
Abstract
In most tissues of the body, cellular ATP production predominantly occurs via mitochondrial oxidative phosphorylation of reduced intermediates, which are in turn derived from substrates such as glucose and fatty acids. In order to maintain ATP homeostasis, and therefore cellular function, the mitochondria require a constant supply of fuels and oxygen. In many disease states, or in healthy individuals at altitude, tissue oxygen levels fall and the cell must meet this hypoxic challenge to maintain energetics and limit oxidative stress. In humans at altitude and patients with respiratory disease, loss of skeletal muscle mitochondrial density is a consistent finding. Recent studies that have used cultured cells and genetic mouse models have elucidated a number of elegant adaptations that allow cells with a diminished mitochondrial population to function effectively in hypoxia. This article reviews these findings alongside studies of hypoxic human skeletal muscle, putting them into the context of whole-body physiology and acclimatization to high-altitude hypoxia. A number of current controversies are highlighted, which may eventually be resolved by a systems physiology approach that considers the time-or tissue-dependent nature of some adaptive responses. Future studies using high-throughput metabolomic, transcriptomic, and proteomic technologies to investigate hypoxic skeletal muscle in humans and animal models could resolve many of these controversies, and a case is therefore made for the integration of resulting data into computational models that account for factors such as duration and extent of hypoxic exposure, subjects' backgrounds, and whether data have been acquired from active or sedentary individuals. An integrated and more quantitative understanding of the body's metabolic response to hypoxia and the conditions under which adaptive processes occur could reveal much about the ways that tissues function in the very many disease states where hypoxia is a critical factor.
Similar content being viewed by others

Introduction
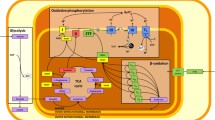
In oxidative tissues of the body, production of cellular energy, in the form of adenosine triphosphate (ATP), occurs primarily via the process of oxidative phosphorylation at the inner mitochondrial membrane. In order to sustain normal cellular function, therefore, the mitochondria require a constant supply of fuels and oxygen (Figure 1). In diseases where oxygen delivery to the peripheral tissues is impaired, through hypoxemia (for example, chronic obstructive pulmonary disease (COPD), cystic fibrosis), decreased oxygen carriage capacity (for example, anaemia), or decreased convective transport (for example, shock, heart failure), or in healthy individuals at altitude, a process of adaptation must occur to maintain cellular energy homeostasis. A compromise in cellular energetics can lead to more rapid fatigue in exercising skeletal muscle, since both crossbridge cycling at the sarcomere during contraction and calcium reuptake to the sarcoplasmic reticulum during relaxation are heavily dependent on ATP hydrolysis. Moreover, in cardiac muscle, energetic impairment has been associated with the pathogenesis of hypertrophic cardiomyopathy and sudden cardiac death [1].

Mitochondrial energy metabolism. Fatty acid β-oxidation and the TCA cycle produce NADH and FADH2, which are oxidized by complexes I and II, respectively, of the electron transport chain. Electrons are transferred through the chain to the final acceptor, O2. The free energy from electron transfer is used to pump H+ out of the mitochondria and generate an electrochemical gradient, ΔμH+, across the inner mitochondrial membrane. This gradient is the driving force for ATP synthesis via the ATP synthase.
The study of how healthy human subjects acclimatize to high altitude is a useful model in which to investigate hypoxic adaptation in the absence of the many confounding factors associated with hypoxic disease states and therapeutic interventions [2]. Indeed, many common features have been noted between COPD in particular and altitude exposure, including similar patterns of muscle wasting, weight loss, and altered cellular metabolism [3, 4]. Individual variability in the process of hypoxic adaptation and performance has been identified in healthy individuals at altitude [5], and similar mechanisms may therefore underlie the observed variations in disease progression and outcome in patients where cellular hypoxia is an important feature, in particular severe respiratory and cardiac disease and critical illness [6].
Physiological adaptations that can improve oxygen delivery in hypoxic individuals are well documented and include increased ventilation rate and cardiac output, erythropoiesis, and possibly enhanced vascularization of tissues [7]. At altitude, however, despite normal oxygen content and delivery up to 7,000 m above sea level [8], exercise capacity is dramatically reduced, and inter-individual variation in oxygen content does not correlate with exercise capacity [7]. These findings support an important role for adaptive responses to a low arterial oxygen (O2) partial pressure at the tissue level. In the hypoxic myocyte, adaptations might aim to improve local O2 delivery by redistributing mitochondria within the cell to minimize O2 diffusion gradients; to limit ATP utilization by switching off non-essential cellular functions; or to enhance ATP synthesis.
The master regulator for many of the body's adaptive responses is hypoxia-inducible factor 1 (HIF-1), a heterodimeri transcription factor comprising HIF-1α and HIF-1β subunits [9]. HIF-1α protein is continuously synthesized, and is predominantly regulated post-transcriptionally by the O2-dependent hydroxylation of two proline residues by the prolyl-hydroxylase enzymes (PHD1-3). Hydroxylation promotes binding of the von Hippel-Lindau protein (VHL), leading to ubiquitination and proteasomal degradation [9, 10]. HIF-1α protein is thus stabilized in low concentrations of O2, and accumulates spontaneously in the hypoxic cell [9]. HIF-1β is constitutively present in the nucleus, and when dimerized with HIF-1α is able to bind to hypoxia response elements (HREs) in the regulatory region of a number of genes [10], thereby activating their transcription (Figure 2). The levels of HIF-target genes are therefore precisely controlled in response to cellular O2 concentrations, and include those associated with improving O2 delivery to muscle, such as vascular endothelial growth factor (VEGF) and erythropoietin (EPO) [11], and many metabolic enzymes or regulators of metabolism, including all glycolytic enzymes, pyruvate dehydrogenase kinase 1 (PDK1), and subunit 4-2 of mitochondrial cytochrome c oxidase (COX).

