
Abstract
The neurexin family of cell adhesion proteins consists of three members in vertebrates and has homologs in several invertebrate species. In mammals, each neurexin gene encodes an α-neurexin in which the extracellular portion is long, and a β-neurexin in which the extracellular portion is short. As a result of alternative splicing, both major isoforms can be transcribed in many variants, contributing to distinct structural domains and variability. Neurexins act predominantly at the presynaptic terminal in neurons and play essential roles in neurotransmission and differentiation of synapses. Some of these functions require the formation of trans-synaptic complexes with postsynaptic proteins such as neuroligins, LRRTM proteins or cerebellin. In addition, rare mutations and copy-number variations of human neurexin genes have been linked to autism and schizophrenia, indicating that impairments of synaptic function sustained by neurexins and their binding partners may be relevant to the pathomechanism of these debilitating diseases.
Similar content being viewed by others


Key aspects of neurexins
Neurexins are transmembrane proteins that function primarily at the cell surface of neurons [1–3]. Neurexin variants are essential for Ca2+-dependent transmission at diverse types of excitatory and inhibitory synapses from the central and peripheral nervous system [4–8], and play additional roles in their formation and differentiation [9–14]. One of the most intensely studied features of neurexins is their ability to bind extracellularly to proteins of other synaptically connected neurons. The first and prototypical interaction partner discovered was postsynaptic neuroligin [15, 16]. However, a number of additional molecules associated with the synaptic cleft have been identified as binding partners, including neurexophilin [17–19], dystroglycan [20], LRRTM proteins [21, 22] and cerebellin [23, 24]. Neurexin isoforms bound to neuroligins, for example, can form trans-synaptic complexes at excitatory and inhibitory synapses that are involved in synapse specification, establishment, maturation and plasticity. Important from a medical point of view, impairments caused by mutations in the neurexin-neuroligin complex [25] lead to an imbalance of excitatory to inhibitory activity in neuronal circuits, which has been implicated in the pathomechanisms of autism spectrum disorders [26] and schizophrenia [27].
Gene organization and evolutionary history
There are three neurexin genes in the mammalian genome [2, 3, 28]. In addition, a member of the Caspr/paranodin/CTNAP family is named ‘neurexin 4’ for historical reasons but in fact contains a domain structure that is only more distantly related [29, 30], and is thus not included in our discussion here. Each neurexin gene encodes two major protein isoforms: the extracellularly long α-neurexin and a short β-neurexin (Figure 1). They are transcribed from independent promoters [1] but share most sequences (Figure 1). β-Neurexins differ by using specific first exons (exon 17 or 18, depending on the nrxn gene; Figure 2a) to encode an atypically long signal peptide and some unique amino-terminal residues, while the carboxy-terminal part is identical to α-neurexins [2]. The genes for neurexin 1 (nrxn1) and 3 (nrxn3) are among the largest in the mammalian genome (Table 1), stretching more than 1 Mbp in mice and humans [30, 31]. They cover nearly 0.1% of the entire human genome [31], and human nrxn3 extends over about 2% of chromosome 14 [30]. It has been suggested that the size of mammalian nrxn genes limits their expression to postmitotic cells such as neurons, or slowly dividing cells such as β-islet cells, because their transcription in rapidly dividing cells would take too long to be completed [31]. A single α-neurexin locus is also present in invertebrates, as has been shown for Drosophila melanogaster, Apis mellifera and Caenorhabditis elegans[30, 32], but the shorter β-isoform has only been confirmed for C. elegans[33]. Consistent with a rapid mitotic cycle, invertebrate neurexins are transcribed from shorter genes with smaller introns and without extensive alternative splicing (Figure 2b).

Domain organization of α-neurexins and β-neurexins. Neurexins are type I transmembrane proteins with a single path transmembrane helix (TM) that separates amino-terminal extracellular from cytosolic intracellular domains. The hallmark of neurexins is a cassette of LNS(green)-EGF(orange)-LNS(green) that is repeated three times in α-neurexin (Nrxn1α), albeit with low sequence conservation (16% identity and 27% homology). β-Neurexin (Nrxn1β) starts with its own exon that encodes a signal peptide (SP) and unique 37 histidine-rich residues (blue). The remainder is identical to the corresponding α-neurexin starting from the last LNS domain. Red symbols indicate positions of up to five canonically conserved splice sites (SS#1 to SS#5), and hexamers point to N-glycosylation sites and O-glycosylation sites. EGF, epidermal growth factor-like domain; LNS, laminin-neurexin-sex hormone binding globulin.

Genomic organization of neurexin genes. (a) Gene organization of mouse neurexins (nrxn) with exons (vertical lines) segregating introns (horizontal lines). The nrxn2 gene is 10 times smaller than nrxn1 or nrxn3 due to shorter introns but the relative positions of transcription starts for α-variants and β-variants (kinked arrows) are similar in all cases. Red numbers indicate alternatively spliced exons, while β-specific exons are in black. The first splice site (SS#1) accepts different inserts derived from combinations of two to four mini exons, whereas others such as SS#2 can also use parts of an insert sequence from one exon. (b) Vertebrate nrxn genes are up to 100 times longer than the single nrxn from invertebrates. The length ratio of Drosophila (dm nrxn) to mouse neurexins (ms nrxn) 2 and 3 is 1:10:100, respectively.
In addition to the two major α-neurexin and β-neurexin variants, vertebrate neurexin genes contain five conserved alternative splice sites in the α-neurexin coding sequence (SS#1 to SS#5) and two in β-neurexin (SS#4 and SS#5) that by permutation allow for about 3,908 possible neurexin variants. For example, the SS#1 of neurexin 1 consists of four mini-exons (2, 3, 4 and 5; Figure 2a) that can be inserted in 24 permutations [30]. In addition, some of the splice events may lead to soluble isoforms lacking the membrane-bound carboxy-terminal part of the protein [28]. Alternative splicing is a hallmark of all neurexin genes [1, 30–32, 34, 35], and has received considerable attention because binding to postsynaptic partners was found to depend on splicing events, at least partially. Some alternatively spliced exons in neurexins are more conserved than exons that are constitutively expressed [30], supporting the idea that long introns with weak splice sites and rare splice events result in higher conservation of the entire inserted DNA, often indicating functionally important protein sequences [36]. In particular, the inserted protein sequences at SS#2 and SS#4 are highly conserved and all known α-neurexin interacting proteins bind to the domains where SS#2 and SS#4 are located (see below).
A phylogenetic tree of the protein family demonstrates that neurexin 1, neurexin 2 and neurexin 3 of the same genome differ more than the same isoform between species (Figure 3). Because of that and since neurexin 1 and 3 are more closely related than either is to neurexin 2, a gene duplication has likely taken place before vertebrates evolved, and each of the three paralogous isoforms has continued to change independently. Other paralogous genes in the vicinity of the genome localization of neurexins in fact indicate an ancient large-scale segmental duplication, but a functional inter-relationship of the genes involved is not obvious [31]. Although nrxn genes differ mostly within a genome, no functional differentiation of neurexin 1, 2 and 3 has been determined so far, consistent with the observation that α-neurexins are able to replace each other in a rescue experiment [37].

Phylogenetic tree of the neurexin protein family. Dendrogram showing the phylogenetic relationships between the vertebrate and invertebrate neurexins. The tree was generated using neurexin amino acid sequences from several vertebrate species and invertebrate homologs, and a gap-free sequence alignment with GeneBee [132]. The neurexin 1 (Nrxn1) family is shown in red, neurexin 2 (Nrxn2) in blue, and neurexin 3 (Nrxn3) in green. The invertebrate sequences are shown in black. Species names and GenBank accession numbers [133] are given for each branch. Cluster distance values indicated at branches represent the amino acid differences for the particular group of sequences. Note that the more distantly related Caspr/paranodin/CTNAP family member ‘neurexin 4’ contains a different domain structure and is not included in the analysis.
Structural features and the splice-code hypothesis
α-Neurexins contain six LNS (laminin-neurexin-sex hormone binding globulin) domains with three epidermal growth factor-like (EGF) domains interspersed (Figure 1, upper panel). The shorter β-neurexins are identical to the carboxyl terminus of α-neurexins starting from αLNS6 but have a unique amino-terminal stretch of 37 histidine-rich residues (Figure 1, lower panel). All neurexins are N-glycosylated and the sequence between αLNS6 and the transmembrane region is characterized by O-glycosylation [2]. The cytosolic domains have a potential endoplasmatic retention signal, a cytoskeleton integrating protein 4.1, and a PDZ-binding motif that is required for trafficking of neurexins [38].
