1-MCP-3-Eotaxin gene cluster influences HIV-1 transmission. Aids. 2003, 17: 2357-2365. 10.1097/00002030-200311070-00011.
Anzala AO, Ball TB, Rostron T, O'Brien SJ, Plummer FA, Rowland-Jones SL: CCR2-64I allele and genotype association with delayed AIDS progression in African women. University of Nairobi Collaboration for HIV Research. Lancet. 1998, 351: 1632-1633. 10.1016/S0140-6736(05)77688-1.
Easterbrook PJ, Rostron T, Ives N, Troop M, Gazzard BG, Rowland-Jones SL: Chemokine receptor polymorphisms and human immunodeficiency virus disease progression. J Infect Dis. 1999, 180: 1096-1105. 10.1086/314997.
Schinkel J, Langendam MW, Coutinho RA, Krol A, Brouwer M, Schuitemaker H: No evidence for an effect of the CCR5 delta32/+ and CCR2b 64I/+ mutations on human immunodeficiency virus (HIV)-1 disease progression among HIV-1-infected injecting drug users. J Infect Dis. 1999, 179: 825-831. 10.1086/314658.
Magierowska M, Theodorou I, Debre P, Sanson F, Autran B, Riviere Y, Charron D, Costagliola D: Combined genotypes of CCR5, CCR2, SDF1, and HLA genes can predict the long-term nonprogressor status in human immunodeficiency virus-1-infected individuals. Blood. 1999, 93: 936-941.
Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, Zhang K, Gumbs C, Castagna A, Cossarizza A, Cozzi-Lepri A, De Luca A, Easterbrook P, Francioli P, Mallal S, Martinez-Picado J, Miro JM, Obel N, Smith JP, Wyniger J, Descombes P, Antonarakis SE, Letvin NL, McMichael AJ, Haynes BF, Telenti A, Goldstein DB: A whole-genome association study of major determinants for host control of HIV-1. Science. 2007, 317: 944-947. 10.1126/science.1143767.
Welzel TM, Gao X, Pfeiffer RM, Martin MP, O'Brien SJ, Goedert JJ, Carrington M, O'Brien TR: HLA-B Bw4 alleles and HIV-1 transmission in heterosexual couples. Aids. 2007, 21: 225-229. 10.1097/QAD.0b013e3280123840.
Gao X, Nelson GW, Karacki P, Martin MP, Phair J, Kaslow R, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, O'Brien SJ, Carrington M: Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N Engl J Med. 2001, 344: 1668-1675. 10.1056/NEJM200105313442203.
Gao X, Bashirova A, Iversen AK, Phair J, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, Altfeld M, O'Brien SJ, Carrington M: AIDS restriction HLA allotypes target distinct intervals of HIV-1 pathogenesis. Nat Med. 2005, 11: 1290-1292. 10.1038/nm1333.
Carrington M, Nelson GW, Martin MP, Kissner T, Vlahov D, Goedert JJ, Kaslow R, Buchbinder S, Hoots K, O'Brien SJ: HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science. 1999, 283: 1748-1752. 10.1126/science.283.5408.1748.
Martin MP, Gao X, Lee JH, Nelson GW, Detels R, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, Trowsdale J, Wilson M, O'Brien SJ, Carrington M: Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet. 2002, 31: 429-434.
Qi Y, Martin MP, Gao X, Jacobson L, Goedert JJ, Buchbinder S, Kirk GD, O'Brien SJ, Trowsdale J, Carrington M: KIR/HLA pleiotropism: protection against both HIV and opportunistic infections. PLoS Pathog. 2006, 2: e79-10.1371/journal.ppat.0020079.
Winchester R, Pitt J, Charurat M, Magder LS, Goring HH, Landay A, Read JS, Shearer W, Handelsman E, Luzuriaga K, Hillyer GV, Blattner W: Mother-to-child transmission of HIV-1: strong association with certain maternal HLA-B alleles independent of viral load implicates innate immune mechanisms. J Acquir Immune Defic Syndr. 2004, 36: 659-670. 10.1097/00126334-200406010-00002.
Aikhionbare FO, Kumaresan K, Shamsa F, Bond VC: HLA-G DNA sequence variants and risk of perinatal HIV-1 transmission. AIDS Res Ther. 2006, 3: 28-10.1186/1742-6405-3-28.
Dommett RM, Klein N, Turner MW: Mannose-binding lectin in innate immunity: past, present and future. Tissue Antigens. 2006, 68: 193-209. 10.1111/j.1399-0039.2006.00649.x.