Mechanisms of hypoxic adaptation. (a) In normoxia, hypoxia inducible factor-1α (HIF-1α) is degraded, following O2-dependent hydroxylation by prolyl hydroxylase (PHD) enzymes. (b) In hypoxia, HIF-1α spontaneously accumulates and combines with HIF-1β in the nucleus to activate the transcription of hypoxia-responsive genes and driving a number of metabolic adaptations: (i) BNIP3 upregulation leads to mitochondrial autophagy; (ii) a subunit switch at cytochrome c oxidase (COX), complex IV of the electron transport chain, increases the efficiency of electron (e-) transfer, and attenuates reactive oxygen species (ROS) production; (iii) glycolytic enzymes and lactate dehydrogenase (LDH) are upregulated, increasing anaerobic ATP production and lactate; (iv) pyruvate dehydrogenase kinase (PDK) enzymes are upregulated, de-activating pyruvate dehydrogenase (PDH) and limiting the conversion of pyruvate to acetyl CoA.
Regulation of mitochondrial volume and redox homeostasis
Mitochondrial volume density is consistently and substantially decreased in the skeletal muscle of climbers acclimatizing to high altitude [12, 13], and was found to be lower in the muscles of Himalayan Sherpas than those of unacclimatized lowlanders [13]. Prolonged exposure to altitude is also associated with accumulation of lipofuscin in skeletal muscle [14], a lipid peroxidation product that may be indicative of mitochondrial damage. Similarly, mitochondrial respiratory capacity is attenuated in the hearts of rats housed in hypoxia chambers [15], limiting total oxidative capacity. The primary mechanism driving the loss of mitochondrial density is likely to be mitochondrial autophagy, brought about by a HIF-1-dependent upregulation of the pro-apoptotic protein BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3) [16], which localizes on mitochondrial membranes [11, 17] (Figure 2(i)). In rat hearts, BNIP3 levels were induced after one hour of hypoxia, and were found to integrate into the mitochondria of hypoxic ventricular myocytes, leading to mitochondrial defects associated with opening of the permeability transition pore, hence causing loss of inner membrane integrity [18].
A further mechanism that restricts mitochondrial respiration in response to hypoxia was elucidated in a renal cancer cell line, where HIF-1-dependent repression of c-Myc decreased expression of the downstream factor peroxisome proliferator-activated receptor γ co-activator-1β (PGC-1β) [19]. PGC-1β, and its homolog, PGC-1α, are abundant in skeletal muscle and stimulate mitochondrial biogenesis via the activation of a number of transcriptional pathways [20, 21]. Overexpression of PGC-1α in mouse skeletal muscle leads to a proliferation of fatigue-resistant type I muscle fibers [22]. Muscle wasting is common in patients with COPD, and studies have frequently reported a preferential loss of the mitochondrial-rich type I and type IIa fibers, with a sparing of glycolytic type IIb fibers [3], which have an enhanced capacity for anaerobic metabolism. In climbers at high altitude, a similar degree of muscle wasting also occurs [23], and has been thought to contribute towards an increased muscle capillary density, although increased vascularization has not always been reported [24]. Unlike COPD patients, however, muscle wasting at altitude does not appear to be associated with the preferential loss of any particular fiber types [23]. Moreover, although a switch in skeletal muscle fiber type towards more glycolytic fibers could theoretically be driven by decreased transcriptional activity of the PGC-1 cofactors, a measurement of decreased PGC-1α levels in a muscle biopsy could in fact be secondary to a loss of the type I fibers in which they are more highly expressed.
An additional related mechanism that might restrict mitochondrial biogenesis in hypoxic skeletal muscle at altitude involves the downregulation of protein synthesis in response to energy deprivation. The AMP-activated serine/threonine protein kinase (AMPK) is a molecular energy sensor that is activated when cellular ATP levels fall, for example, during stresses such as nutrient deprivation and hypoxia [25]. Elevation of AMPK activity thereafter leads to modulation of multiple pathways in order to restore energetic homeostasis, typically involving suppression of biosynthesis and cell growth while stimulating ATP synthesis [25]. One major downstream element negatively regulated by AMPK is the mammalian target-of-rapamycin (mTOR) pathway, which regulates cell growth via activation of protein synthesis [26], and, along with PGC-1α, is a necessary component of a transcriptional complex controlling mitochondrial oxidative function [27]. In the skeletal muscle of lowlanders acclimatized to 4,559 m, levels of mTOR were decreased [28], possibly indicating a suppression of mitochondrial biogenesis via downregulation of PGC-1α transcriptional activity; however, the mechanism is almost certainly more complex than this since AMPK itself activates mitochondrial biogenesis by upregulating PGC-1α expression [29] in order to enhance skeletal muscle ATP production [30]. It is probable, given the profound loss of mitochondrial density at altitude, that the suppression of biogenesis via mTOR downregulation is the more dominant of these stimuli in hypoxic skeletal muscle; however, the consequent effect on cellular energetics both at rest and during exercise remains to be determined.
It is likely that the loss of mitochondria in hypoxic skeletal muscle is an adaptive response, which aims to minimize production of harmful reactive O2 species (ROS) such as superoxide ( ). ROS can be produced by a number of means within the cell, including generation as a by-product during mitochondrial oxidative phosphorylation [31]. The full reduction of O2 to water at complex IV of the electron transport chain, COX, requires the donation of four electrons. Donation of a single electron results in superoxide () formation, whereas hydrogen peroxide (H2O2) can be formed following the donation of two electrons to O2 and its subsequent protonation. ROS are thus generated as intermediates during the sequential donation of electrons to O2, but can also arise in the hypoxic cell due to electron leak from a highly reduced respiratory chain, with complex III implicated as a major source of this leakage [32].