LNS domains in neurexins are characterized by a β-sheet sandwich built by strands β3, β8, β9 and β10, β4, β5, β6 and β7, and an adjacent two-stranded sheet of β2 and β11 (Figure 4). This core fold contains more than 50% of the domain and is structurally similar to the concanavalin A (ConA) fold family [39], although the primary protein sequences vary considerably [40–43]. Due to the family classification, LNS domains are thought to behave like glycan-binding lectins. For example, dystroglycan requires a specific glycosylation to bind to laminin LNS4-5 [44, 45], but a general function of LNS domains as lectins has not been demonstrated so far. All ConA family members bind divalent cations like Ca2+ or Zn2+, and the LNS domains of neurexin, laminin and agrin have similar Ca2+ sites at the rim of the LNS domain (Figure 4). Unlike other Ca2+-binding proteins, this Ca2+ coordination site is rigid and undergoes no conformational change upon calcium binding. Neurexin αLNS2 and αLNS6/βLNS are further distinguished by the presence of hydrophobic residues, and Ca2+ binding to this last LNS domain neutralizes the negatively charged pocket, allowing neuroligin to make mainly hydrophobic contacts with neurexin [46, 47]. Currently, binding partners are known for only αLNS2 and αLNS6/βLNS (Table 2). Interestingly, neuroligin and LRRTM, albeit having non-homologous structures, compete for the same Ca2+-binding epitope on αLNS6 [40–42, 48], while dystroglycan binds Ca2+-dependently to αLNS2 and αLNS6, which have no similar surfaces [46]. Ca2+-dependent binding apparently tolerates shape and sequence variations, while Ca2+-independent binding of neurexophilin and cerebellin requires exclusive features on αLNS2 [17] and αLNS6 + SS#4 [23, 24], respectively.

LNS domains as a versatile toolbox for protein-protein interactions. The diagram shows a ribbon structure of αLNS6 (PDB ID: 2R1D) representing the lowest common denominator of the six neurexin LNS folds; it is used here to highlight specific features among the individual domains. The fold is formed by 14 β-strands (β1 to 14), which are generally tightly connected. In αLNS6/βLNS, β10 (blue) can be displaced by an alternatively spliced insert at SS#4 (red). The synopsis also shows that positions of splice sites SS#2 (green) from αLNS2, SS#3 (orange) from αLNS4, and SS#4 from αLNS6 are all in vicinity of the corresponding Ca2+-binding site. The splice insert in SS#4 participates in Ca2+ coordination, while an insert in the SS#3 domain might prevent Ca2+ association in adjacent αLNS3. In the αLNS3 domain, the β4/β5 loop (magenta) is prolonged and can be interpreted as a permanent splice insert that interacts with the insert in SS#3. These β-loop variations individually shape each LNS domain around the Ca2+-binding site suitable to mediate specific LNS-protein or LNS-glycan interactions. LNS, laminin-neurexin-sex hormone binding globulin.
The binding of some of these proteins to αLNS2 or αLNS6 can be modified by alternative splicing that occurs in a hypervariable region in the vicinity of the Ca2+-binding site (Figure 4). While neurexophilin binds αLNS2 independently of alternative splicing [17], dystroglycan and LRRTM require a splice insert-free LNS domain [20, 48] and cerebellin binds presumably directly to the insert in SS#4 of αLNS6/βLNS [23, 24]. Splice insert dependency of neurexin/neuroligin complex formation is more complicated because neuroligins also have two splice sites, termed A and B. While all neurexins share the five splice sites, the neuroligins differ: neuroligin 1 contains splice sites A and B [16], neuroligin 2 and neuroligin 3 have only splice site A [49] and neuroligin 4 is not alternatively spliced [50]. Co-crystal data exist for the binding interface of neurexin 1αLNS6/βLNS without insert in SS#4 to neuroligin 1 and 4 [40–42], and neuroligin 3 is predicted to form similar complexes [40–42]. In contrast, the proposed binding interface of neuroligin 2 to αLNS6 differs structurally with a G500Q change from neuroligin 1 to 2, which raises the possibility that neuroligin 2 uses an alternative binding epitope [42, 51].
Affinity purification of neuroligin with the extracellular domain of β-neurexin originally suggested that only β-neurexin without an insert in SS#4 (−SS#4) binds neuroligin 1 [16]. This apparent splice insert dependency of neurexin binding to neuroligin then led to the generalized idea of a splice code that classifies specific pairings in the neurexin/neuroligin complex (for neurexins: ±SS#4; for neuroligins: ±A, ±B) according to specific roles at excitatory and inhibitory synapses [13, 15, 16, 52, 53]. Subsequently, it has been shown that also α-neurexins, even with insert in SS#4, bind to neuroligin 1(−B) [15] and neuroligins 2 and 3, albeit with lower affinity than β-neurexins [54, 55]. Biochemical experiments have now established that, with one exception discussed below, any neurexin can bind any neuroligin [54, 56] and that neurexins + SS#4 yield considerable amounts of protein complexes with neuroligins if only the incubation time is long enough [46]. This behavior can be explained by recent crystal structures of β-neurexin + SS#4 that show a remarkable displacement of the inserts at SS#4 [54, 57].
Surface plasmon resonance binding and crystal structures of the β-neurexin/neuroligin complex [40–42, 54] now suggest a dynamic rather than a static splice code, in which β-neurexin + SS#4 assumes an equilibrium between a neuroligin-inactive (non-binding; PDB ID: 2R1B) and an active form (PDB ID: 3 MW2) (Figure 5). In short-term binding studies the amount of active form may be too low for sufficient complex formation, while in overnight incubations all neurexins are transferred into the active form that binds to neuroligin [46, 53]. While all β-neurexins and all α-neurexins-SS#4 bind to all neuroligin variants [15, 46, 54–56, 58, 59], the splice code still restricts α-neurexin + SS#4 binding to neuroligin 1 + B [15], forming the exception mentioned above. Recent crystal structures of α-neurexin extracellular sequences containing the αLNS2-to-αLNS6 [55, 60] and αLNS5-to-LNS6 domains [59] eventually provided an explanation for this restriction by suggesting that the molecular switch of the insert in SS#4, necessary especially for binding of β-neurexin + SS#4 variants to neuroligin 1 + B [54], is sterically inhibited by the spatial orientation of αLNS5 and αEGF3. The fact that α-neurexins + SS#4 still bind to neuroligins without insert B suggests the presence of distorted intermediate conformations of αLNS6 + SS#4 similar to those in βLNS + SS#4/neuroligin 1 + A determined by NMR [61].

Splice insert in SS#4 causes a molecular switch. Splice insert-free βLNS-SS#4 (PDB ID: 3B3Q; left panel) can bind efficiently to neuroligin (Nlgn) and leucine-rich repeat proteins (LRRTM), which have overlapping binding epitopes. The prolonged conformation caused by an insert in SS#4 (orange/red; from PDB ID: 2R1B) blocks binding to Nlgn and LRRTM, and instead allows the binding of cerebellin (Cbln, middle panel). This structure of βLNS + SS#4 is in equilibrium with an additional conformation (PDB ID: 3 MW2), in which β10 (cyan) is replaced by part of the SS#4 insert (orange, right panel). In the latter, Nlgn and LRRTM binding is restored, while interaction with Cbln should be abolished. The diagrams were made using the actual structural coordinates and PyMOL software (Schrödinger, Mannheim, Germany).