Garred P, Larsen F, Madsen HO, Koch C: Mannose-binding lectin deficiency - revisited. Mol Immunol. 2003, 40: 73-84. 10.1016/S0161-5890(03)00104-4.
Kilpatrick DC: Mannan-binding lectin and its role in innate immunity. Transfus Med. 2002, 12: 335-352. 10.1046/j.1365-3148.2002.00408.x.
Turner MW: Mannose-binding lectin (MBL) in health and disease. Immunobiology. 1998, 199: 327-339.
Boniotto M, Crovella S, Pirulli D, Scarlatti G, Spano A, Vatta L, Zezlina S, Tovo PA, Palomba E, Amoroso A: Polymorphisms in the MBL2 promoter correlated with risk of HIV-1 vertical transmission and AIDS progression. Genes Immun. 2000, 1: 346-348. 10.1038/sj.gene.6363685.
Simon JH, Gaddis NC, Fouchier RA, Malim MH: Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat Med. 1998, 4: 1397-1400. 10.1038/3987.
Singh KK, Spector SA: Host genetic determinants of HIV infection and disease progression in children. Pediatr Res. 2009, 65: 55R-63R. 10.1203/PDR.0b013e31819dca03.
Mwapasa V, Rogerson SJ, Kwiek JJ, Wilson PE, Milner D, Molyneux ME, Kamwendo DD, Tadesse E, Chaluluka E, Meshnick SR: Maternal syphilis infection is associated with increased risk of mother-to-child transmission of HIV in Malawi. Aids. 2006, 20: 1869-1877. 10.1097/01.aids.0000244206.41500.27.
Mwapasa V, Rogerson SJ, Molyneux ME, Abrams ET, Kamwendo DD, Lema VM, Tadesse E, Chaluluka E, Wilson PE, Meshnick SR: The effect of Plasmodium falciparum malaria on peripheral and placental HIV-1 RNA concentrations in pregnant Malawian women. Aids. 2004, 18: 1051-1059. 10.1097/00002030-200404300-00014.
StataCorp: Stata Statistical Software: Release 10. 2007, College Station, TX: StataCorp LP
Rothman K, Greenland S: Modern Epidemiology. 1998, Philapdelphia: Lippincott-Raven Publishers, Inc
Gauderman WJ, Morrison JM: QUANTO 1.1: A computer program for power and sample size calculations for genetic-epidemiology studies, 2006. [http://hydra.usc.edu/gxe/]
Thorisson GA, Smith AV, Krishnan L, Stein LD: The International HapMap Project Web site. Genome Res. 2005, 15: 1592-1593. 10.1101/gr.4413105.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007, 81: 559-575. 10.1086/519795.
Ge D, Zhang K, Need AC, Martin O, Fellay J, Telenti A, Goldstein D: WGAViewer: a software for genomic annotation of whole genome association studies. Genome Res. 2008, 18: 640-643. 10.1101/gr.071571.107.
Patterson N, Price AL, Reich D: Population structure and eigenanalysis. PLoS Genet. 2006, 2: e190-10.1371/journal.pgen.0020190.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D: Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006, 38: 904-909. 10.1038/ng1847.
Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG: A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999, 99: 13-22. 10.1016/S0092-8674(00)80058-6.
Liu J, Thorp SC: Cell surface heparan sulfate and its roles in assisting viral infections. Med Res Rev. 2002, 22: 1-25. 10.1002/med.1026.
Crublet E, Andrieu JP, Vives RR, Lortat-Jacob H: The HIV-1 envelope glycoprotein gp120 features four heparan sulfate binding domains, including the co-receptor binding site. J Biol Chem. 2008, 283: 15193-15200. 10.1074/jbc.M800066200.
Tyagi M, Rusnati M, Presta M, Giacca M: Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem. 2001, 276: 3254-3261. 10.1074/jbc.M006701200.
Vidricaire G, Gauthier S, Tremblay MJ: HIV-1 infection of trophoblasts is independent of gp120/CD4 Interactions but relies on heparan sulfate proteoglycans. J Infect Dis. 2007, 195: 1461-1471. 10.1086/515576.
Vives RR, Imberty A, Sattentau QJ, Lortat-Jacob H: Heparan sulfate targets the HIV-1 envelope glycoprotein gp120 coreceptor binding site. J Biol Chem. 2005, 280: 21353-21357. 10.1074/jbc.M500911200.
Saphire AC, Bobardt MD, Zhang Z, David G, Gallay PA: Syndecans serve as attachment receptors for human immunodeficiency virus type 1 on macrophages. J Virol. 2001, 75: 9187-9200. 10.1128/JVI.75.19.9187-9200.2001.