). ROS can be produced by a number of means within the cell, including generation as a by-product during mitochondrial oxidative phosphorylation [31]. The full reduction of O2 to water at complex IV of the electron transport chain, COX, requires the donation of four electrons. Donation of a single electron results in superoxide () formation, whereas hydrogen peroxide (H2O2) can be formed following the donation of two electrons to O2 and its subsequent protonation. ROS are thus generated as intermediates during the sequential donation of electrons to O2, but can also arise in the hypoxic cell due to electron leak from a highly reduced respiratory chain, with complex III implicated as a major source of this leakage [32].
A role for ROS in hypoxic signaling and adaptation has been proposed, with oxidative stress increasing HIF stabilization in cells [32]. The hypoxic mitochondrion might therefore bring about its own destruction via the resulting increase in autophagy, thereby maintaining sufficient O2 supply to the remaining mitochondrial population and thus minimizing ROS production. Correspondingly, PGC-1α activity appears to be tightly coupled to HIF-1 activity in skeletal muscle cells, with an increase in PGC-1α-driven mitochondrial biogenesis causing increased O2 consumption and intracellular hypoxia, leading to HIF-1 stabilization [33]. By shifting the balance between the competing responses of mitochondrial biogenesis and mitochondria-specific autophagy, cellular mitochondrial density, and thus O2 demand, is closely matched to O2 supply.
A further HIF-1-mediated response to hypoxia that restores redox homeostasis occurs at COX, and might also improve cellular energetics. A switching of subunit 4 (COX4) at this complex occurs via the HIF-1-dependent transcription of a COX4-2 subtype, and a mitochondrial protease, LON, which degrades COX4-1 [34]. This subunit switch increases the efficiency of electron donation to O2 at COX under hypoxic conditions, minimizing electron leakage and ROS production at complexes I and III [34] (Figure 2(ii)). This switch would have an additional energetic benefit, as increased proton pumping into the mitochondrial intermembrane space would arise from a decreased electron leak, and thus enhance the efficiency of ATP synthesis for a given O2 consumption.
Despite a loss of mitochondria, there is some evidence that successful acclimatization to high-altitude hypoxia over a period of several weeks can result in normal skeletal muscle energetics at rest and following an exercise challenge [35]. The mechanisms by which a depleted mitochondrial population might be able to generate adequate quantities of high-energy phosphates to support normal cellular function are of direct relevance to basic and clinical physiology in a number of fields, including respiratory, cardiovascular, and fetal medicine; yet these mechanisms, which may involve enhanced function of the remaining mitochondria or increased non-mitochondrial ATP production, remain incompletely resolved.
Substrate switches and anaerobic metabolism
A greater contribution of anaerobic glycolysis to ATP production, particularly during an exercise challenge, is a tempting solution to the problem of maintaining energy homeostasis in hypoxic skeletal muscle. Indeed, a number of genes encoding glycolytic enzymes, including aldolase, phosphoglycerate kinase 1, pyruvate kinase, phosphofructokinase, enolase, and lactate dehydrogenase, have HREs in their regulatory regions [36] (Figure 2(iii)). Furthermore, the glucose transporter, GLUT1, which mediates non-insulin-stimulated glucose uptake by heart and skeletal muscle, is upregulated in hypoxia in a HIF-dependent manner [37], and protein levels of both GLUT1 and the insulin-stimulated glucose transporter, GLUT4, were increased in hypoxic rat skeletal muscle, although mRNA levels were unchanged [38]. Curiously, no HRE has been identified in the GLUT4 gene, although its expression patterns correlate with HIF-1 activity, perhaps suggesting that it is indirectly regulated by HIF-1α [39].
Measurement of metabolic enzyme activities in the skeletal muscle of rats housed in hypoxic-hypobaric chambers has suggested that shifts towards glycolysis are dependent on muscle type as well as activity levels [40]. For example, altitude simulation increased hexokinase activity in soleus and plantaris, whereas lactate dehydrogenase activity increased in plantaris alone [40]. In addition, decreased activity of the fatty acid β-oxidation enzyme hydroxyacyl-CoA dehydrogenase was seen in soleus, though not in plantaris [40], although this may be secondary to an overall loss of mitochondrial mass.
In addition to an enhanced capacity for glycolysis, an active shunting of pyruvate, the end-product of glycolysis, towards lactate production and away from oxidative metabolism in the mitochondrion appears to be upregulated in hypoxia. Induction of PDK1 by HIF-1 deactivates pyruvate dehydrogenase (PDH) [41, 42], preventing the conversion of pyruvate into acetyl-CoA (Figure 2(ii)). Two-dimensional difference in-gel electrophoresis (2D-DIGE) and mass spectrometry analysis of gastrocnemius muscle from chronically hypoxic rats showed downregulation of proteins involved in the tricarboxylic acid (TCA) cycle, ATP production, and electron transport, with upregulation of HIF-1α, glycolytic enzymes and PDK1 [43]. This exclusion of pyruvate from mitochondrial oxidation occurs alongside a HIF-1-dependent upregulation of lactate dehydrogenase [40], which converts pyruvate to lactate. The transport of lactate out of the muscle cell is primarily mediated by the monocarboxylate transporters 1 and 4 (MCT1 and MCT4). MCT4, but not MCT1, was found to be upregulated in hypoxia via HIF-1 induction in a human uterus cancer cell line (HeLa) [44]. Although skeletal muscle levels of neither MCT1 nor MCT4 increased in lowlanders acclimatized to 4,100 m [45], exercise in hypoxia increased muscle mRNA levels of MCT1 [46]. Together with a decrease in mitochondrial density, these studies suggest an active Pasteur effect in which glycolytically derived lactate is expelled from the hypoxic cell [47]. Human biopsy studies at altitude, however, have been less conclusive.