The crystal structures of α-neurexin extracellular domains and electron microscopy studies also highlight important additional features of these molecules (Figure 6). (i) The core structure of αLNS2-to-αLNS5 is relatively rigid and does not change in the presence of Ca2+ or with an insert in SS#3 [55, 60]. Similarly, the splice insert at SS#2 is expected to prolong loop β8/β9 and should also not impact the remaining structure. In contrast, inserts at SS#1 and SS#5 are located in structurally distorted regions. While this permits inserts at SS#1 to increase the distance between αLNS1 and αLNS2 as observed [62], the putative role of a few inserted residues at SS#5 remains unclear at present. (ii) A conformational hinge between αLNS5 and αEGF3 allows a rotation of about 180°, which orients the αLNS2-to-αLNS5 core from a U-form to an elongated, active form parallel to presynaptic and postsynaptic membranes that allows binding to neuroligin [63]. (iii) The smaller β-neurexin assembles in a dense layer in a tetrameric 2:2 complex with neuroligin, while α-neurexin is highly variable in shape due to the hinges and the extended extracellular domain, which requires larger distances between complexes [64]. This scenario provides the first difference between the otherwise identical cytosolic carboxyl termini of α-neurexins and β-neurexins, as they could possibly be distinguished by their intermolecular distances. As a consequence, the spatial organization of proteins interacting with, for example, the identical PDZ-binding motif at the carboxyl terminus could be different for the two isoforms.

Structural models of α-neurexin. The diagram visualizes conformations that the extracellular domain of α-neurexin can assume. In the U-form (modeled from PDB ID: 3R05; left) only cerebellin (Cbln), neurexophilin (Nxph) and dystroglycan (DAG) might bind to LNS6 and LNS2, respectively. After rotation of about 180° in the αLNS5-αLNS6 hinge (modeled using PDB ID: 3ASI and 3R05; right), the core structure and αLNS6 become elongated and accessible to additional ligands, including neuroligins (Nlgn) and leucine-rich repeat molecules (LRRTM). The parentheses indicate the required presence (+) or absence (−) of the splice inserts in αLNS6 (SS#4) or αLNS2 (SS#2). Coordinates for αLNS1 have been modeled by sequence homology to other LNS domains because its electron density map was not resolved in the crystal structure [60]. Intracellularly, cytosolic proteins such as synaptotagmin (Syt), protein 4.1 from brain (4.1 m), CASK, Mint and Veli bind to the disordered carboxy-terminal domain of neurexins. LNS domains, green (numbered 1 to 6); EGF-like domains, yellow; splice inserts at splice sites #1 to #5, red. EGF, epidermal growth factor-like; LNS, laminin-neurexin-sex hormone binding globulin.
Finally, the conservation of the splice insert sequence in SS#4 is in accordance with the conformational switch [54] that (i) increases affinity for Ca2+ binding by positioning an additional Ca2+ coordinating residue [57], and (ii) requires a match to the sequence of β10 that is replaced by the SS#4 insert. However, the reason for the conservation may be different: since the insert sequence at SS#4 itself binds exclusively to cerebellin [23, 24] and cerebellin constitutes an ancestral protein, it can be hypothesized that the interaction of neurexins + SS#4 with cerebellin may be responsible for the evolutionary pressure on the splice insert conservation, rather than the interaction of neurexin with neuroligins that is reduced by the alternative splicing at SS#4.
Localization and function
The discovery of neurexins as a receptor for α-latrotoxin [3], a neurotoxin that causes massive neurotransmitter vesicle release from terminals, has argued in favor of a presynaptic localization. This location has been confirmed by the finding of a prominent presynaptic release phenotype in α-neurexin knockout (KO) mice [6, 65]. Nevertheless, additional postsynaptic defects and localization of transgenically expressed variants may indicate that a small population of postsynaptic neurexins exists [5, 66]. Due to the lack of isoform-specific antibodies for high-resolution morphology, endogenous neurexin proteins have not been mapped systematically to subpopulations of neurons and/or synapses by immunolabeling. Localization patterns have been obtained mostly from mRNA studies [1, 67–69] and by subcellular fractionation [65, 69]. In situ hybridization data reveal that neurexins 1/2 and neurexin 3 may be expressed initially in distinct cell populations [67], whereas in the mature central nervous system the α-neurexin and β-neurexin isoforms are distributed in a partially overlapping, partially differential pattern [1, 67]. In particular, the three β-isoforms show a more unique distribution, in which, for example, neurexin 1β is restricted to cortical layers 2 and 3, thalamus and parts of the hippocampus [1, 67]. Using the regulation by alternative splicing, juvenile neurons in chicken express insert-negative neurexin variants [68]. With progressing neuronal and synaptic development, the number of insert-positive variants increases [68]. Since insert-negative neurexins have the highest potential to bind to known interaction partners (Table 2), these data suggest that maturation is accompanied by reduced binding capacities for neuroligins, LRRTM and dystroglycan. Instead, insert-positive variants at SS#4 favor the binding to cerebellin [24, 70]. Interestingly, in the cerebellum where the cerebellin/GluRδ2 complex is abundantly expressed [24], much higher levels of neurexins lacking all inserts have been found compared with the rest of the brain [1]. These results are consistent with an activity-controlled expression of neurexin + SS#4 and, thereby, a regulated interaction with cerebellin/GluRδ2. Supporting this idea of an activity-dependent ‘splice-code’ that changes the profile of neurexins for binding partners, the generation of different splice variants was shown to be coupled to synaptic activity via the Ca2+/calmodulin-dependent kinase pathway and involves RNA-binding protein SAM68 [71, 72]. For example, it has been shown that the inclusion of a splice insert at SS#3 in neurexin 2 depends on depolarization and Ca2+ influx [73]. Furthermore, the expression of + SS#3/+SS#4-containing variants follows closely the activity rhythm in autonomous oscillating cells of the suprachiasmatic nucleus [71], and + SS#4 expression is reduced in α-neurexin isoforms after applying a learning and memory paradigm [74]. Unfortunately, expression results from different species and different experimental paradigms are sometimes contradictory [68, 75], suggesting that more research is needed to establish the regulated variability of splice variants and to determine which variants are actually realized under which conditions.
Mouse models
KO studies in mice established the importance of α-neurexins as essential because they are required for Ca2+-dependent exocytosis at neuronal synapses [4–7, 11, 37]. For β-neurexins, in contrast, no results from KO studies have been published yet.
The deletion of two or three α-neurexin isoforms resulted in severely impaired spontaneous and evoked neurotransmitter release at excitatory and inhibitory synapses in brainstem and neocortex [5, 6]. Even the deletion of a single isoform, neurexin 1α, resulted in a reduction of spontaneous release from excitatory synapses in hippocampal pyramidal neurons [4], emphasizing the importance of every neurexin for synaptic homeostasis [52]. In addition, the loss of one or more α-neurexin isoforms reduced Ca2+ currents and caused unresponsiveness to specific blockers [6], suggesting that an impaired Ca2+-channel function is part of the process. It remains unclear, however, how the deletion of α-neurexins uncouples N-type and P/Q-type Ca2+ channels from the neurotransmitter release machinery [37, 76]. A direct interaction of the extracellular domains of α-neurexins and the pore-forming subunits of the Ca2+ channels appears unlikely as neurexins are not required for normal Ca2+ currents per se[76], and the surface expression and number of Ca2+ channels were also unchanged in KO neurons [6].
Any mechanistic explanation of the effect of α-neurexins on Ca2+ channels also needs to consider the observation that the carboxyl terminus binds to PDZ-domain proteins such as CASK [77] and Mints [78]. Both, CASK and Mints interact with the β-subunit of N-type Ca2+ channels, while Mints also interact with P/Q-type Ca2+ channels [79]. This complex, in turn, could be coupled to synaptic vesicles by the interaction of α-neurexin with synaptotagmin [80] and/or Mints to Munc18 [78]. Although this molecular pathway provides a possible link between neurexins, Ca2+ channels and the release machinery, the comparatively moderate effect of genetic deletion of CASK and Mint on synaptic transmission [81, 82] does not support a crucial contribution of these molecules. More work needs to be done to integrate α-neurexins into the current view of Ca2+-channel tethering or positioning by synaptotagmins, RIMs, liprins and CAST/ERC/ELKS, which also appears independent of Mint or CASK [83]. In addition, recent advances on the function of Ca2+-channel α2δ subunits as important modulators of synaptic transmission [84] suggest alternative routes to influence Ca2+-channel activity and mobility [85]. This includes the possibility, albeit speculative, of direct or indirect interference with extracellular domains of α-neurexins that could explain why β-neurexins do not rescue the α-neurexin KO phenotype [37].