Slimani H, Charnaux N, Mbemba E, Saffar L, Vassy R, Vita C, Gattegno L: Interaction of RANTES with syndecan-1 and syndecan-4 expressed by human primary macrophages. Biochim Biophys Acta. 2003, 1617: 80-88. 10.1016/j.bbamem.2003.09.006.
Slimani H, Charnaux N, Mbemba E, Saffar L, Vassy R, Vita C, Gattegno L: Binding of the CC-chemokine RANTES to syndecan-1 and syndecan-4 expressed on HeLa cells. Glycobiology. 2003, 13: 623-634. 10.1093/glycob/cwg083.
Tkachenko E, Rhodes JM, Simons M: Syndecans: new kids on the signaling block. Circ Res. 2005, 96: 488-500. 10.1161/01.RES.0000159708.71142.c8.
Sayers EW, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Mizrachi I, Ostell J, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Yaschenko E, Ye J: Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2009, 37: D5-D15. 10.1093/nar/gkn741.
Rozier L, El-Achkar E, Apiou F, Debatisse M: Characterization of a conserved aphidicolin-sensitive common fragile site at human 4q22 and mouse 6C1: possible association with an inherited disease and cancer. Oncogene. 2004, 23: 6872-6880. 10.1038/sj.onc.1207809.
Smith DI, Zhu Y, McAvoy S, Kuhn R: Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 2006, 232: 48-57. 10.1016/j.canlet.2005.06.049.
Nomura N, Zu YL, Maekawa T, Tabata S, Akiyama T, Ishii S: Isolation and characterization of a novel member of the gene family encoding the cAMP response element-binding protein CRE-BP1. J Biol Chem. 1993, 268: 4259-4266.
Dong B, Kim S, Hong S, Das Gupta J, Malathi K, Klein EA, Ganem D, Derisi JL, Chow SA, Silverman RH: An infectious retrovirus susceptible to an IFN antiviral pathway from human prostate tumors. Proc Natl Acad Sci USA. 2007, 104: 1655-1660. 10.1073/pnas.0610291104.
Guay LA, Musoke P, Fleming T, Bagenda D, Allen M, Nakabiito C, Sherman J, Bakaki P, Ducar C, Deseyve M, Emel L, Mirochnick M, Fowler MG, Mofenson L, Miotti P, Dransfield K, Bray D, Mmiro F, Jackson JB: Intrapartum and neonatal single-dose nevirapine compared with zidovudine for prevention of mother-to-child transmission of HIV-1 in Kampala, Uganda: HIVNET 012 randomised trial. Lancet. 1999, 354: 795-802.
Online Mendelian Inheritance in Man (OMIM). [http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim]
Acknowledgements
We would like to acknowledge Kevin Shianna and David Goldstein at Duke University, Institute for Genome Sciences and Policy, for their role in the genotyping and laboratory-based quality control of the data used in this study. Funding for the genotyping was provided by the NIAID Center for Vaccine Immunology grant AI067854. Additional funding was provided by the NIH Virology Training Grant (T32 AI007419, 2007), and the Centers for Disease Control and Prevention Dissertation Award (PAR 07-231, 2008).
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
BRJ completed the statistical analysis, writing of the manuscript, and contributed to the intellectual content of the study. EL contributed to the statistical analysis and intellectual content. NF contributed to the intellectual content and revisions of the manuscript. VM was involved in the original cohort design and data collection. KEN was involved in the intellectual content, statistical analysis, and manuscript revisions. SRM was involved in the original cohort design and data collection, provided project mentorship, and contributed to the intellectual content and manuscript revisions.
An erratum to this article is available at http://dx.doi.org/10.1186/gm197.
Electronic supplementary material
13073_2009_138_MOESM1_ESM.DOCX
Additional file 1: A Word document giving effect estimates for top SNPs of interest, by mode of transmission. The data provided represent the genome-wide association analysis by mode of HIV transmission. (DOCX 15 KB)
13073_2009_138_MOESM2_ESM.DOCX
Additional file 2: A Word document giving effect estimates for SNPs near or within genes associated with HIV/AIDS. The data provided represent the genome-wide association analysis for specific regions that have previously demonstrated association with HIV/AIDS, described in the Introduction section. (DOCX 70 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Joubert, B.R., Lange, E.M., Franceschini, N. et al. A whole genome association study of mother-to-child transmission of HIV in Malawi. Genome Med 2, 17 (2010). https://doi.org/10.1186/gm138
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/gm138