Increased activity of hexokinase was measured in vastus lateralis of human subjects after 3 weeks' residence at 4,300 m, yet decreased activity of phosphofructokinase was also noted, although this had also been recorded within 4 hours of arrival at altitude and may not form part of a longer-term adaptive response [35]. A similar study, however, showed no change in activities of glycogen phosphorylase, hexokinase, lactate dehydrogenase, or malate dehydrogenase in vastus lateralis after 18 days at 4,300 m [48]. Biopsies taken from climbers returning from ascents above 8,000 m, or undergoing simulated ascents over 8,000 m in a hypobaric chamber, showed decreased activities of mitochondrial enzymes [49], including citrate synthase [49, 50], succinate dehydrogenase [49], malate dehydrogenase [50], COX [50], and 3-hydroxyacyl-CoA dehydrogenase [50], although again, these changes may simply represent loss of mitochondrial mass. A dramatic decrease in hexokinase activity in sedentary subjects undergoing a simulated chamber ascent [49] contrasted with the same investigators' findings at 4,300 m [35], perhaps suggesting a combined effect of hypoxia and physical activity. Gel electrophoresis and mass spectrometry of vastus lateralis biopsies taken at sea level and after 7 to 9 days' exposure to 4,500 m, showed downregulation of TCA cycle and oxidative phosphorylation enzymes, but with no change in HIF-1α or PDK1 levels [28]. Moreover, decreased levels of the protein synthesis marker mTOR suggested a global repression of transcription at this early stage of acclimatization, perhaps as part of a program to limit ATP consumption [28]. Indeed, tissue hypoxia induces a specific pattern of chromatin modifications that appear to decrease transcriptional activity independent of HIF-1 regulation or cell type [51].
An apparent, and perhaps counterintuitive, blunting of glycolysis upon acclimatization to chronic hypoxia has also been suggested by the so-called 'lactate paradox' [52]. Acute exposure to high altitude is accompanied by greater blood lactate levels ([Lab]) at any given submaximal exercise workload than in normoxia, although peak [Lab] remains unchanged. In subjects who have acclimatized to altitude over a period of more than 3 weeks, however, exercise at the same absolute workload and maximal exercise result in lower [Lab] compared with the same subjects exercising in the unacclimatized state [35]. This phenomenon, initially seen as paradoxical, suggested that ATP production in chronic hypoxia is perhaps not dependent on increased anaerobic glycolysis but rather that mitochondrial ATP production becomes 'better tuned' to the hypoxic state. Recent studies, however, have suggested that the lactate paradox may only be a transient feature of hypoxic adaptation at altitude, disappearing in lowlanders over durations greater than 6 weeks at altitudes above 5,000 m [53, 54]. Moreover, a reduction in the capacity of muscles to produce lactate following acclimatization has not always been demonstrated [55].
Whether the lactate paradox arises from decreased muscle lactate production due to altered substrate preference, altered lactate handling via MCT1 and MCT4 at the muscle, or a better coupling of pyruvate production and oxidation at the mitochondria, remains to be resolved, along with a clear profile of the conditions under which it occurs. Intriguingly, metabolomic analysis of placentas from high-altitude births showed a blunting of lactate production following a labored delivery compared with sea-level placentas [56], suggesting that an analogous phenomenon to the lactate paradox may occur in tissues other than muscle in response to acute metabolic stress in chronic hypoxia.
Metabolic efficiency in chronic hypoxia
One further possible solution to the problem of maintaining energetic homeostasis in chronic hypoxia might be to alter metabolic pathways to maximize the yield of ATP per mole of O2 consumed. As already discussed, an improvement in O2 efficiency could be achieved by minimizing electron leakage via COX subunit switching. A further increase in oxygen efficiency could be achieved via a switch in substrate preference towards more oxygen-efficient fuels (for example, glucose instead of fatty acids). For instance, stoichiometric calculations predict that complete oxidation of palmitate yields 8 to 11% less ATP per mole of O2 than that of glucose [57]. A metabolic switch towards the exclusive oxidation of carbohydrate is, however, unlikely and unsustainable due to limited muscle and liver glycogen stores and a need to preserve glucose for the brain, which cannot oxidize fat.
In hypoxic epithelial cells, a dramatic reduction in levels of the fatty acid-activated transcription factor peroxisome proliferator-activated receptor (PPAR)α was mediated by HIF-1 [58]. PPARα activation increases the expression of a number of proteins associated with fatty acid oxidation, and is therefore a mechanism for a metabolic shift towards fat metabolism [59]. Downregulation of the PPARα gene regulatory pathway [60] and a number of PPARα target genes, including uncoupling protein 3 (UCP3), occurs in the hypoxic heart [61]. In muscle fibers, however, it appears that the hypoxic response may be critically mediated by an upregulation of PPARα [62], which might promote anaerobic glycolysis by de-activating PDH via the upregulation of another pyruvate dehydrogenase kinase isoform, PDK4 [62]. PPARα activation could, however, increase inefficient fatty acid oxidation. Indeed, lipid metabolism in liver, and fatty acid uptake and oxidation in skeletal muscle increased in rats exposed to 10.5% O2 for 3 months [63].