Neurexins and neuroligins induce synaptic specializations
Studies using co-cultures between primary neurons and non-neuronal cells transfected with neurexins or neuroligins have uncovered their ability to stimulate the de novo formation of functional synapses by clustering presynaptic or postsynaptic proteins [12, 14]. Surface expression of neurexins induces clusters of PSD95 and gephyrin at excitatory and inhibitory postsynapses of contacting dendrites [10, 13]. Expression of neuroligins, in turn, induces clustering of presynaptic marker proteins on contacting axons [10] and different neuroligin isoforms appear to trigger differentiation of excitatory versus inhibitory terminals [9, 53, 86]. Interestingly, this strong synaptogenic effect of overexpressed neurexins and neuroligins observed in these cell culture assays has not been matched by prominently reduced numbers of excitatory and inhibitory synapses in loss-of-function mouse models [6, 11, 87, 88]. For example, the multiple KO of α-neurexins leads to a moderate reduction of symmetric, presumably inhibitory, synapses and leaves excitatory synapse density unscathed that at the same time displays a severely impaired neurotransmitter release [5, 6, 11]. For neuroligins that have served as the prototypical synaptogenic molecule in vitro[14], there are no visible effects on synapse numbers in multiple or single KO mice [87, 88]. Overexpression versus deletion strategies cannot be the sole reason for these differences because lentiviral-mediated expression of neurexins has failed to elevate synapse numbers [8] and transgenic overexpression of neurexin in mice does not increase mini frequencies above wild-type levels [37]. Since RNAi-mediated knock-down of neurexins, in turn, can lower the numbers of excitatory and inhibitory synapses in cultured neurons [86], it is clear that more research is needed to define the role of the neurexin/neuroligin complex in synapse formation.
Synapse formation assays have also been used to decipher the putative splice code for preferred binding between neurexins and neuroligins, and to other partners. Most studies using neurexins have been performed with overexpressed β-neurexin variants that represent the best binding partner for all neuroligin isoforms regardless of alternative splice inserts in either protein [15, 46, 54, 89], as also discussed above (Structural features and the splice-code hypothesis). Accordingly, β-neurexin instantly reaches the maximal synaptogenetic effect [90], and optimizing binding to neuroligin by deglycosylation or removal of the B insert does not significantly increase clustering of synaptic proteins [89]. In contrast, only a few cell culture studies have been performed with α-neurexins [12, 75, 89]. These were limited to α-neurexin + SS#4 variants that bind reliably only to neuroligins without insert B [15] but do not reach the complex forming capacity of β-neurexin + SS#4 to neuroligin 1-B [89]. Since neuroligin 1-B was shown to cluster and bind α-neurexins, it is not surprising that most synaptogenic effects of overexpressed α-neurexins have been observed at inhibitory synapses [12, 75]. This is because inhibitory synapses contain mostly neuroligin 2 [12, 91], which has similar biochemical binding properties to neuroligin 1-B [54]. As α-neurexins look more diffusibly distributed on the axonal surface [92] but are clustered by neuroligin 2/neuroligin 1-B [89], it can be hypothesized that α-neurexins are the more potent variants for dynamic adaptations that may be particularly relevant for inhibitory synapses.
Neurexins and psychiatric diseases
The observation that neuroligin 1 is more abundant at excitatory and neuroligin 2 at inhibitory synapses has led to the hypothesis that β-neurexin/neuroligin 1 + B and α-neurexin/neuroligin 2 are molecular determinants of the excitatory (E) and inhibitory (I) synaptic input, respectively (Figure 7). While the role of α-neurexins is not restricted to inhibitory synapses [5, 6] and β-neurexins may also affect inhibitory transmission [8], it appears that GABAergic transmission plays a particularly important role in the so-called excitatory/inhibitory balance (E/I balance) at synapses (for example, [52, 93, 94]). It has become widely accepted that impairments in neurexins and neuroligins caused by mutations may disturb the balance between excitatory and inhibitory activity that is thought to be critical for the pathomechanisms in autism spectrum disorders (ASDs) and schizophrenia [25, 26, 95].

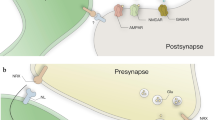
Trans-synaptic neurexin-neuroligin complexes shape excitatory and inhibitory synapses. Presynaptic α-neurexins or β-neurexins (red) can interact with dimeric neuroligins (green) across the synaptic cleft to regulate important aspects of establishment, differentiation and maturation of synapses. Isoforms and splice variants of both molecules have been proposed to be differentially distributed at excitatory or inhibitory synapses to establish specificity. Note that presence of β-neurexins (β-Nrxn) at inhibitory terminals is unclear, while for neuroligins (Nlgn), Nlgn2 and Nlgn4 show quite specific localization and roles at inhibitory synapses. Intracellularly, the cytosolic domains of Nrxn and Nlgn are able to cluster components of the presynaptic release machinery and of postsynaptic signaling pathways and transmitter receptors (R). The clustering ability of Nrxn and Nlgn variants at excitatory or inhibitory synapses is mostly derived from cell culture assays. AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; GABA, γ-aminobutyric acid; NMDAR, N-methyl-d-aspartate receptor; PSD95, postsynaptic density protein-95; VGat, vesicular GABA transporter; VGlu, vesicular glutamate transporter.
The outcome of the autism genome-wide association study projects surprisingly revealed only weak correlations for ASD to common genetic variants, but identified genes with rare single nucleotide polymorphisms (SNPs) or copy number variations that have a considerable impact [96]. Such rare mutations have been found in the α-neurexin coding region of nrxn1[97–99], nrxn3[100] and the signal peptide of β-neurexins [101]. An excess of mutations in these genes is found in patients with ASD [27, 102], schizophrenia [103, 104] and substance abuse and impulsive behavior [105]. Historically, the neuroligin 3 single mutation R451C has been the first SNP of a protein gene associated with ASD [106] but other molecules such as nrxn1, nrxn3, nlgn3, nlgn4, shank2, shank3 and genomic regions at 1q21.1 and 16p11.2 are now accepted as bona fide ASD risk loci [100]. Some of the single site mutations found in patients have been introduced in mouse models, such as neuroligin 3 R451C [93, 107] and neuroligin 4 R704C [108]. Interestingly, analysis of mutations in mice also demonstrates converging phenotypes of different risk loci [109]. As might be expected, the mouse models recapitulate some but not all aspects of the diseases: for example, repetitive grooming as stereotype behavior in neurexin 1α KO, but not the social disabilities [4]. When tested in cell culture or biochemical assays, most mutations cause a complete loss of expression or largely reduced trafficking of the defective protein to synapses [109–111]. These observations highlight the central role of neurexins and neuroligins at the synapse and have prompted new research into the protein interaction network across the synaptic cleft that may provide insights into higher cognitive functions at the molecular level.
Neurexins in C. elegans and D. melanogaster
Invertebrate models have already proven excellent systems to study multiple mutations in neurexin and neuroligin genes that are impossible to obtain in mice [112] or to follow effects on synaptic cell adhesion by imaging in live animals [113]. Due to the sequence conservation of neurexin and neuroligin throughout the animal kingdom, identification of mutations and binding partners in one species facilitates the finding of orthologs, and allows the description of a canonical protein network. For example, binding to neuroligin is blocked in all species investigated by a synthetic aspartate to alanine mutation in the neurexin αLNS6 domain that corresponds to the essential Ca2+-binding residue D137 of β-neurexin [41, 46, 114]. In addition, mutations Y189H, L319SSM and L849Q, which inhibit neuroligin function in Drosophila[115], can be readily localized on the mammalian neuroligin crystal structure [41] and are likely to destabilize the fold of the extracellular (Y85, L235) or the transmembrane domain (L712). This could explain the reduced level of neuroligin reaching the postsynapse [115], similar to other ASD mutations in mammals [97–101]. Finally, the fact that a synthetic D356R mutation in Drosophila neuroligin 1 rescues the KO phenotype [115] suggests neurexin-independent functions of neuroligin, as the corresponding mutation D271R in rat neuroligin 1 was found to inhibit neurexin binding [46].
Unlike these structural similarities, any functional comparisons need to keep in mind that mostly presynaptic α-neurexins interact with postsynaptic neuroligin in vertebrates, as discussed above. In C. elegans, in contrast, neurexin and also neuroligin are expressed presynaptically and postsynaptically [33, 113] and retrograde trans-synaptic signaling from the postsynapse to the presynapse in the worm is modulated by an interaction in trans and cis simultaneously [116]. It is also important to realize that while C. elegans expresses a β-neurexin with a yet unresolved function [113], flies rely on a single α-neurexin alone [35, 117]. It is therefore not surprising that the functional phenotypes in vertebrate and invertebrate neurexin mutant animals share similarities but can also differ considerably (reviewed in detail in [118]). For example, analyses of Drosophila loss-of-function mutants of α-neurexins have described effects on synapse ultrastructure [35, 117] that are absent from the mouse KOs [6, 11], whereas both model systems suffer from impaired neurotransmission. These limitations notwithstanding, the recent finding of a triple complex of α-neurexin/syd-1/liprin-α at the active zone of neuromuscular junctions in flies [119], for another example, will encourage the search for a similar complex in mammals that might help to solve the question why and how α-neurexins couple Ca2+ channels to release sites.
Non-neuronal functions of neurexins
In addition to synapses of the central nervous system, neurexin isoforms have been reported to act in smooth muscle cells [116, 120, 121], pancreatic β-islet cells [122–124], melanotrophs of the hypophysis [76] and endothelial cells [125]. For example, α-neurexins and neuroligins modulate Ca2+-triggered exocytosis from melanotrophs in the hypophysis [76] and from insulin-secreting β cells in the endocrine pancreas’s islets of Langerhans [124]. In β cells, the cytosolic domain of α-neurexins is essential for insulin granule docking through an indirect interaction with granuphilin, which lines vesicles to the cell surface membrane that are ready for fusion [122]. In this process, the number of release-ready vesicles is homeostatically regulated by neurexin or granuphilin, while the reduction of either protein increases glucose-sensitive fusion. Interestingly, granuphilin is selectively expressed in β cells and melanotrophs, which might explain why α-neurexins function in both cell types. Since the granuphilin homolog Rab3A plays a similar role in the docking of synaptic vesicles in neurons, canonical protein complexes consisting of α-neurexins-CASK-Mint1/2-Rab3a/Granuphilin-Munc18 have been suggested [122].
Frontiers
The neurexin/neuroligin pair most likely represents one of the best characterized protein complexes at the neuronal synapse. Its modulation due to alternative splicing and isoform pairings is remarkable and its roles in synaptic function and differentiation are essential. However, important issues remain to be addressed.
First, it is incompletely understood if α-neurexins and β-neurexins have overlapping [126] or different functions at the synapse. Rescue experiments have suggested that their functions are non-redundant [37], but analysis of multiple β-neurexin KOs and comparative knock-down studies will be necessary to address this issue directly.
Second, the apparent preference of α-neurexins for GABAergic synapses as observed in some assays [10, 12, 13, 75] needs to be reconciled with the KO mouse phenotype that is characterized by a dramatic release impairment that affects both excitatory and inhibitory synapses [4, 6].
Third, neurexins act at the synapse but only little is known about how they are transported to the presynaptic terminal during intracellular trafficking. It has been shown that neurexin targeting requires a PDZ-binding motif interaction in mouse neurons [38] and a Syd-1/RhoGAP100F-dependent delivery in Drosophila[119]. However, the characteristics of the vesicular pathways responsible and the dynamics of the transport are unclear.
Fourth, most known interacting proteins of neurexins bind to the last LNS domain of α-neurexin/the single LNS domain of β-neurexin, and only neurexophilin and dystroglycan are known to bind to αLNS2 (Table 2). It needs to be studied if the additional domains in α-neurexin simply act as spacers or if they provide additional sites for binding partners that have yet to be discovered.
Fifth, the early expression and the preference of juvenile neurons for neurexins without splice inserts [67, 68] suggest an additional role of some neurexin variants in developmental processes such as neurite growth [11, 127] that needs to be explored in more detail.
Finally, human genetic work and mouse models have linked the neurexin/neuroligin complex to synapse-related neuropsychiatric disorders such as autism and schizophrenia [25]. It will be one of the most challenging tasks ahead of us to unravel the underlying cellular mechanisms that explain, for example, why mutations in the same molecules lead to diverse symptoms, a prerequisite to develop more causative therapeutic strategies.
Abbreviations
- ASD:
-
Autism spectrum disorder
- ConA:
-
Concanavalin A
- EGF:
-
Epidermal growth factor-like
- LNS:
-
Laminin-neurexin-sex hormone binding globulin
- RNAi:
-
RNA interference
- SNP:
-
Single nucleotide polymorphism.
References
Ullrich B, Ushkaryov YA, Südhof TC: Cartography of neurexins: more than 1000 isoforms generated by alternative splicing and expressed in distinct subsets of neurons. Neuron. 1995, 14: 497-507.
Ushkaryov YA, Hata Y, Ichtchenko K, Moomaw C, Afendis S, Slaughter CA, Südhof TC: Conserved domain structure of beta-neurexins. Unusual cleaved signal sequences in receptor-like neuronal cell-surface proteins. J Biol Chem. 1994, 269: 11987-11992.
Ushkaryov YA, Petrenko AG, Geppert M, Südhof TC: Neurexins: synaptic cell surface proteins related to the alpha-latrotoxin receptor and laminin. Science. 1992, 257: 50-56.
Etherton MR, Blaiss CA, Powell CM, Südhof TC: Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci U S A. 2009, 106: 17998-18003.
Kattenstroth G, Tantalaki E, Südhof TC, Gottmann K, Missler M: Postsynaptic N-methyl-d-aspartate receptor function requires alpha-neurexins. Proc Natl Acad Sci U S A. 2004, 101: 2607-2612.
Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Südhof TC: Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature. 2003, 423: 939-948.
Sons MS, Busche N, Strenzke N, Moser T, Ernsberger U, Mooren FC, Zhang W, Ahmad M, Steffens H, Schomburg ED, Plomp JJ, Missler M: alpha-Neurexins are required for efficient transmitter release and synaptic homeostasis at the mouse neuromuscular junction. Neuroscience. 2006, 138: 433-446.
Zhang C, Atasoy D, Arac D, Yang X, Fuccillo MV, Robison AJ, Ko J, Brunger AT, Südhof TC: Neurexins physically and functionally interact with GABA(A) receptors. Neuron. 2010, 66: 403-416.
Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, Südhof TC: Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. 2007, 54: 919-931.
Dean C, Scholl FG, Choih J, DeMaria S, Berger J, Isacoff E, Scheiffele P: Neurexin mediates the assembly of presynaptic terminals. Nat Neurosci. 2003, 6: 708-716.
Dudanova I, Tabuchi K, Rohlmann A, Südhof TC, Missler M: Deletion of alpha-neurexins does not cause a major impairment of axonal pathfinding or synapse formation. J Comp Neurol. 2007, 502: 261-274.
Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM: Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004, 119: 1013-1026.
Nam CI, Chen L: Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc Natl Acad Sci U S A. 2005, 102: 6137-6142.
Scheiffele P, Fan J, Choih J, Fetter R, Serafini T: Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000, 101: 657-669.
Boucard AA, Chubykin AA, Comoletti D, Taylor P, Südhof TC: A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron. 2005, 48: 229-236.
Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Südhof TC: Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell. 1995, 81: 435-443.
Missler M, Hammer RE, Südhof TC: Neurexophilin binding to alpha-neurexins. A single LNS domain functions as an independently folding ligand-binding unit. J Biol Chem. 1998, 273: 34716-34723.
Missler M, Südhof TC: Neurexophilins form a conserved family of neuropeptide-like glycoproteins. J Neurosci. 1998, 18: 3630-3638.
Petrenko AG, Ullrich B, Missler M, Krasnoperov V, Rosahl TW, Südhof TC: Structure and evolution of neurexophilin. J Neurosci. 1996, 16: 4360-4369.
Sugita S, Saito F, Tang J, Satz J, Campbell K, Südhof TC: A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol. 2001, 154: 435-445.
de Wit J, Sylwestrak E, O’Sullivan ML, Otto S, Tiglio K, Savas JN, Yates JR, Comoletti D, Taylor P, Ghosh A: LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron. 2009, 64: 799-806.
Ko J, Fuccillo MV, Malenka RC, Südhof TC: LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron. 2009, 64: 791-798.
Matsuda K, Yuzaki M: Cbln family proteins promote synapse formation by regulating distinct neurexin signaling pathways in various brain regions. Eur J Neurosci. 2011, 33: 1447-1461.
Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, Taguchi R, Sakimura K, Mishina M: Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell. 2010, 141: 1068-1079.
Südhof TC: Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008, 455: 903-911.