Furthermore, PPARα activation could activate mitochondrial uncoupling in hypoxic skeletal muscle by upregulation of UCP3, leading to relatively inefficient metabolism, and a recent study has shown that UCP3 is upregulated in hypoxic skeletal muscle via another PPARα-independent mechanism [64]. Mitochondrial uncoupling by UCP3 can be activated by superoxide and may be an additional mechanism of antioxidant defense in the hypoxic cell [65], but at the cost of decreased metabolic efficiency, as protons re-enter the mitochondrial matrix independent of ATP synthesis. The regulation of mitochondrial efficiency, however, may occur independently of gene transcription mechanisms, and liver mitochondria from non-hypoxic acclimatized rats were found to have an improved phosphorylation efficiency and depressed uncoupling when respiration was measured under hypoxic conditions [66].
Whether changes in oxygen efficiency in the hypoxic mitochondrion translate into altered exercise economy at the whole-body level remains controversial. A number of studies from independent groups have reported improvements in exercise economy following acclimatization of between 3% and 10% (reviewed in [67]); however, other investigations have shown no change in economy [68]. The choice of subjects and the varying methodologies of these studies may explain the discrepancies. For example, studies in which no changes in efficiency are shown have often reported data from highly trained athletes, who have high efficiency (and low UCP3 levels) at baseline [69]. Clearly, more studies are required at the tissue level to characterize whether mitochondrial efficiency is altered in chronically hypoxic skeletal muscle, and the physiological significance of this.
Future directions: towards an integrative and quantitative approach
The study of healthy humans at altitude has translational implications for many human diseases characterized by tissue hypoxia. In particular, parallels have been noted between muscle wasting and metabolic adaptation at altitude and in patients with COPD [3, 4]. While many facets of hypoxic adaptation, particularly those driven by HIF-1, have come to light over the past decade, much of this work has been carried out in cultured cells and animal models. The integrated response to hypoxic challenge in man is much less well understood and a number of controversies exist regarding the timings of such adaptations, the degree of the hypoxia in which they occur, and the tissue specificity of alterations in gene expression and metabolism.
The emergence of new technologies that enable relatively inexpensive, comprehensive and high-throughput analysis of gene expression, protein levels, and metabolic markers has the potential to contribute much to this area of research. Currently, very few investigators have applied these technologies to the study of humans at altitude and in hypoxia chambers, and only then under a limited number of hypoxic conditions and durations, and with small sample sizes. This may, at least in part, be due to the logistical difficulties of collecting and preserving biopsy samples in the high-altitude environment while preventing loss of oxygen-sensitive factors in the tissue. There is a clear need for technologists and high-altitude researchers to collaborate towards a resolution of these logistical difficulties in order to apply high-throughput analysis techniques to this area of research.
Such techniques can contribute much to our understanding of the adaptations in structure and function of cells, tissues, organs, and the whole organism in the hypoxic environment. While this review has focused primarily on the metabolic response of skeletal muscle to hypoxia, it is essential that the adaptations discussed are placed firmly within the context of the whole organism, and therefore the information generated by 'omics' technologies should, by necessity, form part of an integrative and quantitative physiological approach [70]. The use of computational models, combining experimental and theoretical methods, greatly aids the understanding of complex biological systems and the response of such systems to stress [71]; for example, the integration of metabolomic and transcriptomic data with measures of exercise efficiency and gas exchange within such a model could reveal much about how metabolic adaptations drive changes in performance at high altitude. Moreover, this approach would be particularly beneficial to physiologists concerned with hypoxic adaptation, since a powerful model could incorporate data pertaining to differing degrees of hypoxia and time courses of exposure.
A strong case is therefore emerging for a coordinated approach in study design between the many groups currently involved in altitude research worldwide, and for an increased use of open access databases to disseminate findings. The potential for accumulating a significant body of genomic, proteomic, and metabolomic data from subjects at altitude is a tremendously exciting prospect, but also one that runs the risk of occurring in a haphazard manner. Inconsistencies in study design between different groups are all too abundant in this field, with differences in ascent profiles, durations of exposure, experimental details, and timings of physiological measurements and biopsy sampling, as well as variations in subjects' genetic backgrounds, ages, fitness levels, and pre-assessed ability to perform at altitude, often muddying the interpretation of results. In addition, while comparisons between sedentary subjects in hypoxic chambers and relatively active participants in the field have been revealing, the ability to infer biological responses that might be either common or specific to these particular stresses is limited if the study design and subject selection is not appropriately coordinated.
A final consideration concerns the logistical difficulties and significant financial cost of mounting large-scale research expeditions to altitude. The benefits of comprehensive, collaborative studies that bring together researchers with a wide range of expertise, both from the established altitude research community and from other research backgrounds, are clear and greatly outweigh the potential difficulties of conducting research in this way [72]; however, the frequency at which such studies can occur is inevitably limited. It is imperative, therefore, when planning field studies at altitude, that consideration is given to the particulars of how data generated by 'omic' technologies, as well as from physiological studies, can best be incorporated into accessible and universal models of hypoxic acclimatization. Such a coordinated approach could not only prove to be a powerful means of resolving the current controversies regarding metabolic adaptation at altitude, but could reveal much about the ways in which tissues respond to hypoxia in many disease states.
Author information
Andrew Murray is a Research Councils UK academic fellow and a lecturer in Integrative Mammalian Physiology at the University of Cambridge. He is also a member of the Caudwell Xtreme Everest Research Group.
Abbreviations
- AMPK:
-
AMP-activated serine/threonine protein kinase
- ATP:
-
adenosine triphosphate
- BNIP3:
-
BCL2/adenovirus E1B 19 kDa interacting protein
- COPD:
-
chronic obstructive pulmonary disease
- COX:
-
cytochrome c oxidase
- EPO:
-
erythropoietin
- HIF:
-
hypoxia inducible factor
- HRE:
-
hypoxia response element
- [Lab]:
-
blood lactate concentration
- MCT:
-
monocarboxylate transporter
- mTOR:
-
mammalian target of rapamycin
- PDH:
-
pyruvate dehydrogenase
- PDK1/4:
-
pyruvate dehydrogenase kinase
- PGC-1α/β:
-
peroxisome proliferator-activated receptor γ co-activator 1α/β
- PHD:
-
prolyl hydroxylase
- PPAR:
-
peroxisome proliferator-activated receptor
- ROS:
-
reactive oxygen species
- TCA:
-
tricarboxylic acid
- UCP3:
-
uncoupling protein 3
- VEGF:
-
vascular endothelial growth factor
- VHL:
-
von Hippel-Lindau.
References
Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, Salmon A, Ostman-Smith I, Watkins H: Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet. 2001, 10: 1215-1220. 10.1093/hmg/10.11.1215.
Grocott M, Montgomery H, Vercueil A: High-altitude physiology and pathophysiology: implications and relevance for intensive care medicine. Crit Care. 2007, 11: 203-10.1186/cc5142.
Raguso CA, Guinot SL, Janssens JP, Kayser B, Pichard C: Chronic hypoxia: common traits between chronic obstructive pulmonary disease and altitude. Curr Opin Clin Nutr Metab Care. 2004, 7: 411-417. 10.1097/01.mco.0000134372.78438.09.
Khosravi M, Grocott M: Mountainside to bedside: reality or fiction?. Expert Rev Resp Med. 2009, 3: 561-565.
Thompson J, Raitt J, Hutchings L, Drenos F, Bjargo E, Loset A, Grocott M, Montgomery H: Angiotensin-converting enzyme genotype and successful ascent to extreme high altitude. High Alt Med Biol. 2007, 8: 278-285. 10.1089/ham.2007.1044.
Hajiro T, Nishimura K, Tsukino M, Ikeda A, Oga T: Stages of disease severity and factors that affect the health status of patients with chronic obstructive pulmonary disease. Respir Med. 2000, 94: 841-846. 10.1053/rmed.2000.0804.
Sutton JR, Reeves JT, Wagner PD, Groves BM, Cymerman A, Malconian MK, Rock PB, Young PM, Walter SD, Houston CS: Operation Everest II: oxygen transport during exercise at extreme simulated altitude. J Appl Physiol. 1988, 64: 1309-1321.
Grocott MP, Martin DS, Levett DZ, McMorrow R, Windsor J, Montgomery HE: Arterial blood gases and oxygen content in climbers on Mount Everest. N Engl J Med. 2009, 360: 140-149. 10.1056/NEJMoa0801581.
Semenza GL: Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007, 2007: cm8-10.1126/stke.4072007cm8.
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ: Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001, 292: 468-472. 10.1126/science.1059796.
Fedele AO, Whitelaw ML, Peet DJ: Regulation of gene expression by the hypoxia-inducible factors. Mol Interv. 2002, 2: 229-243. 10.1124/mi.2.4.229.
Ferretti G: Limiting factors to oxygen transport on Mount Everest 30 years after: a critique of Paolo Cerretelli's contribution to the study of altitude physiology. Eur J Appl Physiol. 2003, 90: 344-350. 10.1007/s00421-003-0923-2.
Howald H, Hoppeler H: Performing at extreme altitude: muscle cellular and subcellular adaptations. Eur J Appl Physiol. 2003, 90: 360-364. 10.1007/s00421-003-0872-9.
Martinelli M, Winterhalder R, Cerretelli P, Howald H, Hoppeler H: Muscle lipofuscin content and satellite cell volume is increased after high altitude exposure in humans. Experientia. 1990, 46: 672-676. 10.1007/BF01939930.
Zungu M, Young ME, Stanley WC, Essop MF: Expression of mitochondrial regulatory genes parallels respiratory capacity and contractile function in a rat model of hypoxia-induced right ventricular hypertrophy. Mol Cell Biochem. 2008, 318: 175-181. 10.1007/s11010-008-9867-5.
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL: Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008, 283: 10892-10903. 10.1074/jbc.M800102200.
Zhang HM, Cheung P, Yanagawa B, McManus BM, Yang DC: BNips: a group of pro-apoptotic proteins in the Bcl-2 family. Apoptosis. 2003, 8: 229-236. 10.1023/A:1023616620970.
Regula KM, Ens K, Kirshenbaum LA: Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res. 2002, 91: 226-231. 10.1161/01.RES.0000029232.42227.16.
Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL: HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007, 11: 407-420. 10.1016/j.ccr.2007.04.001.
Meirhaeghe A, Crowley V, Lenaghan C, Lelliott C, Green K, Stewart A, Hart K, Schinner S, Sethi JK, Yeo G, Brand MD, Cortright RN, O'Rahilly S, Montague C, Vidal-Pugh AJ: Characterization of the human, mouse and rat PGC1 beta (peroxisome-proliferator-activated receptor-gamma co-activator 1 beta) gene in vitro and in vivo. Biochem J. 2003, 373: 155-165. 10.1042/BJ20030200.
Puigserver P, Spiegelman BM: Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003, 24: 78-90. 10.1210/er.2002-0012.
Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM: Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002, 418: 797-801. 10.1038/nature00904.
Hoppeler H, Kleinert E, Schlegel C, Claassen H, Howald H, Kayar SR, Cerretelli P: Morphological adaptations of human skeletal muscle to chronic hypoxia. Int J Sports Med. 1990, 11 (suppl 1): S3-9. 10.1055/s-2007-1024846.
Lundby C, Pilegaard H, Andersen JL, van Hall G, Sander M, Calbet JA: Acclimatization to 4100 m does not change capillary density or mRNA expression of potential angiogenesis regulatory factors in human skeletal muscle. J Exp Biol. 2004, 207: 3865-3871. 10.1242/jeb.01225.
Shaw RJ: LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf). 2009, 196: 65-80. 10.1111/j.1748-1716.2009.01972.x.