Bourgeron T: A synaptic trek to autism. Curr Opin Neurobiol. 2009, 19: 231-234.
Reichelt AC, Rodgers RJ, Clapcote SJ: The role of neurexins in schizophrenia and autistic spectrum disorder. Neuropharmacology. 2012, 62: 1519-1526.
Ushkaryov YA, Südhof TC: Neurexin III alpha: extensive alternative splicing generates membrane-bound and soluble forms. Proc Natl Acad Sci U S A. 1993, 90: 6410-6414.
Missler M, Südhof TC: Neurexins: three genes and 1001 products. Trends Genet. 1998, 14: 20-26.
Tabuchi K, Südhof TC: Structure and evolution of neurexin genes: insight into the mechanism of alternative splicing. Genomics. 2002, 79: 849-859.
Rowen L, Young J, Birditt B, Kaur A, Madan A, Philipps DL, Qin S, Minx P, Wilson RK, Hood L, Graveley BR: Analysis of the human neurexin genes: alternative splicing and the generation of protein diversity. Genomics. 2002, 79: 587-597.
Biswas S, Russell RJ, Jackson CJ, Vidovic M, Ganeshina O, Oakeshott JG, Claudianos C: Bridging the synaptic gap: neuroligins and neurexin I in Apis mellifera. PLoS One. 2008, 3: e3542-
Haklai-Topper L, Soutschek J, Sabanay H, Scheel J, Hobert O, Peles E: The neurexin superfamily of Caenorhabditis elegans. Gene Expr Patterns. 2011, 11: 144-150.
Rissone A, Monopoli M, Beltrame M, Bussolino F, Cotelli F, Arese M: Comparative genome analysis of the neurexin gene family in Danio rerio: insights into their functions and evolution. Mol Biol Evol. 2007, 24: 236-252.
Zeng X, Sun M, Liu L, Chen F, Wei L, Xie W: Neurexin-1 is required for synapse formation and larvae associative learning in Drosophila. FEBS Lett. 2007, 581: 2509-2516.
Keren H, Lev-Maor G, Ast G: Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet. 2010, 11: 345-355.
Zhang W, Rohlmann A, Sargsyan V, Aramuni G, Hammer RE, Südhof TC, Missler M: Extracellular domains of alpha-neurexins participate in regulating synaptic transmission by selectively affecting N- and P/Q-type Ca2+ channels. J Neurosci. 2005, 25: 4330-4342.
Fairless R, Masius H, Rohlmann A, Heupel K, Ahmad M, Reissner C, Dresbach T, Missler M: Polarized targeting of neurexins to synapses is regulated by their C-terminal sequences. J Neurosci. 2008, 28: 12969-12981.
Rudenko G, Hohenester E, Muller YA: LG/LNS domains: multiple functions - one business end?. Trends Biochem Sci. 2001, 26: 363-368.
Arac D, Boucard AA, Ozkan E, Strop P, Newell E, Südhof TC, Brunger AT: Structures of neuroligin-1 and the neuroligin-1/neurexin-1 beta complex reveal specific protein-protein and protein-Ca2+ interactions. Neuron. 2007, 56: 992-1003.
Chen X, Liu H, Shim AH, Focia PJ, He X: Structural basis for synaptic adhesion mediated by neuroligin-neurexin interactions. Nat Struct Mol Biol. 2008, 15: 50-56.
Fabrichny IP, Leone P, Sulzenbacher G, Comoletti D, Miller MT, Taylor P, Bourne Y, Marchot P: Structural analysis of the synaptic protein neuroligin and its beta-neurexin complex: determinants for folding and cell adhesion. Neuron. 2007, 56: 979-991.
Rudenko G, Nguyen T, Chelliah Y, Südhof TC, Deisenhofer J: The structure of the ligand-binding domain of neurexin Ibeta: regulation of LNS domain function by alternative splicing. Cell. 1999, 99: 93-101.
Wizemann H, Garbe JH, Friedrich MV, Timpl R, Sasaki T, Hohenester E: Distinct requirements for heparin and alpha-dystroglycan binding revealed by structure-based mutagenesis of the laminin alpha2 LG4-LG5 domain pair. J Mol Biol. 2003, 332: 635-642.
Yoshida-Moriguchi T, Yu L, Stalnaker SH, Davis S, Kunz S, Madson M, Oldstone MB, Schachter H, Wells L, Campbell KP: O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 2010, 327: 88-92.
Reissner C, Klose M, Fairless R, Missler M: Mutational analysis of the neurexin/neuroligin complex reveals essential and regulatory components. Proc Natl Acad Sci U S A. 2008, 105: 15124-15129.
Striegel AR, Biela LM, Evans CS, Wang Z, Delehoy JB, Sutton RB, Chapman ER, Reist NE: Calcium binding by synaptotagmin’s C2A domain is an essential element of the electrostatic switch that triggers synchronous synaptic transmission. J Neurosci. 2012, 32: 1253-1260.
Siddiqui TJ, Pancaroglu R, Kang Y, Rooyakkers A, Craig AM: LRRTMs and neuroligins bind neurexins with a differential code to cooperate in glutamate synapse development. J Neurosci. 2010, 30: 7495-7506.
Ichtchenko K, Nguyen T, Südhof TC: Structures, alternative splicing, and neurexin binding of multiple neuroligins. J Biol Chem. 1996, 271: 2676-2682.
Bolliger MF, Frei K, Winterhalter KH, Gloor SM: Identification of a novel neuroligin in humans which binds to PSD-95 and has a widespread expression. Biochem J. 2001, 356: 581-588.
Koehnke J, Jin X, Budreck EC, Posy S, Scheiffele P, Honig B, Shapiro L: Crystal structure of the extracellular cholinesterase-like domain from neuroligin-2. Proc Natl Acad Sci U S A. 2008, 105: 1873-1878.
Hussain NK, Sheng M: Neuroscience. Making synapses: a balancing act. Science. 2005, 307: 1207-1208.
Chih B, Gollan L, Scheiffele P: Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron. 2006, 51: 171-178.
Koehnke J, Katsamba PS, Ahlsen G, Bahna F, Vendome J, Honig B, Shapiro L, Jin X: Splice form dependence of beta-neurexin/neuroligin binding interactions. Neuron. 2010, 67: 61-74.
Miller MT, Mileni M, Comoletti D, Stevens RC, Harel M, Taylor P: The crystal structure of the alpha-neurexin-1 extracellular region reveals a hinge point for mediating synaptic adhesion and function. Structure. 2011, 19: 767-778.
Leone P, Comoletti D, Ferracci G, Conrod S, Garcia SU, Taylor P, Bourne Y, Marchot P: Structural insights into the exquisite selectivity of neurexin/neuroligin synaptic interactions. EMBO J. 2010, 29: 2461-2471.
Shen KC, Kuczynska DA, Wu IJ, Murray BH, Sheckler LR, Rudenko G: Regulation of neurexin 1beta tertiary structure and ligand binding through alternative splicing. Structure. 2008, 16: 422-431.
Comoletti D, Flynn RE, Boucard AA, Demeler B, Schirf V, Shi J, Jennings LL, Newlin HR, Südhof TC, Taylor P: Gene selection, alternative splicing, and post-translational processing regulate neuroligin selectivity for beta-neurexins. Biochemistry (Mosc). 2006, 45: 12816-12827.
Tanaka H, Nogi T, Yasui N, Iwasaki K, Takagi J: Structural basis for variant-specific neuroligin-binding by alpha-neurexin. PLoS ONE. 2011, 6: e19411-
Chen F, Venugopal V, Murray B, Rudenko G: The structure of neurexin 1alpha reveals features promoting a role as synaptic organizer. Structure. 2011, 19: 779-789.
Koehnke J, Jin X, Trbovic N, Katsamba PS, Brasch J, Ahlsen G, Scheiffele P, Honig B, Palmer AG, Shapiro L: Crystal structures of beta-neurexin 1 and beta-neurexin 2 ectodomains and dynamics of splice insertion sequence 4. Structure. 2008, 16: 410-421.
Comoletti D, Miller MT, Jeffries CM, Wilson J, Demeler B, Taylor P, Trewhella J, Nakagawa T: The macromolecular architecture of extracellular domain of alphaNRXN1: domain organization, flexibility, and insights into trans-synaptic disposition. Structure. 2010, 18: 1044-1053.