Wullschleger S, Loewith R, Hall MN: TOR signaling in growth and metabolism. Cell. 2006, 124: 471-484. 10.1016/j.cell.2006.01.016.
Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P: mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007, 450: 736-740. 10.1038/nature06322.
Vigano A, Ripamonti M, De Palma S, Capitanio D, Vasso M, Wait R, Lundby C, Cerretelli P, Gelfi C: Proteins modulation in human skeletal muscle in the early phase of adaptation to hypobaric hypoxia. Proteomics. 2008, 8: 4668-4679. 10.1002/pmic.200800232.
Lee WJ, Kim M, Park H-S, Kim HS, Jeon MJ, Oh KS, Koh EH, Won JC, Kim M-S, Oh GT, Yoon M, Lee K-U, Park J-Y: AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARα and PGC-1. Biochem Biophys Res Commun. 2006, 340: 291-295. 10.1016/j.bbrc.2005.12.011.
Bonen A: PGC-1alpha-induced improvements in skeletal muscle metabolism and insulin sensitivity. Appl Physiol Nutr Metab. 2009, 34: 307-314. 10.1139/H09-008.
Giordano FJ: Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005, 115: 500-508.
Guzy RD, Schumacker PT: Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 2006, 91: 807-819. 10.1113/expphysiol.2006.033506.
O'Hagan KA, Cocchiglia S, Zhdanov AV, Tambuwala MM, Cummins EP, Monfared M, Agbor TA, Garvey JF, Papkovsky DB, Taylor CT, Allan BB: PGC-1alpha is coupled to HIF-1alpha-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc Natl Acad Sci USA. 2009, 106: 2188-2193. 10.1073/pnas.0808801106.
Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL: HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007, 129: 111-122. 10.1016/j.cell.2007.01.047.
Green HJ, Sutton JR, Wolfel EE, Reeves JT, Butterfield GE, Brooks GA: Altitude acclimatization and energy metabolic adaptations in skeletal muscle during exercise. J Appl Physiol. 1992, 73: 2701-2708.
Semenza GL, Roth PH, Fang HM, Wang GL: Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994, 269: 23757-23763.
Semenza GL: Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999, 15: 551-578. 10.1146/annurev.cellbio.15.1.551.
Xia Y, Warshaw JB, Haddad GG: Effect of chronic hypoxia on glucose transporters in heart and skeletal muscle of immature and adult rats. Am J Physiol. 1997, 273: R1734-1741.
Silva JL, Giannocco G, Furuya DT, Lima GA, Moraes PA, Nachef S, Bordin S, Britto LR, Nunes MT, Machado UF: NF-kappaB, MEF2A, MEF2D and HIF1-a involvement on insulin- and contraction-induced regulation of GLUT4 gene expression in soleus muscle. Mol Cell Endocrinol. 2005, 240: 82-93. 10.1016/j.mce.2005.05.006.
Bigard AX, Brunet A, Guezennec CY, Monod H: Skeletal muscle changes after endurance training at high altitude. J Appl Physiol. 1991, 71: 2114-2121.
Kim JW, Tchernyshyov I, Semenza GL, Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3: 177-185. 10.1016/j.cmet.2006.02.002.
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC: HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3: 187-197. 10.1016/j.cmet.2006.01.012.
De Palma S, Ripamonti M, Vigano A, Moriggi M, Capitanio D, Samaja M, Milano G, Cerretelli P, Wait R, Gelfi C: Metabolic modulation induced by chronic hypoxia in rats using a comparative proteomic analysis of skeletal muscle tissue. J Proteome Res. 2007, 6: 1974-1984. 10.1021/pr060614o.
Ullah MS, Davies AJ, Halestrap AP: The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem. 2006, 281: 9030-9037. 10.1074/jbc.M511397200.
Juel C, Lundby C, Sander M, Calbet JA, Hall G: Human skeletal muscle and erythrocyte proteins involved in acid-base homeostasis: adaptations to chronic hypoxia. J Physiol. 2003, 548: 639-648. 10.1113/jphysiol.2002.035899.
Zoll J, Ponsot E, Dufour S, Doutreleau S, Ventura-Clapier R, Vogt M, Hoppeler H, Richard R, Fluck M: Exercise training in normobaric hypoxia in endurance runners. III. Muscular adjustments of selected gene transcripts. J Appl Physiol. 2006, 100: 1258-1266. 10.1152/japplphysiol.00359.2005.
Simon MC: Coming up for air: HIF-1 and mitochondrial oxygen consumption. Cell Metab. 2006, 3: 150-151. 10.1016/j.cmet.2006.02.007.
Young AJ, Evans WJ, Fisher EC, Sharp RL, Costill DL, Maher JT: Skeletal muscle metabolism of sea-level natives following short-term high-altitude residence. Eur J Appl Physiol Occup Physiol. 1984, 52: 463-466. 10.1007/BF00943381.
Green HJ, Sutton JR, Cymerman A, Young PM, Houston CS: Operation Everest II: adaptations in human skeletal muscle. J Appl Physiol. 1989, 66: 2454-2461.
Howald H, Pette D, Simoneau JA, Uber A, Hoppeler H, Cerretelli P: Effect of chronic hypoxia on muscle enzyme activities. Int J Sports Med. 1990, 11 (suppl 1): S10-14. 10.1055/s-2007-1024847.
Johnson AB, Denko N, Barton MC: Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat Res. 2008, 640: 174-179.
West JB: Lactate during exercise at extreme altitude. Fed Proc. 1986, 45: 2953-2957.