Reissner C, Missler M: Unveiled alpha-neurexins take center stage. Structure. 2011, 19: 749-750.
Tanaka H, Miyazaki N, Matoba K, Nogi T, Iwasaki K, Takagi J: Higher-order architecture of cell adhesion mediated by polymorphic synaptic adhesion molecules neurexin and neuroligin. Cell Rep. 2012, 2: 101-110.
Geppert M, Khvotchev M, Krasnoperov V, Goda Y, Missler M, Hammer RE, Ichtchenko K, Petrenko AG, Südhof TC: Neurexin I alpha is a major alpha-latrotoxin receptor that cooperates in alpha-latrotoxin action. J Biol Chem. 1998, 273: 1705-1710.
Taniguchi H, Gollan L, Scholl FG, Mahadomrongkul V, Dobler E, Limthong N, Peck M, Aoki C, Scheiffele P: Silencing of neuroligin function by postsynaptic neurexins. J Neurosci. 2007, 27: 2815-2824.
Püschel AW, Betz H: Neurexins are differentially expressed in the embryonic nervous system of mice. J Neurosci. 1995, 15: 2849-2856.
Patzke H, Ernsberger U: Expression of neurexin Ialpha splice variants in sympathetic neurons: selective changes during differentiation and in response to neurotrophins. Mol Cell Neurosci. 2000, 15: 561-572.
Berninghausen O, Rahman MA, Silva JP, Davletov B, Hopkins C, Ushkaryov YA: Neurexin Ibeta and neuroligin are localized on opposite membranes in mature central synapses. J Neurochem. 2007, 103: 1855-1863.
Yasumura M, Yoshida T, Lee SJ, Uemura T, Joo JY, Mishina M: Glutamate receptor delta1 induces preferentially inhibitory presynaptic differentiation of cortical neurons by interacting with neurexins through cerebellin precursor protein subtypes. J Neurochem. 2012, 121: 705-716.
Shapiro-Reznik M, Jilg A, Lerner H, Earnest DJ, Zisapel N: Diurnal rhythms in neurexins transcripts and inhibitory/excitatory synapse scaffold proteins in the biological clock. PLoS One. 2012, 7: e37894-
Iijima T, Wu K, Witte H, Hanno-Iijima Y, Glatter T, Richard S, Scheiffele P: SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell. 2011, 147: 1601-1614.
Rozic-Kotliroff G, Zisapel N: Ca2+-dependent splicing of neurexin IIalpha. Biochem Biophys Res Commun. 2007, 352: 226-230.
Rozic G, Lupowitz Z, Piontkewitz Y, Zisapel N: Dynamic changes in neurexins’ alternative splicing: role of Rho-associated protein kinases and relevance to memory formation. PLoS One. 2011, 6: e18579-
Kang Y, Zhang X, Dobie F, Wu H, Craig AM: Induction of GABAergic postsynaptic differentiation by alpha-neurexins. J Biol Chem. 2008, 283: 2323-2334.
Dudanova I, Sedej S, Ahmad M, Masius H, Sargsyan V, Zhang W, Riedel D, Angenstein F, Schild D, Rupnik M, Missler M: Important contribution of alpha-neurexins to Ca2+-triggered exocytosis of secretory granules. J Neurosci. 2006, 26: 10599-10613.
Hata Y, Butz S, Südhof TC: CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J Neurosci. 1996, 16: 2488-2494.
Biederer T, Südhof TC: Mints as adaptors. Direct binding to neurexins and recruitment of munc18. J Biol Chem. 2000, 275: 39803-39806.
Maximov A, Südhof TC, Bezprozvanny I: Association of neuronal calcium channels with modular adaptor proteins. J Biol Chem. 1999, 274: 24453-24456.
O'Connor VM, Shamotienko O, Grishin E, Betz H: On the structure of the ‘synaptosecretosome’. Evidence for a neurexin/synaptotagmin/syntaxin/Ca2+ channel complex. FEBS Lett. 1993, 326: 255-260.
Atasoy D, Schoch S, Ho A, Nadasy KA, Liu X, Zhang W, Mukherjee K, Nosyreva ED, Fernandez-Chacon R, Missler M, Kavalali ET, Südhof TC: Deletion of CASK in mice is lethal and impairs synaptic function. Proc Natl Acad Sci U S A. 2007, 104: 2525-2530.
Ho A, Liu X, Südhof TC: Deletion of Mint proteins decreases amyloid production in transgenic mouse models of Alzheimer’s disease. J Neurosci. 2008, 28: 14392-14400.
Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Südhof TC: RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell. 2011, 144: 282-295.
Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA: alpha2delta expression sets presynaptic calcium channel abundance and release probability. Nature. 2012, 486: 122-125.
Di Biase V, Tuluc P, Campiglio M, Obermair GJ, Heine M, Flucher BE: Surface traffic of dendritic CaV1.2 calcium channels in hippocampal neurons. J Neurosci. 2011, 31: 13682-13694.
Chih B, Engelman H, Scheiffele P: Control of excitatory and inhibitory synapse formation by neuroligins. Science. 2005, 307: 1324-1328.
Poulopoulos A, Soykan T, Tuffy LP, Hammer M, Varoqueaux F, Brose N: Homodimerization and isoform-specific heterodimerization of neuroligins. Biochem J. 2012, 446: 321-330.
Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Südhof TC, Brose N: Neuroligins determine synapse maturation and function. Neuron. 2006, 51: 741-754.
Lee H, Dean C, Isacoff E: Alternative splicing of neuroligin regulates the rate of presynaptic differentiation. J Neurosci. 2010, 30: 11435-11446.
Futai K, Kim MJ, Hashikawa T, Scheiffele P, Sheng M, Hayashi Y: Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95-neuroligin. Nat Neurosci. 2007, 10: 186-195.
Varoqueaux F, Jamain S, Brose N: Neuroligin 2 is exclusively localized to inhibitory synapses. Eur J Cell Biol. 2004, 83: 449-456.
Fu Y, Huang ZJ: Differential dynamics and activity-dependent regulation of alpha- and beta-neurexins at developing GABAergic synapses. Proc Natl Acad Sci U S A. 2010, 107: 22699-22704.
Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Südhof TC: A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007, 318: 71-76.
Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY: Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010, 468: 263-269.
Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K: Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011, 477: 171-178.
Anney R, Klei L, Pinto D, Almeida J, Bacchelli E, Baird G, Bolshakova N, Bolte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Casey J, Conroy J, Correia C, Corsello C, Crawford EL, de Jonge M, Delorme R, Duketis E, Duque F, Estes A, Farrar P, Fernandez BA, Folstein SE, Fombonne E, Gilbert J, Gillberg C, Glessner JT, Green A, et al: Individual common variants exert weak effects on the risk for autism spectrum disorderspi. Hum Mol Genet. 2012, 21: 4781-4792.
Kim HG, Kishikawa S, Higgins AW, Seong IS, Donovan DJ, Shen Y, Lally E, Weiss LA, Najm J, Kutsche K, Descartes M, Holt L, Braddock S, Troxell R, Kaplan L, Volkmar F, Klin A, Tsatsanis K, Harris DJ, Noens I, Pauls DL, Daly MJ, MacDonald ME, Morton CC, Quade BJ, Gusella JF: Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet. 2008, 82: 199-207.
Kirov G, Rujescu D, Ingason A, Collier DA, O’Donovan MC, Owen MJ: Neurexin 1 (NRXN1) deletions in schizophrenia. Schizophr Bull. 2009, 35: 851-854.
Ching MS, Shen Y, Tan WH, Jeste SS, Morrow EM, Chen X, Mukaddes NM, Yoo SY, Hanson E, Hundley R, Austin C, Becker RE, Berry GT, Driscoll K, Engle EC, Friedman S, Gusella JF, Hisama FM, Irons MB, Lafiosca T, LeClair E, Miller DT, Neessen M, Picker JD, Rappaport L, Rooney CM, Sarco DP, Stoler JM, Walsh CA, Wolff RR, et al: Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am J Med Genet B Neuropsychiatr Genet. 2010, 153B: 937-947.
Vaags AK, Lionel AC, Sato D, Goodenberger M, Stein QP, Curran S, Ogilvie C, Ahn JW, Drmic I, Senman L, Chrysler C, Thompson A, Russell C, Prasad A, Walker S, Pinto D, Marshall CR, Stavropoulos DJ, Zwaigenbaum L, Fernandez BA, Fombonne E, Bolton PF, Collier DA, Hodge JC, Roberts W, Szatmari P, Scherer SW: Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am J Hum Genet. 2012, 90: 133-141.
Feng J, Schroer R, Yan J, Song W, Yang C, Bockholt A, Cook EH, Skinner C, Schwartz CE, Sommer SS: High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci Lett. 2006, 409: 10-13.
Ey E, Leblond CS, Bourgeron T: Behavioral profiles of mouse models for autism spectrum disorders. Autism Res. 2011, 4: 5-16.
Doherty JL, O’Donovan MC, Owen MJ: Recent genomic advances in schizophrenia. Clin Genet. 2012, 81: 103-109.
Levinson DF, Shi J, Wang K, Oh S, Riley B, Pulver AE, Wildenauer DB, Laurent C, Mowry BJ, Gejman PV, Owen MJ, Kendler KS, Nestadt G, Schwab SG, Mallet J, Nertney D, Sanders AR, Williams NM, Wormley B, Lasseter VK, Albus M, Godard-Bauché S, Alexander M, Duan J, O’Donovan MC, Walsh D, O’Neill A, Papadimitriou GN, Dikeos D, Maier W, et al: Genome-wide association study of multiplex schizophrenia pedigrees. Am J Psychiatry. 2012, 169: 963-973.
Stoltenberg SF, Lehmann MK, Christ CC, Hersrud SL, Davies GE: Associations among types of impulsivity, substance use problems and neurexin-3 polymorphisms. Drug Alcohol Depend. 2011, 119: e31-e38.
Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T: Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003, 34: 27-29.
Etherton M, Foldy C, Sharma M, Tabuchi K, Liu X, Shamloo M, Malenka RC, Südhof TC: Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc Natl Acad Sci U S A. 2011, 108: 13764-13769.
Etherton MR, Tabuchi K, Sharma M, Ko J, Südhof TC: An autism-associated point mutation in the neuroligin cytoplasmic tail selectively impairs AMPA receptor-mediated synaptic transmission in hippocampus. EMBO J. 2011, 30: 2908-2919.
Arons MH, Thynne CJ, Grabrucker AM, Li D, Schoen M, Cheyne JE, Boeckers TM, Montgomery JM, Garner CC: Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. J Neurosci. 2012, 32: 14966-14978.
Chih B, Afridi SK, Clark L, Scheiffele P: Disorder-associated mutations lead to functional inactivation of neuroligins. Hum Mol Genet. 2004, 13: 1471-1477.
Comoletti D, De Jaco A, Jennings LL, Flynn RE, Gaietta G, Tsigelny I, Ellisman MH, Taylor P: The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J Neurosci. 2004, 24: 4889-4893.
Sun M, Xing G, Yuan L, Gan G, Knight D, With SI, He C, Han J, Zeng X, Fang M, Boulianne GL, Xie W: Neuroligin 2 is required for synapse development and function at the Drosophila neuromuscular junction. J Neurosci. 2011, 31: 687-699.
Feinberg EH, Vanhoven MK, Bendesky A, Wang G, Fetter RD, Shen K, Bargmann CI: GFP Reconstitution Across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron. 2008, 57: 353-363.
Graf ER, Kang Y, Hauner AM, Craig AM: Structure function and splice site analysis of the synaptogenic activity of the neurexin-1 beta LNS domain. J Neurosci. 2006, 26: 4256-4265.
Banovic D, Khorramshahi O, Owald D, Wichmann C, Riedt T, Fouquet W, Tian R, Sigrist SJ, Aberle H: Drosophila neuroligin 1 promotes growth and postsynaptic differentiation at glutamatergic neuromuscular junctions. Neuron. 2010, 66: 724-738.
Hu Z, Hom S, Kudze T, Tong XJ, Choi S, Aramuni G, Zhang W, Kaplan JM: Neurexin and neuroligin mediate retrograde synaptic inhibition in C. elegans. Science. 2012, 337: 980-984.
Li C, Han D, Zhang F, Zhou C, Yu HM, Zhang GY: Preconditioning ischemia attenuates increased neurexin-neuroligin1-PSD-95 interaction after transient cerebral ischemia in rat hippocampus. Neurosci Lett. 2007, 426: 192-197.
Knight D, Xie W, Boulianne GL: Neurexins and neuroligins: recent insights from invertebrates. Mol Neurobiol. 2011, 44: 426-440.
Owald D, Khorramshahi O, Gupta VK, Banovic D, Depner H, Fouquet W, Wichmann C, Mertel S, Eimer S, Reynolds E, Holt M, Aberle H, Sigrist SJ: Cooperation of Syd-1 with Neurexin synchronizes pre- with postsynaptic assembly. Nat Neurosci. 2012, 15: 1219-1226.
Bottos A, Rissone A, Bussolino F, Arese M: Neurexins and neuroligins: synapses look out of the nervous system. Cell Mol Life Sci. 2011, 68: 2655-2666.
Occhi G, Rampazzo A, Beffagna G, Antonio Danieli G: Identification and characterization of heart-specific splicing of human neurexin 3 mRNA (NRXN3). Biochem Biophys Res Commun. 2002, 298: 151-155.
Mosedale M, Egodage S, Calma RC, Chi NW, Chessler SD: Neurexin-1alpha contributes to insulin-containing secretory granule docking. J Biol Chem. 2012, 287: 6350-6361.
Suckow AT, Comoletti D, Waldrop MA, Mosedale M, Egodage S, Taylor P, Chessler SD: Expression of neurexin, neuroligin, and their cytoplasmic binding partners in the pancreatic beta-cells and the involvement of neuroligin in insulin secretion. Endocrinology. 2008, 149: 6006-6017.
Suckow AT, Zhang C, Egodage S, Comoletti D, Taylor P, Miller MT, Sweet IR, Chessler SD: Transcellular neuroligin-2 interactions enhance insulin secretion and are integral to pancreatic beta cell function. J Biol Chem. 2012, 287: 19816-19826.
Bottos A, Destro E, Rissone A, Graziano S, Cordara G, Assenzio B, Cera MR, Mascia L, Bussolino F, Arese M: The synaptic proteins neurexins and neuroligins are widely expressed in the vascular system and contribute to its functions. Proc Natl Acad Sci U S A. 2009, 106: 20782-20787.
Aoto J, Martinelli DC, Malenka RC, Tabuchi K, Südhof TC: Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell. 2013, 154: 75-88.
Gjorlund MD, Nielsen J, Pankratova S, Li S, Korshunova I, Bock E, Berezin V: Neuroligin-1 induces neurite outgrowth through interaction with neurexin-1beta and activation of fibroblast growth factor receptor-1. FASEB J. 2012, 26: 4174-4186.
Beglopoulos V, Montag-Sallaz M, Rohlmann A, Piechotta K, Ahmad M, Montag D, Missler M: Neurexophilin 3 is highly localized in cortical and cerebellar regions and is functionally important for sensorimotor gating and motor coordination. Mol Cell Biol. 2005, 25: 7278-7288.
Owen MJ, Williams HJ, O’Donovan MC: Schizophrenia genetics: advancing on two fronts. Curr Opin Genet Dev. 2009, 19: 266-270.
Zhou H, Xu Y, Yang Y, Huang A, Wu J, Shi Y: Solution structure of AF-6 PDZ domain and its interaction with the C-terminal peptides from Neurexin and Bcr. J Biol Chem. 2005, 280: 13841-13847.
Clarris HJ, McKeown S, Key B: Expression of neurexin ligands, the neuroligins and the neurexophilins, in the developing and adult rodent olfactory bulb. Int J Dev Biol. 2002, 46: 649-652.
Brodskii LI, Ivanov VV, Kalaidzidis lL, Leontovich AM, Nikolaev VK, Feranchuk SI, Drachev VA: GeneBee-NET: an internet based server for biopolymer structure analysis. Biokhimiia. 1995, 60: 1221-1230.
GenBank. [http://ncbi.nlm.nih.gov/genbank/]
Acknowledgements
This work is supported by Deutsche Forschungsgemeinschaft grant number SFB629, TPB11 (MM).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Reissner, C., Runkel, F. & Missler, M. Neurexins. Genome Biol 14, 213 (2013). https://doi.org/10.1186/gb-2013-14-9-213
Published:
DOI: https://doi.org/10.1186/gb-2013-14-9-213