Lundby C, Saltin B, van Hall G: The 'lactate paradox', evidence for a transient change in the course of acclimatization to severe hypoxia in lowlanders. Acta Physiol Scand. 2000, 170: 265-269. 10.1046/j.1365-201x.2000.00785.x.
van Hall G, Calbet JA, Sondergaard H, Saltin B: The re-establishment of the normal blood lactate response to exercise in humans after prolonged acclimatization to altitude. J Physiol. 2001, 536: 963-975. 10.1111/j.1469-7793.2001.00963.x.
van Hall G, Lundby C, Araoz M, Calbet JA, Sander M, Saltin B: The lactate paradox revisited in lowlanders during acclimatization to 4100 m and in high-altitude natives. J Physiol. 2009, 587: 1117-1129. 10.1113/jphysiol.2008.160846.
Tissot van Patot MC, Murray AJ, Beckey V, Cindrova-Davies T, Johns J, Zwerdlinger L, Jauniaux ER, Burton GJ, Serkova NJ: Human placental metabolic adaptation to chronic hypoxia, high altitude: Hypoxic pre-conditioning. Am J Physiol Regul Integr Comp Physiol. 2009.
Hinkle PC, Kumar MA, Resetar A, Harris DL: Mechanistic stoichiometry of mitochondrial oxidative phosphorylation. Biochemistry. 1991, 30: 3576-3582. 10.1021/bi00228a031.
Narravula S, Colgan SP: Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor alpha expression during hypoxia. J Immunol. 2001, 166: 7543-7548.
Gilde AJ, van Bilsen M: Peroxisome proliferator-activated receptors (PPARS): regulators of gene expression in heart and skeletal muscle. Acta Physiol Scand. 2003, 178: 425-434. 10.1046/j.1365-201X.2003.01161.x.
Huss JM, Levy FH, Kelly DP: Hypoxia inhibits the peroxisome proliferator-activated receptor alpha/retinoid × receptor gene regulatory pathway in cardiac myocytes: a mechanism for O2-dependent modulation of mitochondrial fatty acid oxidation. J Biol Chem. 2001, 276: 27605-27612. 10.1074/jbc.M100277200.
Essop MF, Razeghi P, McLeod C, Young ME, Taegtmeyer H, Sack MN: Hypoxia-induced decrease of UCP3 gene expression in rat heart parallels metabolic gene switching but fails to affect mitochondrial respiratory coupling. Biochem Biophys Res Commun. 2004, 314: 561-564. 10.1016/j.bbrc.2003.12.121.
Aragones J, Schneider M, Van Geyte K, Fraisl P, Dresselaers T, Mazzone M, Dirkx R, Zacchigna S, Lemieux H, Jeoung NH, Lambrechts D, Bishop T, Lafuste P, Diez-Juan A, Harten SK, Van Noten P, De Bock K, Willam C, Tjwa M, Grosfeld A, Navet R, Moons L, Vandendriessche T, Deroose C, Wijeyekoon B, Nuyts J, Jordan B, Silasi-Mansat R, Lupu F, Dewerchin M, et al: Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008, 40: 170-180. 10.1038/ng.2007.62.
Ou LC, Leiter JC: Effects of exposure to a simulated altitude of 5500 m on energy metabolic pathways in rats. Respir Physiol Neurobiol. 2004, 141: 59-71. 10.1016/j.resp.2004.04.001.
Lu Z, Sack MN: ATF-1 is a hypoxia-responsive transcriptional activator of skeletal muscle mitochondrial-uncoupling protein 3. J Biol Chem. 2008, 283: 23410-23418. 10.1074/jbc.M801236200.
Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD: Superoxide activates mitochondrial uncoupling proteins. Nature. 2002, 415: 96-99. 10.1038/415096a.
Gnaiger E, Mendez G, Hand SC: High phosphorylation efficiency and depression of uncoupled respiration in mitochondria under hypoxia. Proc Natl Acad Sci USA. 2000, 97: 11080-11085. 10.1073/pnas.97.20.11080.
Gore CJ, Clark SA, Saunders PU: Nonhematological mechanisms of improved sea-level performance after hypoxic exposure. Med Sci Sports Exerc. 2007, 39: 1600-1609. 10.1249/mss.0b013e3180de49d3.
Lundby C, Calbet JA, Sander M, van Hall G, Mazzeo RS, Stray-Gundersen J, Stager JM, Chapman RF, Saltin B, Levine BD: Exercise economy does not change after acclimatization to moderate to very high altitude. Scand J Med Sci Sports. 2007, 17: 281-291.
Mogensen M, Bagger M, Pedersen PK, Fernstrom M, Sahlin K: Cycling efficiency in humans is related to low UCP3 content and to type I fibres but not to mitochondrial efficiency. J Physiol. 2006, 571: 669-681. 10.1113/jphysiol.2005.101691.
Noble D: The Music of Life: Biology Beyond the Genome. 2006, Oxford: Oxford University Press
Beard DA, Bassingthwaighte JB, Greene AS: Computational modeling of physiological systems. Physiol Genomics. 2005, 23: 1-3. 10.1152/physiolgenomics.00117.2005. discussion 4.
Grocott M, Richardson A, Montgomery H, Mythen M: Caudwell Xtreme Everest: a field study of human adaptation to hypoxia. Crit Care. 2007, 11: 151-10.1186/cc5921.
Acknowledgements
The author wishes to thank Dr Mike Grocott, Professor Hugh Montgomery, Dr Denny Levett, Dr Daniel Martin, and other members of the Caudwell Xtreme Everest Research Group for many fascinating discussions on this topic.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author has no competing interests.
Authors' contributions
AJM researched and wrote the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Murray, A.J. Metabolic adaptation of skeletal muscle to high altitude hypoxia: how new technologies could resolve the controversies. Genome Med 1, 117 (2009). https://doi.org/10.1186/gm117
Published:
DOI: https://doi.org/10.1186/gm117